


 , Yun-Wen Zheng 1,2,3,4,*
, Yun-Wen Zheng 1,2,3,4,*1 Institute of Regenerative Medicine, and Department of Dermatology, Affiliated Hospital of Jiangsu University, Jiangsu University, 212001 Zhenjiang, Jiangsu, China
2 Guangdong Provincial Key Laboratory of Large Animal Models for Biomedicine, and South China Institute of Large Animal Models for Biomedicine, School of Biotechnology and Health Sciences, Wuyi University, 529020 Jiangmen, Guangdong, China
3 Center for Stem Cell Biology and Regenerative Medicine, Institute of Medical Science, The University of Tokyo, 108-8639 Tokyo, Japan
4 Department of Medicinal and Life Sciences, Faculty of Pharmaceutical Sciences, Tokyo University of Science, 278-8510 Noda, Japan
†These authors contributed equally.
Abstract
Establishing reliable and reproducible animal models for disease modelling, drug screening and the understanding of disease susceptibility and pathogenesis is critical. However, traditional animal models differ significantly from humans in terms of physiology, immune response, and pathogenesis. As a result, it is difficult to translate laboratory findings into biomedical applications. Although several animal models with human chimeric genes, organs or systems have been developed in the past, their limited engraftment rate and physiological functions are a major obstacle to realize convincing models of humans. The lack of human transplantation resources and insufficient immune tolerance of recipient animals are the main challenges that need to be overcome to generate fully humanized animals. Recent advances in gene editing and pluripotent stem cell-based xenotransplantation technologies offer opportunities to create more accessible human-like models for biomedical research. In this article, we have combined our laboratory expertise to summarize humanized animal models, with a focus on hematopoietic/immune system and liver. We discuss their generation strategies and the potential donor cell sources, with particular attention given to human pluripotent stem cells. In particular, we discuss the advantages, limitations and emerging trends in their clinical and pharmaceutical applications. By providing insights into the current state of humanized animal models and their potential for biomedical applications, this article aims to advance the development of more accurate and reliable animal models for disease modeling and drug screening.
Keywords
- humanized animal
- human pluripotent stem cells
- immunodeficient animal
- xenotransplantation
Animal models as surrogates of human biology avoid logistical and ethical restrictions of working with cells and tissue samples from human donor. Studying the effects and side effects of therapeutic drugs or transplantation in animal models first is an important step before translation to human studies, particularly for safety consideration. With the advantages of small size, rapid reproduction, ease of handling, sharing of genomic and physiological properties with humans and ability to be easily genetically edited, small animals such as mice and rats are widely used nowadays. However, there are huge differences between humans and rodents in size, behavior, lifespan, living conditions, ecological niches and immune systems, for instance, human blood is neutrophil rich (50–70% neutrophils, 30–50% lymphocytes) whereas mouse blood has a strong preponderance of lymphocytes [1, 2]; unlike human endothelia cells expressing both major histocompatibility complex (MHC) classes I and II molecules, rodent endothelial cells barely express MHC class II [3]. In fact vascularized grafts were found tolerable in many rodent models, while in humans they were frequently subject to rapid rejection [4], indicating great differences in immune responses between humans and mice.
Human-animal gaps also affect the acute presentation of human diseases and drug responses. The experimental conclusions drawn by ordinary animals are often inconsistent with the results of clinical trials, which hinders the transformation towards clinical. Diabetes candidate drug C-peptide has been shown to have good beneficial effects on the development of neuropathy, retinopathy and nephropathy in a variety of diabetes rat and mouse models, but C-peptide replacement therapy failed in Type I diabetes (T1D) adults [5]. In model of asthma, plasma exudation dominates in bronchial asthma and allergic rhinitis in humans, rather than little plasma exudation in mouse airways, since the peripheral lungs of mice lack mast cells [6]. In mice, most of the lung parenchyma is maintained by low-pressure and high-flow pulmonary circulation, which is largely different from human whose vascular supply of lung tumors mainly comes from high-pressure and high-flow systemic circulation, contributing an important impact for modeling lung tumors [7]. Exposing mice to cigarette smoke to simulate emphysema to induce chronic obstructive pulmonary disease required a much longer time, while only producing partial symptoms of human diseases; meanwhile, the pathological changes did not progress after the stimulation stopped [7, 8]. Since traditional animal experiments have seriously ignored the possibility that the human internal environment contributes to the response and therapeutic effects, it is increasingly emphasized by contemporary pharmacology to study the effect of intervention measures in humanized models.
Using primates is believed to greatly reduce the gap between animal models and disease models, however, primates are expensive and require a long cultivation time, thus difficult to use in large quantities to date, as a novel alternative choice, humanized animal models draw much attentions, and generation and applications of humanized animal models has achieved impressive progresses in the past two decades. Genomically humanized animals are generated by introducing specific human genomic regions into the host genome. These animals serve as valuable tools for advancing drug development and studying human diseases, and enable the robust production of humanized antibodies by introducing human immunoglobulin loci [9, 10]. They can accurately mimic human disease conditions associated with genetic factors, such as aneuploidy syndromes [11], and facilitate the prediction of drug pharmacokinetics and toxicokinetics in humans through CYP3A or UGT2 transchromosomic models [12]. In addition, they provide insights into specific single nucleotide polymorphisms (SNPs) in drug metabolism-related genes [13]. However, challenges arise from the influence of host-dependent factors on transgene and protein expression, limiting the achievement of intended functionality [14]. On the other hand, cell-humanized animal models involve xenotransplantation of human cells or tissues into animal hosts, providing a more comprehensive human physiological environment by incorporating various human cellular components with a complete set of human genetic information. This approach provides a more reliable platform for studying diseases, drug responses, and immune reactions, especially when multiple genes or gene networks are involved in specific physiological processes, disease progression, and metabolism, and is attracting widespread attention. As hottest and most enduring subject of study, hematopoiesis-immune humanization in mice has been realized through human hematopoietic stem cell (HSC) transplantation, which permitted continuous producing of human innate immune cells and establishment of adaptive immunity [15]. Liver humanization has been achieved by transplantation of human hepatocyte, with human specific albumin secretion and drug metabolic kinetics and metabolites [16]. Xenografting of human tumors had become a mainstream approach in cancer research, the graft maintained the histological and cytological characteristics of the original tumor [17]. Despite of these encouraging successes, the shortage of primary tissue or cell sources, the variance between different donors and ethical issues markedly hinder the further development. In recent years, induced pluripotent stem cell (iPSC) and their derivation has received considerable attentions in the fields of xenotransplantation and humanization. They have been reportedly differentiated and transplanted into animals to construct humanization in specific organ or system and model certain physiological functions, organogenesis and diseases. For instance, iPSC-derived HSCs [18] to model human hematopoiesis and immunity; iPSC-derived HSCs to model human drug metabolism; iPSC-derived dopamine neurons to model human extrapyramidal system regeneration [19]. iPSC-derived lung epithelial progenitor to model human pulmonary regeneration [20]. With the development of precise modulation of cell differentiation fate, increasing cell types are expected to be generated which would extensively broad the modeling of specific human organs or systems [21, 22, 23, 24]. Up-to-now, there remains critical problems to be addressed, including physiological maturation, engraft efficiency, long-term survival and homing, vascularization and integration with host organ microenvironment after transplantation.
With human-specific anatomical features, metabolic profiles, immune system and stress response, humanized models are expected to dramatically improve etiological accuracy from physiologically relevant disease induction and presentation capability to reproduce human disease characteristics of animal models for human disease studies and drug development, thus bridging the gap to preclinical experiments. With rapidly developed techniques for generating immunodeficient and human xenotransplantation-assured animals, as well as ready-to-use donors like iPSC-derived cells or tissues, humanized animals are believed to be widely used in biomedical research.
Immunodeficient animals derives the primary requirement for generating humanized
animals. As earliest immunodeficient mice, the nude mice, lacked T cells due to
the Foxn1 gene mutation, while retained B cells and natural killer (NK)
cells, supported engraftment of most primary human solid tumors and even human
hematopoietic cells [25]. Later, the Prkdc gene mutation was established
in server combined immune-deficiency (SCID) mice with both B and T lymphocytes
deficient, which indicated a fundamental breakthrough in the history of
immunodeficient animals. However, the human cell repopulation rate in SCID mice
was still limited, which might be related to the remaining activity of natural
immune cells (including NK cells and myeloid cells) and the gradual development
of T and B cells [26]. Another major breakthrough is the mutation of the
Interleukin-2 (IL-2) receptor, which resulted in the complete loss of NK cells
and elimination of the leaky development of T and B cells. To this regard,
investigators introduced Interleukin-2 receptor
The development of targeted gene editing in animals paved the way for
constructing specific disease models that permitted the generation of local or
systemic humanization. Fumarylacetoacetate hydrolase (Fah) gene knockout induced
controllable liver injury [31, 32], and provide the model of metabolic liver
disorder and HT-1 (Hereditary Tyrosinemia type I) [33], which in turn facilitated
xenotransplantation of human hepatocytes under regeneration signals in host liver
[34]. In early study in Rag1 gene knockout rat model, combined with
thymectomy and anti-asialo GM1 antibody treatment, the transplantation of human
hepatocyte reached approximately 17% repopulation rate [35]. Furthermore,
combined with Rag2 and Il2rg double knockout, robust engraftment (up to 90%) of
human hepatocytes from was achieved in this
Fah
Advances in gene-editing technology, particularly CRISPR/Cas9, have opened up a wide range of possibilities for generating immune and function deficient animals by precise and targeted gene knockout or knock-in, the emergence of liver humanized animals provides a more convenient platform for liver disease modeling, drug screening and regenerative studies. Despite of the progressive development of mice models since 1990’s, rat model, holding advantages in terms of size, surgery, and rich drug testing profiles, achieving first success in liver humanization since 2021 (Fig. 1). Currently, small animal models such as mice and rats remain the predominant models for studies. Despite the advantages of efficient reproduction, they cannot be used to assess secondary pathologies that may occur years after xenotransplantation and have shown limitations in generating clinically relevant data for human diseases such as cancer, neurodegenerative diseases, diabetes and stroke [43]. In additional, large animal models (including dogs, pigs and non-human primates), which have greater similarities to human anatomy and immunology, can be studied over longer timescales with clinical dosing regimens [44].
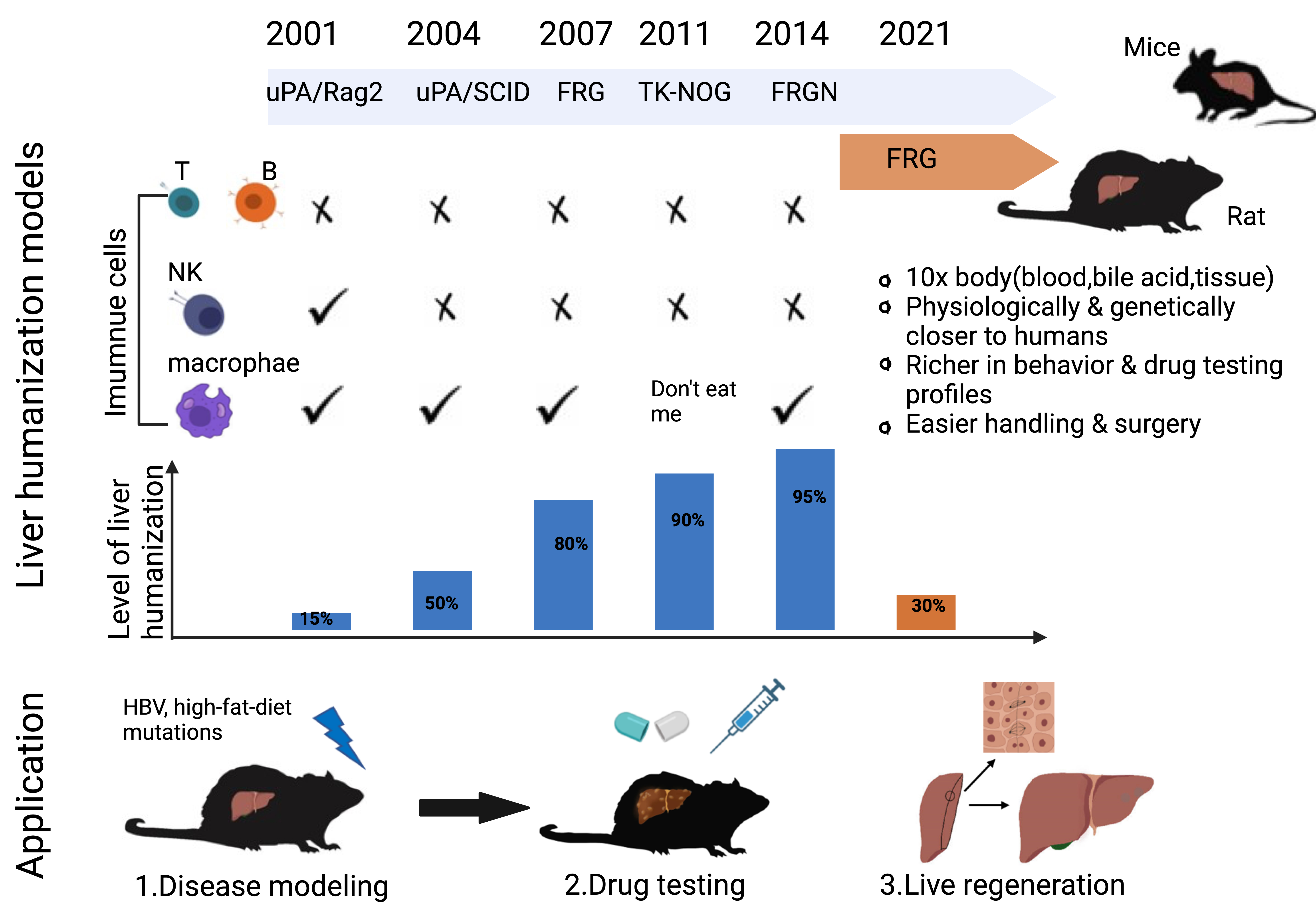 Fig. 1.
Fig. 1.Development and application of mice and rat models for liver humanization. In the past two decades, many types of mice has been developed for generating liver humanization, such as uPA/rag2, uPA/SCID, FRG, TK-NOG and FRGN. However, until 2021, the first rat model of FRG was achieved. Compared to mice, rats are much bigger in shape and easier for surgery, have richer drug testing profiles, and more in line with the needs of pharmaceutical and clinical trials. Liver humanized rats may provide better platform for liver disease modelling, drug testing and liver regenerative studies.
Xenotransplantation of human cells or tissues into immunodeficient animals is a fundamental approach for generating humanized animal models. Human primary cells, including adult stem cells and terminal functional cells, have been extensively investigated for generating human-specific organs or systems. Here, we focus on the humanization of the blood/immune system and liver, which have been widely studied worldwide over the past few decades (Table 1, Ref. [15, 37, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93]). We summarize and discuss the development of donor sources used to achieve these goals.
| Humanized system/organ | Donor cells & isolation method | Transplantation route | Mouse or rat host genetic background | Applications |
| Immune and hematopoietic systems | HSCs/CD34 |
Intravenous | NOD/SCID | Model of skin xenograft rejection reaction, 2006 [15] |
| Via tail vein | BALB/c-Rag1 |
Model of Zika virus infection, 2018 [45] | ||
| Intravenous | NOD.CB17-Prkdc |
Model of pancreatic cancer, 2019 [46] | ||
| Via liver | NSG | Model of evaluation for CAR-T cell therapy in acute myeloid leukemia, 2021 [47] | ||
| Via liver, vein or bone marrow | SCID | Model of HIV infection and therapies evaluation, 2021 [48] | ||
| Intravenous | NSG | Model of sepsis, 2021 [49] | ||
| Via retro-orbital vein | NSG | Model of human gamma delta T cell-HIV interactions in vivo, 2022 [50] | ||
| Intravenous | NSG | Model of tumor xenotransplantation, 2022 [51] | ||
| HSCs from cord blood by MACS or FACS | Intravenous | BALB/c-Rag2 |
Model of patient derived tumor xenograft and anti-PD-1 immunotherapy, 2019 [52] | |
| Intravenous | NSG™, NSG™-SGM3 | Patient-derived xenograft model for study immune-oncology, 2019 [53] | ||
| Intravenous | NSG | Patient-derived xenograft model for evaluation of lung cancer immune responses, 2019 [54] | ||
| Intravenous | NOD/SCID/IL2r |
Model of HIV infection and persistence, 2023 [55] | ||
| Via retro-orbital vein | NBSGW | Patient-derived xenograft model of ovarian cancer, 2023 [56] | ||
| HSCs from bone marrow by MACS or FACS | Intravenous | NSG-SGM3 | Patient-derived xenograft model of estrogen-independent, hormone receptor positive metastatic breast cancer, 2021 [57] | |
| HSCs from CD34 |
Via liver, vein or bone marrow | RG, NSG, NOG | Model for vaccine assessment, 2013 [58] | |
| Intravenous or intrafemoral | RG, NOG, NSG | Model of human immune responses and generating human monoclonal antibodies, 2014 [59] | ||
| HPSCs from CD34 |
Intravenous | NSG | Model of evaluation for PD-1-targeted cancer immunotherapy, 2018 [60] | |
| Via retro-orbital vein | NSG | Model of virus dissemination, 2019 [61] | ||
| Via tail vein | NOG | Model of study on communication mechanism of HIV in brain, 2021 [62] | ||
| HPCs from iPSC/iPSC-teratoma by FACS | Subcutaneous injection/via bone marrow | NSG | Patient-derived model of chronic myelomonocytic leukemia and therapeutic drug identification, 2018 [63] | |
| CD34 |
Via posterior orbital vein | NSG | Evaluation of the possibility of using iPSC-HSCs to resistant HIV, 2021 [64] | |
| HSPCs from umbilical cord blood by MACS and FACS | Via liver | Rag2 |
Model of sustained Plasmodium vivax blood-stage infections, 2022 [65] | |
| Reprogramed HSPCs from haemogenic endothelium by ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1 and PI1 | Via bone marrow | NSG | Model of patient derived tumor xenograft, 2017 [66] | |
| Reprogramed HSPCs from iPSC-CD34 |
Via the temporal vein | NSG | Model of patient derived tumor xenograft, 2018 [67] | |
| HSPCs by converting the SCD allele (HBB |
Via tail vein | NOD.Cg-Kit |
Providing treatment methods for sickle cell disease based on genome editing, 2021 [68] | |
| HSPCs from transduction of CXCR4, CXCL12, KIT and ITGA4 in peripheral blood cells | Via bone marrow | Hyper IgM syndrome | Providing a valuable strategy for hematopoietic stem/progenitor cell gene therapy, 2022 [69] | |
| RBCs from iPSCs, PHHs | Via vein | NOD-FRG (FRGN) | Model of pre-erythrocytic stages of Plasmodium falciparum, 2018 [70] | |
| Liver | Perfused PHHs from healthy donor | Via spleen | FRG | Evaluation of clinically relevant adeno-associated viral variants, 2014 [71] |
| Via inferior vena cava | NSG | Testing of hepatocyte transplantation therapies for | ||
| Via spleen | TK-NOG | Model for testing organophosphate and carbamate pesticide exposure, 2018 [73] | ||
| Via spleen | FRG | Model of malaria infection, 2018 [74] | ||
| Via spleen | FRGN | Model of acetaminophen-induced acute liver failure, 2019 [75] | ||
| Via spleen | uPA-SCID/Beige | Model for hepatotropic virus infection, 2020 [76] | ||
| Via spleen | SCID, uPA-SCID, TK-NOG, FRG, NSG | Model of HBV infection, 2021 [77] | ||
| Via portal vein | Rat/FRG | Drug testing, modeling of human hepatitis virus infection, 2021 [37] | ||
| Via tail vein | Rat/FRG | Model of xenotransplantation and viral transduction, 2022 [78] | ||
| Via tail vein, spleen and inferior vena cava | FRG, TK-NOG, uPA-SCID | Model of metabolic disorders, 2022 [79] | ||
| Via tail vein, spleen and inferior vena cava | FRG | Model of acute liver injury through alcohol binging, 2022 [80] | ||
| Via spleen | TK-NOG | Model of cytochrome P450-dependent drug oxidation activities, 2022 [81] | ||
| Via spleen | TK-NOG | Model of human drug metabolism, 2022 [82] | ||
| Via spleen | TK-NOG | Model of metabolites of benzbromarone, 2022 [83] | ||
| Via spleen | FRGN | Model of nonalcoholic steatohepatitis, 2022 [84] | ||
| Perfused PHHs from patient’s hepatocellular carcinoma | Via abdominal cavity or liver | NSG | Model of evaluation for chemic antigen receptor T cells target glypican 3, 2020 [85] | |
| Perfused PHHs from CPS1 deficient patient | Via spleen | FRGN | Model of CPS1 deficiency, 2019 [86] | |
| Hepatic progenitors from FLCs by centrifuging at low speed or MACS | Via liver | NSG | Model for HBV infection, 2015 [87] | |
| Via liver | HLA-A2 NOD-SCID Il2rg |
Model for HCV infection, 2016 [88] | ||
| Transdifferentiated HLCs from mesenchymal stem cells | Via spleen | Rat/WT | Model of acute human liver failure, 2022 [89] | |
| HLCs from iPSCs | Via spleen | FRG | Model of HBV infection, 2018 [90] | |
| Via liver | Nonhuman primate/WT | Model of cell therapy for liver fibrosis, 2020 [91] | ||
| Reprogramed hepatocytes from fibroblasts by transduction of FOXA3, GATA4, HNF1B, HNF4A, HHEX, PROX1, C/EBP |
Via spleen | Fah |
Model of drug metabolism, 2014 [92] | |
| Reprogramed liver progenitor-like cells from hepatocytes by transduction of SIRT1 | Via spleen | Fah |
Model of hepatotropic pathogens, 2019 [93] |
Abbreviations: CSP1D, carbamoyl phosphate synthetase 1 deficiency; FACS, fluorescence-activated cell sorting; FLCs, fetal liver cells; FRG,
Fah
HSCs are the core elements for blood and immune humanization, they are typically
derived from fetus, bone marrow and umbilical cord blood. When transplanted into
the irradiated immunodeficient mice, these primary human HSCs rapidly developed
into T, B and NK cells [94], and could sustain for several months [95, 96].
However, the limited in vitro expansion capacity of HSCs presents a
significant obstacle to their broad use in xeno-transplantation. Recently, a
breakthrough study has identified chemical agonists and a caprolactam-based
polymer that allow for long-term ex vivo expansion of human HSCs. However,
despite achieving a 75-fold expansion of total cells and a 55-fold expansion of
cord blood CD34
In recent years, chemical compounds have emerged as a potential alternative approach with unique advantages in cell fate conversion and expansion [102]. Aryl hydrocarbon receptor (AhR) antagonists and notch ligand agonists activate cord blood cells expansion in vitro [103], Combinations of UM171, 740Y-P and butanamide, and caprolactam based polymers completely replace exogenous cytokines and albumin to achieve long-term in vitro expansion of human HSCs [97]. While, a cocktail of Bix01294, RG108, VPA and PD0325901 successfully converted human cord blood erythroblasts into induced megakaryocytes, which could produce proplatelets and release functional platelets [104]. More key small molecules and their combinations are under in-depth investigation to promote the production of human HSPCs from hiPSC with higher efficiency, shorten time and lower cost.
Apart from HSCs, numerous terminally differentiated immunocytes have been generated from hiPSCs, and may benefit the studies of specific immune responses from particular immune cell types in humanized models, holding great potential for biomedical applications. hiPSC induced natural killer (NK) cells reportedly showed similar metabolic characteristics and gene expression profiles to adaptive NK cells induced by cytomegalovirus, which owned excellent anti-tumor activity [105], maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti-PD-1 therapy [106], iPSC-NK cells containing chimeric antigen receptors (CAR) provided better options for anti-cancer immunotherapy [107]; hiPSCs induced macrophages displayed similar surface and intracellular markers of human macrophages, as well as functional activities [108], iPSCs-derived CAR-expressing macrophage cells enhanced phagocytosis of tumor cells, and in vivo anticancer cell activity [109], genetically modified iPSC-derived macrophages facilitates drug screening [110]; hiPSCs induced red blood cells were found to express mature adult globin [111], progress in vitro red blood cell generation is expected to address the shortage of blood demand for transfusion application [112]. Moreover, “Designed red cells” with custom phenotypes (Rh invalid, GPB invalid and XK invalid/KEL low) have recently been generated from hiPSCs using CRISPR/Cas9, although the problems of low erythroblast production and poor enucleation rate still existed [113]. Of note, Nakamura et al. [114] transfected and generated a self-renewing and immortalized megakaryocyte line from hiPSC-derived hematopoietic progenitor cells, which permitted massive producing of platelet of clinical quality and quantity. With the development of personalized iPSC bankers, once the differentiated cells (including HSC and their derivations) were used to establish humanized animals, it may bring great opportunity to model hematological disease and cancer for identifying novel diagnostic and targeted therapeutic markers in individual patient.
To generate humanized liver, various human hepatic sources have been tested. Primary human hepatocytes (PHH) have long been considered as the most preferred source for repopulation in the host liver in view of efficiency and function, over 90% repopulation rate has been reported in humanized mouse in the past decade [115]; while in rats, over 30% was achieved by our group very recently [37]. In view of the shortage of PHH, alternative hepatic sources such as fetal liver cells and their derived hepatic stem cells (isolated according to specific surface markers such as) had been explored, these cells permitted quite robust expansion in vivo, over 70% repopulation could be achieved in a relatively short time around 2 months, however the human mature hepatic markers expression were extremely lower, imply the immature of the engrafted cells [16, 116]. Recently, the explore of cell signal modulators, particularly TGFb and GSK3 inhibitors, had paved the way to convert primary hepatocyte, which is normally considered not expandable in vitro, into expandable progenitor state [117, 118]. Of note, in one study, it was reported that when transplanted in vivo, these cells could achieve repopulation level comparable with primary hepatocyte [116]. However, to our practical experience, the expanded cells quickly loss the maturation and transplantation potentials after 3–5 passages. Moreover, the development of differentiation and reprogramming strategy have enabled generation of a large number of donor hepatocytes from pluripotent stem cells, and meanwhile permitted personalized liver disease modeling and drug screening, deserved great promises. Numerous reports have demonstrated that hepatocyte-like cells (HLCs) directedly differentiated from hiPSC could repopulate the damaged mouse liver [119]. Meanwhile hiHeps transdifferentiated from fibroblasts hold similar potentials [92]. A comparation between these two strategies from the same donor identified differences in gene expression for phase II drug metabolism and lipid accumulation, Importantly, both cell types markedly elevated hepatic function following transplantation in vivo, suggesting plasticity advantage for generating functional liver [119]. However, much lower efficiency of repopulation rate (less than 5%) and human albumin secretion (around 100 ng/mL) remained major limitations. Similarly, hepatic stem cells were induced with refined transcription factors combinations. Importantly, they held expansion capability and meantime owned dual-differentiation capability towards both hepatocytes and cholangiocytes, which were supposed to meet the diversity of cellular components in the humanized liver [120]. In our recent study, iPSC-derived hepatic progenitor cells were induced with combination of CHIR99021, A83-101 and FGF2, and expanded in vitro over 20 passages for billions-fold increase, without significant decrease of maturation potential [121], holding great promise for generating functional humanized liver in larger animals beyond rodents.
Although primary human cells, including HSCs and hepatocytes remain the first choice for building effective humanized animals, the shortage of sources, ethical issues, and differences in the genetic background in different sources make them difficult to use in a stable and large scale. Alternatively, iPSC source is considered nearly infinite, with highly homogeneity in the same batch. Furthermore, constant breakthroughs have been made in using in human iPSCs derived cells other than liver and hematopoietic system, such as neurons [122], beta cells [123], alveolar type 2 cells [124], intestinal cell [125], and keratinocytes [79, 126]. Undoubtedly this progress will pave the way to generate more specific and complex humanized models in the near future.
The development of humanized animal models allows more closely resembling of human organs and systems, has the potential to greatly improve our understanding of how diseases work and how they can be treated, as well as accelerate the development of new drugs and therapies. The commonly used humanized animals, mainly rodents, are summarized along with their advantages and limitations in Table 2 (Ref. [18, 19, 37, 39, 66, 67, 78, 92, 93, 100, 120, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143, 144, 145, 146, 147]), which highlights the need for further improvements of current models.
| Humanized system/organ | Host background | Immunodeficiency and other modifications | Advantages and/or Limitations | Year |
| Hematopoietic system | BRG line (BALG: Rag1/2 |
Lack of T, B and NK cells | Advantages: myeloid reconstitution improved engraftment and maintenance of human hematopoiesis; improved lifespan of myeloid cell; without anemia or thrombo-cytopenia; Limitations: impaired hematopoietic niche formation. | 2020 [139] |
| NOD/SCID, NOD/SCID/JAK3 |
Lack of effective T, B cell | Advantages: iPSC-HSC could migrate from teratoma to bone marrow and peripheral blood; multilineage and long-term hematopoietic system was recon-structed; Limitations: teratoma formation. | 2013 [18] | |
| NSG | Lack of B, T, NK cells, and cellular complement, deficient macrophages, dendritic cell | Advantages: bone marrow-like structure in teratoma could generate HPSC; Limitations: teratoma formation. | 2013 [129] | |
| NOD/SCID-IL2R |
Lack of B, T, NK cells, and cellular complement, deficient macrophages, dendritic cell | Advantages: repeatable short-term implantation; bone marrow and red blood cells were engrafted; Limitations: express embryonic and fetal hemoglobin, not adult hemoglobin. | 2013 [100] | |
| Immune system | NOD/SCID | Lack of effective T, B cell, and thymus, low levels of NK cells | Advantages: human thymic microenvironment supported the development and selection of human T cells; improved the survival rate of human HSCs transplantation; Limitations: host T cells remained; higher incidence of GVHD. | 2022 [140] |
| NSG (engrafted with CD34 |
Lack of T, B and NK cells, and cellular complement, deficient macrophages, dendritic cell | Advantages: mediate human cellular and humoral immune responses; strong proinflammatory response to T-cell receptors; the engraft functions: inhibit allograft tumor growth and promote effective Ig class conversion; Limitations: host immune cells remained. | 2022 [140] | |
| C.B-17 SCID | Lack of effective T, B cell, thymus, lymph nodes and spleen atrophy | Advantages: human immunoglobulin secretion; specific human antibody reaction; Limitations: graft versus host disease (GVHD). | 2022 [127] | |
| MISTRG (Rag2 |
Lack of T and B cells, express M-CSF, IL-3, GM-CSF, TPO; MISTRG mice also expressed hSIRP |
Advantages: high efficiency of human hematopoietic cell engraftment; robust development of diverse subsets of human innate immune cells, including NK and macrophage; Limitations: weak human adaptive immune responses; with low cytotoxic and humoral immune responses. | 2021 [141] | |
| NSG | Lack of T, B and NK cells | Advantages: eliminate the autologous white blood cells of mice; Limitations: human immune cells are immature; without strong T cell-mediated immune response. | 2021 [142] | |
| Transgenic HLA-expressing NOG | Lack of T cells and B cells, NK maturation disorder; express human HLA | Advantages: human HLA restricted T cell response; Limitations: remain innate immunity. | 2019 [143] | |
| NSG | Lack of B, T, NK cells, and cellular complement, deficient macrophages, dendritic cell. | Advantages: B and T cells and myeloid cells implant; no bone marrow bias; Limitations: more unstable genome than the original HSPCs; susceptible to leukemia after long-term transplantation. | 2018 [67] | |
| NSG engrafted with fetal tissue | Lack of T, B, NK cells, and cellular complement, deficient macrophages, dendritic cell | Advantages: robust human T cell mediated immune response, can’t mounting an effective alloimmune response, unable to fully reject differentiated hESC-EC grafts; Limitations: form teratoma. | 2017 [128] | |
| NSG | Lack of B, T, NK cells, and cellular complement, deficient macrophages, dendritic cell | Advantages: implant B and T cells, myeloid cells are mature and functional and implant; Limitations: without clear and efficient induction protocol. | 2017 [66] | |
| HLA-A2 |
Lack of T, B, NK cells and no residual cytolytic activity; Lack of murine MHC; HLA transgene expression | Advantages: high lymphocytes engraftment rate without GVHD; Limitations: hematopoietic and immune systems not reconstituted. | 2017 [132] | |
| NOD-SCID-SIRP |
Lack of effective T, B cell and thymus; low levels of murine NK cells; inhibit phago-cytosis of macro-phages | Advantages: enhancement of human PBMCs engraftment; Limitations: Preservation of the activity of NK cells and other intrinsic cells; spontaneous thymic lymphoma; Short-term graft survival and failure of immune and haematopoietic reconstitution. | 2007 [39] | |
| Liver | Fah |
Fah deficiency, lack of T B NK cell | Advantages: feasible to serial transplantation; the repopulation rate reached 80%; Limitations: could not provide a growth advantage for immature liver progenitor cells. | 2022 [78] |
| Fah |
Fah deficiency, lack of T, B and NK cells | Advantages: high survival rate with stable repopulation; Limitations: repopulation rate limited to 30%. | 2021 [37] | |
| Fah |
Fah deficiency, lack T and B cells | Advantages: supported infection or reactivation of hepatitis B virus; improved the liver functions of F/R mice and their survival; Limitations: does not reconstitute liver parenchymal cells. | 2019 [93] | |
| Alb-TRECK/SCID | Expressed human heparin-binding epidermal growth factor-like receptor (HB-EGF) | Advantages: functional humanized liver, repopulation rate reached nearly 100% in some cases; Limitations: recipient liver recovered quickly by host cells. | 2017 [144] | |
| Fah |
Fah deficiency | Advantages: restored the liver function without teratoma; Limitations: low survival rate after 9 weeks. | 2014 [92] | |
| Fah |
Fah deficiency | Advantages: efficient derivation of both hepatocytic and cholangiocyte lineages; Limitations: low survival rate after 8 weeks. | 2013 [120] | |
| Brain | YAC128 transgenic Huntington’s disease | Non immune deficiency | Advantages: differentiate into neurons, oligodendrocytes and astrocytes, improve motor and cognitive functions; Limitations: Motor and cognitive functions are not at normal levels. No intact humanized nervous system. | 2021 [145] |
| NOD/SCID | Lack of T and B cells, hSIRP |
Advantages: iPSC-derived neural precursor cells could integrate into mouse neural projections; Limitations: without functional verification and without fully humanized nervous system. | 2020 [136] | |
| NOD/SCID | Lack of T, B and NK cells, hSIR |
Advantages: implant into cortex, differentiate into pyramidal neurons and gradually mature; Limitations: without functional verification and without fully humanized nervous system. | 2020 [146] | |
| MITRG mice: Rag2 |
Lack of T, B and NK cells, expressed hCSF | Advantages: differentiated into functional microglia and other central nervous system macrophages; responsive to both acute and chronic insults; Limitations: without functional verification and without fully humanized nervous system. | 2019 [137] | |
| Cynomolgus monkey | Non-human primate, no immune deficiency | Advantages: implant into putamen, improve functional motor improvement of Parkinson’s disease (PD) model; Limitations: without functional verification and without fully humanized nervous system. | 2015 [19] | |
| NOD-SCID | Lack of TB cells, hSIRP |
Advantages: integrate into neural network and vascular, showed neuronal activity, synaptic connection; Limitations: form glioma. | 2018 [138] | |
| Islet | NOD-SCID IL2R |
Lack of T, B and NK cells, hSIRP |
Advantages: grafts matured into functional |
2015 [134] |
| NOD-Rag1 |
Lack of T, B and NK cells; hSIRP |
Advantages: permitted to restore normal blood glucose in vivo; Limitations: 60% recipients occurred xenogeneic rejection. | 2010 [135] | |
| Alopecia areata | NSG | Lack of T, B and NK cells, hSIRP |
Advantages: effectively mimicked the immune effector activity of human alopecia areata; avoid of permanent scarring alopecia produced by graft-versus-host reactions; Limitations: rejection; cytokines affect the accuracy of the model. | 2020 [131] |
| Androgen alopecia, immune system | NSG | Lack of T, B and NK cells, hSIRP |
Advantages: humanized hematopoietic system facilitated human skin engraftment; Limitations: could not reflect the clinical complexity of human androgen alopecia. | 2016 [130] |
| Melanocyte | KSN/Slc nude | Lack of T, B and thymus gland | Advantages: displayed melanin deposition; melanocytes in dermis could migrate to epidermis; Limitations: rejection present, product contains low levels of melanin. | 2018 [133] |
| Melanomas, immune system | NOD-SCID IL2R |
Lack of T, B and NK cells, hSIRP |
Advantages: high-affinity T cell receptor with allogeneic reactivity was abolished; Limitations: Unable to simulate full human skin. | 2021 [147] |
Abbreviations: ESCs, embryonic stem cell; HSCs, hematopoietic stem cells; HSPC,
hematopoietic stem progenitor cells; iPSCs, induced pluripotent stem cells; NOD,
nonobese diabetic mice; SCID, Severe combined immunodeficiency; NOG,
NOD/Shi-SCID/IL2r
The idea of constructing a transient human immune system in mice dates back to
the 1970s. Early experiments sparked interest in modifying the mouse immune
system to accommodate cells and tissues from the human immune system. Human
peripheral blood mononuclear cells (PBMCs), or human lymphocytes has been
isolated from lymph nodes/spleen and injected into the immunodeficient mice,
However, these early attempts were limited by the short lifespan of human
lymphocytes in the mouse peripheral blood [148]. Subsequently, HSCs
transplantation was considered as more effective approach to realized
humanization. However, initial attempts to engraft HSCs into irradiated CB17-SCID
mice were limited by the high activity of host NK cells and spontaneously
generated B and T cells, leading to low levels of human cell engraftment and a
risk of graft versus host disease (GVHD) [127, 149, 150]. Advances in mouse
strain mutations, including the development of the IL2
The transplantation of human fetal liver and thymus fragments has been a
promising approach to create the BLT (bone marrow, liver, thymus) model, which
enables the development of human hematopoiesis and functional human immune system
in mice [15]. The BLT models provided a human thymic microenvironment that
allowed the sustained development of human hematopoiesis and functional human
immune system, supporting the development of human T cells and their selection on
human MHC molecules, and eventually mediate strong immune responses in
vivo [15, 154]. However, bone marrow microenvironment required for innate immune
cell development was still lack, the incidence of GVHD was higher than CD34
To address these limitations, researchers have explored alternative approaches,
such as hu-SRC and hu-BLT mice, which do not reject human ESCs transplantation
but suffer from inadequate reconstitution of the innate immune system and the
development of a wasting disease-like syndrome that renders them inadequate for
long-term transplantation [128]. Recent studies have shown that hiPSC-thymus
organoid can support the de novo generation of a diverse population of functional
human T cells, potentially overcoming the limitations for immune cell maturation
after transplantation [128]. Additionally, in the latest study of human
hepatocyte transplantation, it was revealed that xenografts recruited macrophages
and neutrophils that cleared themselves. Therefore, the elimination or modulation
of macrophage activities was suggested to support the long-term repopulation
[38]. Future studies should focus on the mechanisms of immune recognition of
xenografts, such as the SIRP
In recent years, research on human iPSCs and transdifferentiated HSCs have attracted much attention. iPSC-HSCs were confirmed to migrate from teratomas into the mice bone marrow (BM) after transplantation. and sustain multi-lineage and long-term reconstructive ability when re-transplanted into secondary mice [18]. However, iPSC-HSCs avoid of teratoma phase have not yet differentiated successfully. Although the efficiency of direct reprogramming method of HSCs increases rapidly, it is still unable to generate functional and transplantable HSCs. Transcription factor combinations SOX4, MYB, HOXA9, ERG, and RORA have been identified to specify myeloid-restricted precursors into short-term transplantable pluripotent progenitor cells with low engraftment level and a strong tendency towards bone marrow differentiation, while absent of adult hemoglobin expression [100]. On the other hand, seven factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1 and SPI1) reportedly convert hematopoietic endothelial cells into long-term transplantable HSPCs that could develop into functional red blood cells, B cells and T cells in vivo, although the rate of red lineage denuclearization was still very limited [66]. Furthermore, BM differentiation bias remains a major obstacle to the derivation of functional HSPCs from hiPSCs. A single transcription factor MLL-AF was found to induce iPSC as a source of highly transplantable HSPC without lineage bias, allowing for the complete reconstruction of the human hematopoietic system, However, since the genome was unstable compared to the primary HSPC, the risks of leukemia transformation increased [67].
However, there are still significant challenges ahead, such as how to completely replace mouse MHCs with diverse HLA molecules and express human-derived growth factors and cytokines [92]. Rebuilding red blood cells and granulocytes, as well as prolonging the maintenance duration, are also yet to be addressed [93, 94]. Further investigation of the hematopoietic developmental signals and the identification of critical gene network regulators may help overcome these limitations.
The development of immune humanized models has significantly advanced our understanding of immune system development and related diseases [155, 156]. Combined transplantation of HSC with islet cells facilitated the modeling of autoimmune diseases type 1 diabetes (T1D) [157]; meanwhile, the human immunity provided an unprecedented platform for antiviral research, including the transmission and pathogenesis of pathogen, such as human immunodeficiency virus and human herpesvirus 4 [158, 159]. Additionally, novel tumor immunotherapy strategies are also being evaluated in immunized humanized mouse models [158, 160, 161]. These models have greatly improved our understanding of immune processes at the cellular and molecular levels, accelerating the current development of translational medicine. So far, neither BM or umbilical cord blood has yet achieved the immune functions as well as BLT in humanized mice [118], partly due to the deficiency of thymic microenvironment essential for innate immune development. iPSCs are considered as attractive and promising source to address these issues, although direct differentiation of functional HSCs was not succeeded yet [18, 129]. How to completely replace mouse MHCs with diverse HLA molecules and express human derived growth factors and cytokines remains large challenges ahead [162], while rebuilding red blood cells and granulocytes, as well as prolonging the maintenance duration are yet unsettled [163]. Further investigation of the hematopoietic developmental signals and the identification of the critical gene network regulators may help overcome these limitations.
By introducing human liver genes into the mouse genome, researchers have
generated a series of liver gene-humanized mice, including mice expressing human
exogenous receptors, drug-metabolizing enzymes and transporters, as well as
combinatorial models [164]. However, transgenic mice do not possess a complete
“humanized metabolic network” within the liver and was not sufficient for
studying human liver metabolism. Animal liver with human hepatocyte replacement
was considered much similar to human liver with respects to liver zonation, and
phase I, II and III enzymes expression, thus particularly suited for studying
drug metabolism and pharmacokinetics, drug-drug interactions and hepatotoxicity
[165]. The chimeric liver is a mixture of endogenous mouse hepatocytes and human
hepatocytes, and the level of human liver characteristics were greatly depended
on the human hepatocyte repopulation ratio [79]. Currently, PHHs are the most
widely used source for liver humanization. In FRG mice model, PHH could
stably achieve an impressive 80% repopulation rate [37], ALB TRECK/SCID
mice also allowed efficient repopulation of human primary fetal liver
cells and hepatic stem cells, with a near 100% in some cases. While adult human
hepatocytes failed to survive in this mouse model [16]. Although humanized liver
models have been successfully generated in mice, practical applications,
particularly in the pharmaceutical industry, require larger animals such as rats.
Very recently, our team made a remarkable breakthrough in generating humanized
liver in a rat model using similar gene knockout strategy as for FRG mice. Unlike
the robust repopulation in mice (usually took 3–4 month to achieve over 70%),
liver humanization in rat took around 7 months to reach a maximum level of around
30%, suggesting that generating humanized liver in larger animals other than
mice is challenging. Since most applications, particularly drug testing requires
humanization level over 30% in animal liver, transplantation of much larger
amount of human hepatocyte was required to achieve efficient liver humanization
[37]. While, PHHs have been shown to be the best source for in vivo
repopulation and functionality, their limited availability, genetic and
phenotypic diversity, and rapid differentiation in culture greatly limit their
applications. The discovery of PSCs and their potential to differentiate into
hepatocytes has drawn much attention as an alternative. Recent research has
focused on increasing the repopulation efficiency and enhancing the hepatic
functions to be closer as PHHs. Co-culture of mesenchymal and endothelial cells
has been shown to significantly promote differentiation and engraftment of
hiPSC-derived endodermal progenitor cells in vivo, achieving 70%
repopulation rate in IL2rg
Integration of human immunity may potentially lead to more accurate modeling of viral infections. Moreover, these models are also beneficial in drug research as they can study human-specific metabolites that are not present in other species, providing insights into human-specific drug responses. Particularly, a promising technique for personalized disease modeling and drug testing is the use of iPSC-derived humanized liver models. These models are thought to better replicate patient-specific liver metabolism and drug response. However, controlling the degree of humanization is challenging. Additionally, remaining mouse hepatocytes in the model may unexpectedly recover or have a higher affinity for certain drugs than their human counterparts [102]. Moreover, without humanization in other metabolic organs like the intestine, it is impossible to identify human-specific extrahepatic drug metabolism and interactions in these humanized mice [103]. Therefore, a more comprehensive and systematic approach to humanization is necessary to achieve better human metabolism modeling.
Mice with humanized skin are commonly used to study transplantation rejection and infectious diseases, by surgically grafting human skin onto mice, researchers can visualize and assess skin diseases. Currently these mice have been widely used to investigate autoimmune and inflammatory skin diseases such as pemphigus vulgaris, GVHD and genetic skin diseases. The human atopic dermatitis (AD) mouse model and humanized alopecia areata (AA) model are both induced lesions in mice transplanted with human skin. However, due to the differences in skin structure and immune function between mice and humans, and the different pathogenesis of various AD subtypes, this model can only simulate acute AD not other complex AD dermatitis [130, 131, 132, 173]. A systemic lupus erythematosus model was successfully generated after transplanting human PBMC from active patients into the dorsal intradermal region of BALB/c nude mice, followed by daily ultraviolet radiation B, resulting in persistent and stable patchy erythema with scaling in the implanted area [174]. However, the expression patterns of adhesion molecules in grafted skin differed from those in original grafts, which significantly affected the homing and recirculation of human lymphocytes in the graft, directly affecting the observations of immunoreaction [175]. Therefore, the construction of humanized animals for skin and immunity is highly in line with the research needs.
Human iPSC-derived skin cells are also increasingly used in the construction of humanized animals. Melanocyte derived from hiPSCs permitted to induce melanosis in nude mice skin where melanocytes aggregated [133]. The hiPSC-derived dermal papilla could partially mimic the role in hair follicle development, and when combined with keratinocytes, it could simulate the three-dimensional assembly of nature hair follicle structure in vitro [176]. An in vitro 3D iPSC-skin organ system with mature hair follicle and sebaceous gland was transplanted into the wound of mouse skin, and it displayed the most skin functions [177]. However, the construction of complete skin organs from iPSCs remains a major obstacle. So far, only melanocyte, keratinocyte, and 3D skin organ system have been successfully induced from iPSC, while it is currently unavailable to simulate complete skin functions. Additionally, different parts of the skin have different thicknesses, with or without hairs, and accessory organs such as sweat glands are different, so establishing partial and localized skin humanization may not thoroughly reflect all aspects of human skin.
In a study, human PSC-derived pancreatic endoderm cells developed into islet-like structures that produced human insulin and maintained normal glycemia when transplanted. Immunotherapies targeting T cell costimulatory pathways attenuated the rejection of xenograft, suggesting that PSC-derived islets may serve as a potential substitute for T1D with proper immunological interventions [122].
To facilitate diabetes drug screening and disease modeling, researchers have
been developing functional pancreatic
To determine the in vivo differentiation potential and functionality,
In clinical experiment, T1D patients indeed restore glucose homeostasis
following islet transplantation, but most patients lose islet function due to
allograft rejection Human islet xenografts were found to have a 60% occurrence
of rejection, which affected the long-term observation of graft outcome [135].
The construction of human islets with human immune system in mice is an ideal
model to study the mechanism of xenograft rejection and to develop therapeutic or
preventive strategy. NSG mouse model reconstituted by CD34
Although the technology for constructing humanized pancreatic islets seems relatively mature, immune rejection remains the main obstacle to the long-term existence and functions of humanized pancreatic islets. The breakthrough in pancreatic humanized mice mainly depends on progress in establishing immune tolerance or immune-humanization.
The newly developed tools such as brain organoids, while a promising tool for studying the human brain, lack the full complexity of in vivo systems, including microenvironments, neuronal circuits, vascular circulation, and immune systems. As a result, they cannot fully capture the complexity of brain development, function, and dysfunction, which limits their use in precise disease modeling or drug screening applications [182, 183]. Humanized animal of brain may better assist in preclinical drug testing of brain-like organs from patient under physiological conditions.
While humanized animal models of the brain have successfully shown the integration of human cells and functional neural networks, questions remain about their ability to accurately simulate aging and dementia in the brain. Nonetheless, the integration of human brain organoids in immunodeficient animals, such as intracerebral integration into NOD-SCID mice, has been achieved, where neurons gradually mature, synapse occur, and functional neural networks form. Additionally, transplantation of induced pluripotent stem cell-derived dopaminergic progenitor cells in a monkey model of Parkinson’s disease expressed markers of dopaminergic neurons in the midbrain, and human iPSC-derived neural precursor cells implanted into the cortex of newborn mice showed potential. Encouragingly, engrafted human microglia into the central nervous system through in vivo differentiation in mice brain responded dynamically to local and peripheral injury.
Since primary brain cells are not readily available, currently induction from hPSCs is the main strategy to obtain neurons, astrocytes and oligodendrocytes for neurodegenerative and myelin diseases studies. In a crab-eating monkey model with Parkinson’s disease (PD), induced by the neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), hiPSC-derived dopaminergic progenitor cells expressed markers of dopaminergic neuron in the midbrain after transplanted in vivo [184]. Moreover, hiPSC-derived neural precursor cells could implant into the cortex of newborn mice [136]. Functional and healthy midbrain dopamine neurons cells from iPSC can effectively restore motor function and re-operate the host brain after transplanting into the rodent model of PD [19]. Followed immune humanization by iPSC-derived hematopoietic progenitor cell transplantation, human microglia could be engrafted into central nervous system though in vivo differentiation in mice brain, and responded dynamically to local and peripheral injury [137]. Encouragingly, up to 80% microglial chimeras were achieved under optimized differentiation and xenotransplantation methods [185]. Nonetheless, the integration of human brain organoids in immunodeficient animals, such as intracerebral integration has been achieved in NOD-SCID mice, where neurons gradually mature, synapse occur, and functional neural networks formed [138]. However, neurons derived from PSCs have significant differences from primary cells, despite sharing the same genotypes and exhibiting similar electrophysiological properties [186]. Neurons derived from fetal iPSC could not simulate the neurodegenerative diseases in the elderly, while the majority of iPSC-neurons generated by direct differentiation were prone to be fetal or neonatal cell-like [187]. These differences arise from variations in epigenetic profiles, which result in significant differences in gene expression. The modification of the epigenetic characteristics of reprogrammed cells is an important obstacle to realizing human neurodegeneration, not to mention the importance of disease epigenetics for disease modeling [188, 189]. It is still doubtful whether iPSC-derived neurons and other central nervous cells can accurately simulate aging and dementia in the brain. Major concerns raised how these differences would affect when hiPSC-derived neurons are used as predictors of human neural responses upon chemical or pathological challenges [190].
While mixed cortical neurons from iPSC have been successfully established, the identification of specific cortical subtypes remains a challenging task [191]. The generation of these subtypes from hiPSCs with correct transcriptome and epigenomic characteristics is crucial to improving the validity and accuracy of disease modeling. However, nervous system is a complex and interconnected integrator, in this regard, animals brain replaced with human brain organoid provide the platform for evaluating the differentiation, axonal growth, and synaptic integration of neural organs from patient specific iPSCs under physiological conditions in vivo, promoting research on the pathogenesis of neurodevelopment, neuropsychology, and neurodegenerative diseases.
Human lung, intestinal and cancer cells have also been successfully transplanted into mice to generate humanized models. Wahl et al. [192] transplanted up to 40 cell types (including non-hematopoietic cells) from human lung tissue into immunocompromised mice to form highly vascularized lung grafts. In this BLT-based humanized mice, the lung grafts and autologous human hematopoietic cells mediated robust humoral and T-cell responses, making it suitable model for studying pathogenic infections in vivo [192]. Human colonic and intestinal epithelial organoids have also been used to reconstruct intestinal epithelial cells in mice, providing a technical basis for organoid transplantation in case of colonic injury [193]. Additionally, transplantation of patient-derived tumor tissue or cells has been widely used to develop individualized xenograft models for studying cancer biology, metastasis, and new therapies.
With the rapid development of transplantation sources and humanization strategies, gene editing techniques have further advanced the creation of humanized models, overcoming limitations inherent in conventional models. For instance, transgenic mice expressing human MHC class I and HLA II were reportedly compatible with human T cells engraftment [194, 195], while mice expressing human p53, a tumor suppressor gene, are ideal for cancer cell engraftment [196]. In mice expressing urokinase fibrinogen activator, which inhibits mouse hepatocyte function, drug metabolism occurs exclusively through engrafted human hepatocytes, which may facilitate the monitoring of human-specific drug pharmacokinetics [197].
With the development of in vitro differentiation strategy, human PSCs-derived function cells have been extensively used in disease modeling, drug screening, and transplantation therapy trails, providing significant successes for building humanized animal models [131, 132]. Patient-derived induced pluripotent stem cells (iPSCs) have a distinct advantage over traditional animal models, particularly in complex diseases such as neurodegenerative and HBV-induced liver diseases, as they not only originate from human cells but also possess unique genetic backgrounds [198]. To date, PSC-derived cells show great promise in the field of refractory diseases and injuries, particularly for patients who do not respond to standard drugs or medications, such as spinal cord injury, heart failure and diabetes [199, 200, 201].
Although PSCs provide an attractive source for xenotransplantation due to their ease of accessibility, high proliferation rate, and homogeneity, three significant challenges remain: tumorigenicity, maturity, and immunogenicity (Fig. 2). The unlimited proliferative potential of PSCs increases the risk of tumorigenesis, such as teratoma formation [202]. Furthermore, retention of undifferentiated or immature cells in the terminal population, integrated transcriptional factors activity, and mitochondrial gene mutations obtained during passages pose potential risks [180]. To address these challenges, genetic stability and purity of the terminally differentiated cells must be considered, particularly when billions of cells are required. While it may be impossible to produce iPSCs completely free of mutations, measures such as using hematopoietic cells from umbilical cord blood with the least pre-existing variation [203], non-virus integrated reprogramming methods, hypoxia culture to minimize DNA oxidative damage, and clustering passage to prevent malignant mutation can minimize mutation rates [204, 205]. For immaturity of iPSCs, with an increasingly comprehensive understanding of gene regulation network involved in cell differentiation, gene manipulation has significantly addressed the functional deficiencies in PSC-derived cells [66, 120], enabling them to reach a level comparable to primary cells. In addition, the emerging role of chemical compounds in cell fate conversion and functional maturation offers a complementary approach for generating functional cell types with high efficiency and cost-effectiveness [102]. For immunogenicity, ischemia-reperfusion [206] and inflammatory factors [207, 208] are known causes of clearance of implanted cells, 3D culture can reduce the expression of dedifferentiated inflammatory factors [38], immunosuppressive pretreatment before transplantation also can promote the engraftment of human cells and reduce immune rejection reactions in vivo [209].
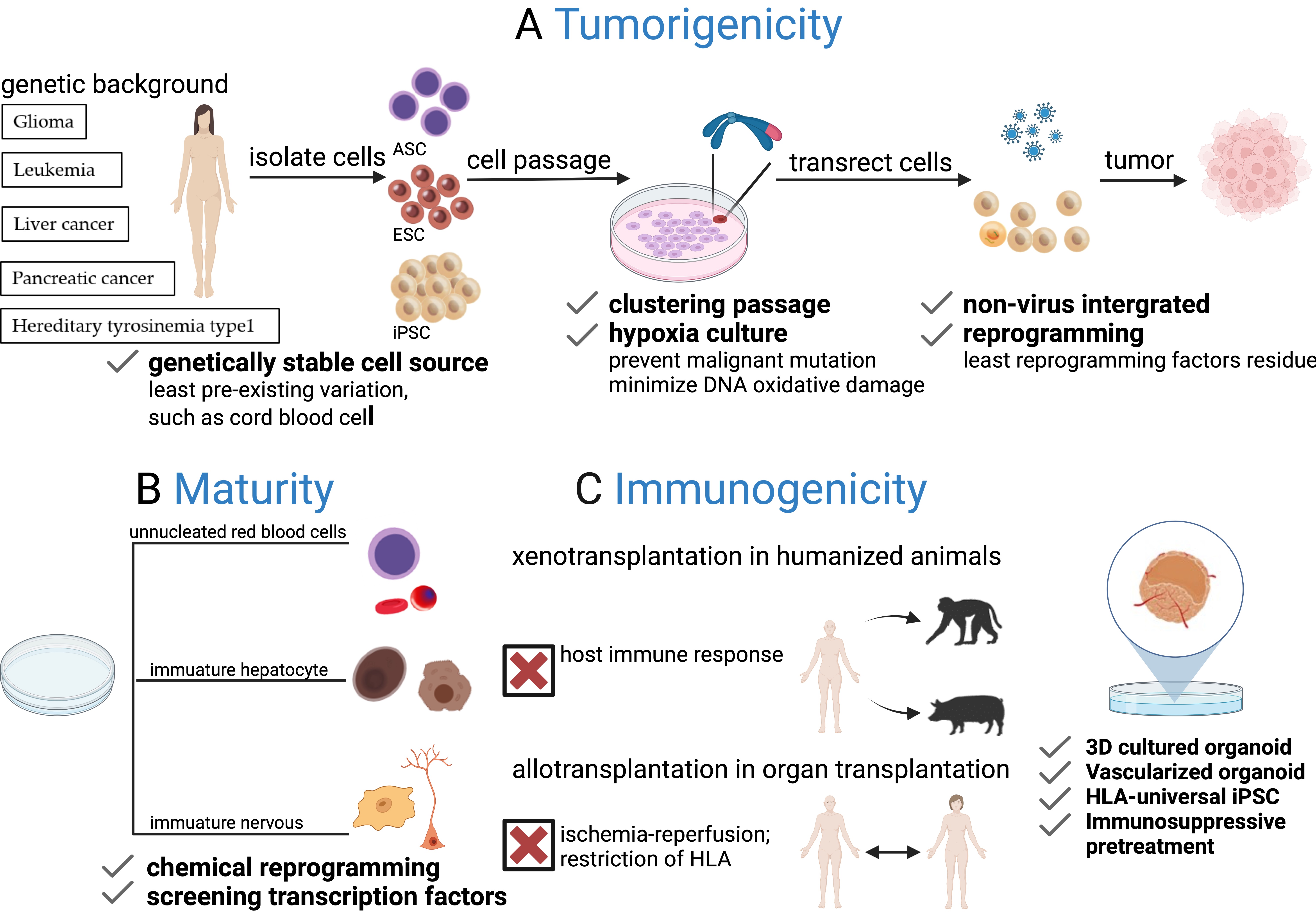 Fig. 2.
Fig. 2.Challenges of PSCs for generating humanized animals. (A) Risk of tumorigenicity: The risk of tumorigenicity arises from undifferentiated PSCs and chromosome abnormalities during in vitro culture, together with integrated reprogramming factors that may lead to tumor formation after transplantation. Safety can be improved by using genetically stable cell sources, non-viral integrated reprogramming strategies, clustered passages, and hypoxic culture conditions. (B) Lack of maturation: PSCs often differentiate into immature states, producing cells such as unnucleated erythrocytes and progenitor-like hepatocytes and neurons, due to limitations in differentiation approaches. Overcoming this challenge will require further exploration of specific small molecules and transcription factors to promote cell maturation. (C) The issue of immunogenicity: Graft clearance is closely related to the host immune response, ischemia-reperfusion, and HLA restrictions. Strategies such as the transplantation of 3D cultured vascularized organoids can attenuate inflammatory factors. In addition, HLA-universal iPSCs and immunosuppressive pre-treatment offer potential solutions to reduce graft immunogenicity and minimize immune rejection.
Notably, our on-going studying in humanized have highlighted the critical
barrier of macrophages phagocytosis in the case of hepatocyte xenotransplantation
into rat, indicating the significant importance of remolding the macrophage
activity, particularly in bigger animals other than mice [40]. Transgenic
expression of human SIRP
Taken together, a comprehensive understanding of the PSC differentiation and maturation route, as well as advanced genetic manipulation of tumorigenesis and immunogenicity, is necessary to obtain large amounts of pure, safe, and functionally mature cell sources for practical use of iPSCs in humanized animal models.
In conclusion, the use of humanized animal models has provided invaluable insights into human disease progression and drug development. While mice have traditionally been the most widely used model in the past, it is believed that bigger models like rats are more important and urgent for biomedical and pharmaceutical research and applications. With our increasing understanding of immune rejection and tolerance mechanisms, as well as the advancement of gene editing technology, even larger humanized animal models such as pigs and monkeys are expected to be established in the future.
Human iPSCs have emerged as a promising source for humanized models and shown great potential due to their unlimited proliferative potential and ability to provide personalized platforms for drug and therapy innovation. However, challenges remain in achieving complete functionalities and maturity of induced cells without tumorigenesis risks through differentiation strategies. Further breakthroughs may be made through the combination of gene editing and chemical-defined modulation, multilineage organogenesis, optimization of culture microenvironment, to extend the number, functionality, and quality of desired cell types. Multi-organ humanized animal model is expected to be made to achieve complete human physiologic functions, such as metabolism, in animals. This will require a concerted effort to integrate different organ systems and establish crosstalk between them. Continued research in these areas will lead to a better understanding of human physiology and disease progression, ultimately leading to the development of more effective treatments and therapies for patients.
YWZ, JYG and YML designed the study. MM, JYG and YZN acquired and interpreted data; MM and JYG drafted the manuscript. MM, JYG, YWZ and YZN revised and advised on the manuscript. MM illustrated. All authors contributed to editorial revisions of the manuscript. All authors read and approved the final version of the manuscript. All authors have agreed to be accountable for all aspects of the work.
Not applicable.
We thank Li-Ping Liu, Hang Zhou and Min Ding for their research assistance and discussions.
This research was funded partly by the National Natural Science Foundation of China (82070638, 82270697 and 82370517), Jiangsu Provincial Medical Key Discipline Cultivation Unit (JSDW202229), the Science and Technology Planning Project of Guangdong Province (2021B1212040016), Japan Society for the Promotion of Science (JSPS), KAKENHI (JP18H02866), and the Grant for International Joint Research Project of the Institute of Medical Science, the University of Tokyo.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.