








1 Department of General Practice, Yangpu Hospital, School of Medicine, Tongji University, 200090 Shanghai, China
2 Department of Spinal Surgery, Shanghai East Hospital, School of Medicine, Tongji University, 200092 Shanghai, China
Abstract
Background: Epigenetic modifications, such as transcription, DNA repair, and replication significantly influence tumour development. Aberrant gene expression and modifications can have a crucial impact on the initiation and progression of tumours. The minichromosome maintenance (MCM) protein family, which is responsible for DNA synthesis, plays a crucial role in tumorigenesis and chemotherapy resistance by regulating the cell cycle and DNA replication stress. Recent studies have shown that dysregulation of the MCMs can lead to these negative outcomes. This study aimed to examine the role of the MCM proteins in DNA synthesis in 33 types of cancers. Methods: Various public databases were used to examine the expression, methylation regulation, mutations, and functions of eight MCM proteins (MCM2–9) in pan-cancer. The study investigated the correlation between abnormal MCM expression and clinical outcomes, including prognosis and drug response. The microRNA–mRNA network upstream of the MCM genes and the downstream signalling pathways were extensively investigated to determine the molecular mechanisms that drive tumour development. Results: The study found that the MCM gene expressions differed depending on the type of cancer; high MCM gene expression was linked to poor overall survival in most cancers. Additionally, MCM gene expression was associated with various immunological features and drug sensitivity. These findings offer important insights for the development of targeted cancer therapies. Conclusions: Altogether, this study reveals that the MCM genes are differentially expressed across various cancers and are associated with clinical prognoses. These genes may influence the occurrence and development of tumours through several pathways, including the PI3K–AKT, PAS/MAPK and TSC/mTOR signalling pathways and immune-related pathways.
Keywords
- minichromosome maintenance family
- tumour immunity
- methylation
- pan-cancer
- multi-omics
- cancer biomarkers
Abnormal DNA replication in cells is a crucial factor in the development of
tumours, thereby making it a significant area of focus in cancer research. The
first minichromosome maintenance (MCM) protein was identified in Saccharomyces
cerevisiae and is considered crucial for maintaining extrachromosomal DNA
replication [1]. The MCM2–7 complex possesses helicase activity, thereby playing
a key role in the formation of the pre-replication complex. Additionally, it is
responsible for recruiting DNA polymerase during DNA unwinding and eventually
initiates both DNA replication and elongation [2]. The MCM2–7 complex is
composed of six nuclear proteins that belong to the MCM family [3]. MCM8 and MCM9
are homologous to MCM2–7 and are capable of repairing DNA double-strand breaks
in cells [4]. However, any changes in the MCM protein functions can contribute
towards tumour development. Various studies have found that the MCM
genes are significantly expressed in various types of cancers and result in
disruptions to the cell cycle [5]. The aberrant expression of the MCM
genes contributes to the initiation and progression of various types of cancers,
such as gastrointestinal, lung, brain, kidney, breast, ovarian, and
haematological malignancies [6, 7, 8, 9, 10, 11, 12]. MCM proteins play a significant role in the
regulation of cell cycle progression and cell proliferation by interacting with
various proteins. Specifically, the interaction between MCM7 and the
retinoblastoma (Rb) protein controls cell cycle progression [13, 14]. miR-885-5p
has the ability to hinder the growth of neuroblastoma cells by binding to the
MCM5 3
RNA sequencing and single nucleotide variation (SNV) data were extracted from The Cancer Genome Atlas (TCGA) database (https://gdc.cancer.gov/). In addition, publicly available data on copy number variation (CNV), methylation, and clinical characteristics of patients were collected. To perform differential expression analysis of MCM family members in various types of cancer, normalized and batch-corrected RSEM mRNA expression data were used. Analysis was conducted on over 10 paired tumours and normal samples from TCGA. Clinical data were obtained from tumour samples of nine cancer types and used to investigate gene expression changes associated with different subtypes. The gene set variation analysis (GSVA) score was used as a measure of gene set expression since it is positively correlated with gene expression. It was calculated using the GSVA R package (version 4.1.1, R Foundation for Statistical Computing, Vienna, Austria). To compare GSVA scores between groups, the Wilcoxon test was used in cases where the number of subtype groups was 2, and ANOVA was used in cases where the number of subtype groups exceeded 2. GSEA was implemented using the fgsea R package (version 4.1.1, R Foundation for Statistical Computing, Vienna, Austria) to determine the overexpression levels of a gene set at the top or bottom of the list of genes. This analysis was based on the gene expression fold change (FC) values between the tumour samples and normal samples.
The GSCALite platform was used for the genome-wide analysis of the MCM genes [17]. Differential expression of MCM family members was evaluated using RSEM-normalised RNA-seq data. A heatmap and waterfall plot were constructed to visualise the SNV and CNV frequencies of the MCM genes. The CNV data were obtained from 11,495 samples in the TCGA database, while GISTIC2.0 was used to identify regions of significant amplification or deletion in various patient groups [18].
The DNA methylation data (Illumina Human Methylation 450k level 3) were extracted from the TCGA database for 14 cancer types. More than 10 pairs of tumours and adjacent normal tissues were analysed. Multiple methylation sites were typically present in a single gene, each with its tag storing the methylation level.
In this study, we collected clinical data from 33 different types of cancers for the purpose of conducting survival analysis. To integrate methylation and clinical survival data, sample barcodes were used, and tumour samples were categorized into high- and low-methylation groups based on the median methylation level. In this study, we utilized the survival package in R (version 4.1.1, R Foundation for Statistical Computing, Vienna, Austria) to build Cox proportional hazards models and conduct log-rank tests on MCM genes across various types of cancers. A p-value of less than 0.05 was deemed statistically significant.
Pathway activity scores (PAS) for cancer-related pathways were calculated using RPPA data extracted from the TCPA (The Cancer Proteome Atlas, http://www.tcpaportal.org) database. In order to estimate differences in PASs between groups, the Student’s t-test was utilized, with p-values being adjusted through the False Discovery Rate (FDR) method. Any FDRs that were equal to or less than 0.05 were considered to indicate significant differences [19, 20].
The IC
The study utilized the immune infiltration and GSVA scoring modules to investigate the correlation between gene expression and immune cell infiltration. Gene set expression was assessed by estimating GSVA scores, which were positively correlated with gene set expression.
The study utilized Student’s t-test to determine variations in the MCM
family expression and methylation levels in tumour tissues compared to their
levels in corresponding normal tissues. The log-rank test was utilized to compare
survival curves, while Pearson or Spearman analyses were utilized to estimate
correlation coefficients. A p-value of less than 0.05 was deemed
statistically significant. All statistical analyses were conducted using
SangerBox database (http://www.sangerbox.com/, Sanger Box 3.0; Hangzhou, China)
[21] and ChiPlot (https://www.chiplot.online/, Shantou, China), free online
platforms for data analysis (*, p
Paired tumour and normal tissue gene expression data from 33 types of cancers in the TCGA database were used to analyse the differential expression of the MCM genes. The results revealed that MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 were upregulated in most tumour tissues, whereas MCM8 and MCM9 were downregulated in certain types of tumours. Specifically, MCM8 was downregulated in kidney renal clear cell carcinoma (KIRC) and thyroid carcinoma (THCA), whereas MCM9 was downregulated in kidney chromophobe (KICH), KIRC, and kidney renal papillary cell carcinomas (KIRP) (Fig. 1A). GSVA was used to determine the gene set expression. The GSVA scores for bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), oesophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), KICH, KIRC, KIRP, liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), stomach adenocarcinoma (STAD), and THCA were significantly higher than in the corresponding adjacent normal tissues (Fig. 1B). The expressions of MCM2–8 have been found to be significantly correlated with BRCA subtypes. Notably, there was a significant difference in the expressions of MCM2–8 proteins between the subtypes of luminal A and luminal B, thereby suggesting that they could potentially be used as biomarkers to differentiate between the two subtypes (Fig. 1C, Supplementary Fig. 1). An assessment of the standardised enrichment scores (NESs) revealed that the MCM gene set was significantly enriched in several types of cancers, including HNSC, ESCA, COAD, LUSC, LIHC, BLCA, STAD, BRCA, and LUAD (Fig. 1D), especially in HNSC and ESCA (Fig. 1E,F). These findings indicate that irregular expression of the MCM genes plays a role in the onset of multiple forms of cancer.
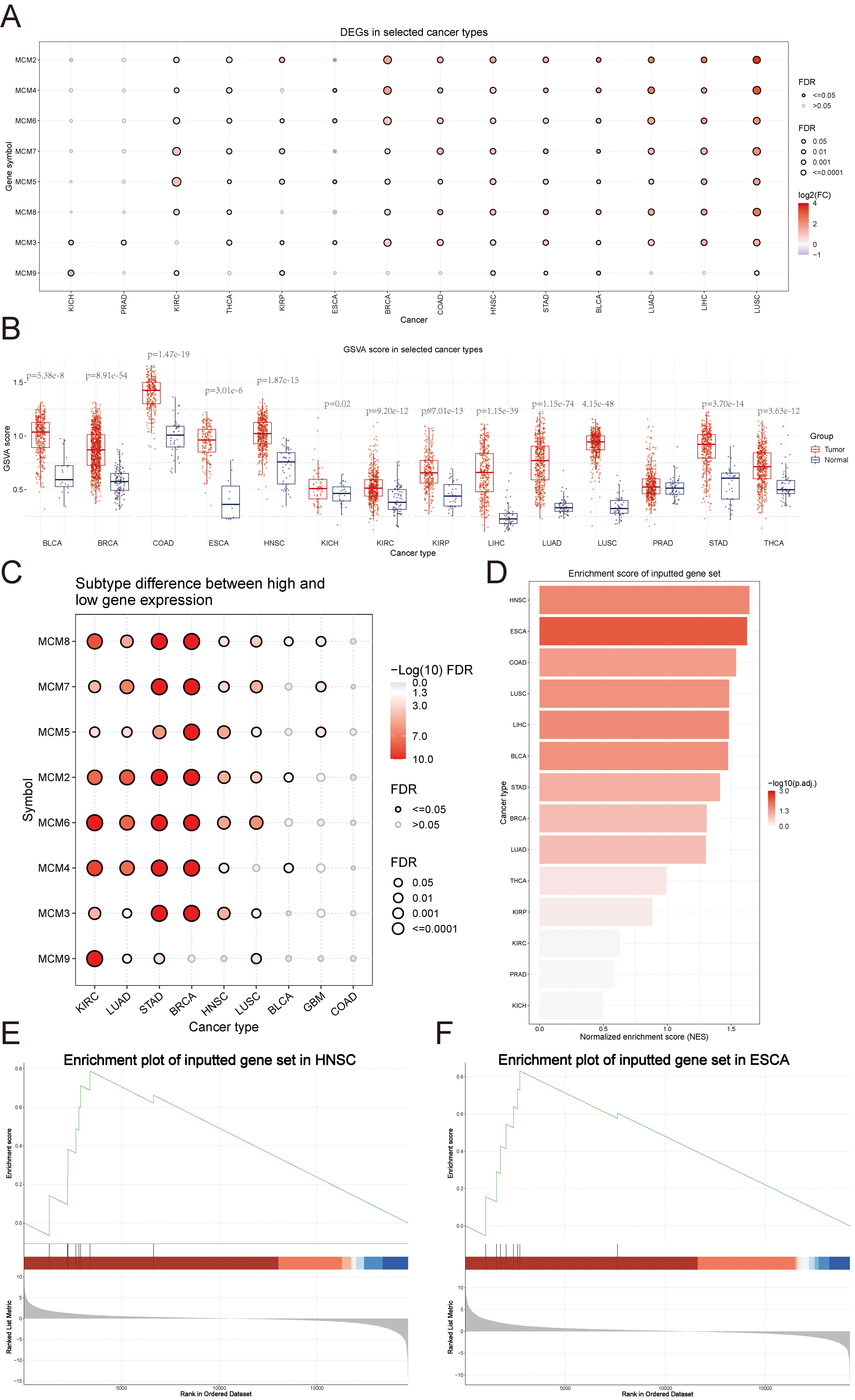 Fig. 1.
Fig. 1.Comparison of the Minichromosome Maintenance Proteins (MCM)
family mRNA expression in various types of cancer. (A) Differential expression
of MCM genes between 14 paired normal and tumour tissues. Purple to red
represents the fold change between tumour tissues and normal tissues. Red dots
indicate that gene expression is higher in tumour tissues than in normal tissues,
and blue dots indicate that gene expression is lower in tumour tissues than in
normal tissues. The size of the dots indicates significance based on the False
Discovery Rate (FDR), while the FDR value of each dot is shown on the right. (B)
The gene set variation analysis (GSVA) score was calculated by comparing the
molecular characterization maps (MCMs) between 14 paired normal and tumour
tissues. (C) Relationship between the MCM gene expressions and subtypes.
FDR values are represented by the bubble’s colours and sizes in the graph. Rows
represent gene set symbols, whereas columns represent selected cancer types.
Significance based on FDR values is indicated by the colour of the bubbles, with
white indicating low significance and red indicating high significance. Bubble
size positively correlates with significance. (D) Summary of the enrichment
scores (ES) of MCM genes in the selected cancers. The score indicates
the extent to which a set of genes is overrepresented at either the top (ES
To identify the various genetic alterations in the MCM genes, the
frequency and types of SNVs were evaluated across the 33 types of cancers.
Uterine corpus endometrial carcinoma (UCEC) had the highest SNV frequency
(198%), followed by skin cutaneous melanoma (SKCM) (110%), STAD (77%), LUAD
(67%), COAD (66%), BLCA (63%), LUSC (56%), HNSC (40%), BRCA (32%),
glioblastoma multiforme (GBM) (26%), LIHC (25%), cervical squamous cell
carcinoma and endocervical adenocarcinoma (CESC) (22%), rectum adenocarcinoma
(READ) (19%), OV (15%), KIRC (12%), prostate adenocarcinoma (PRAD) (12%), and
brain low-grade glioma (LGG) (12%). The SNV frequency was
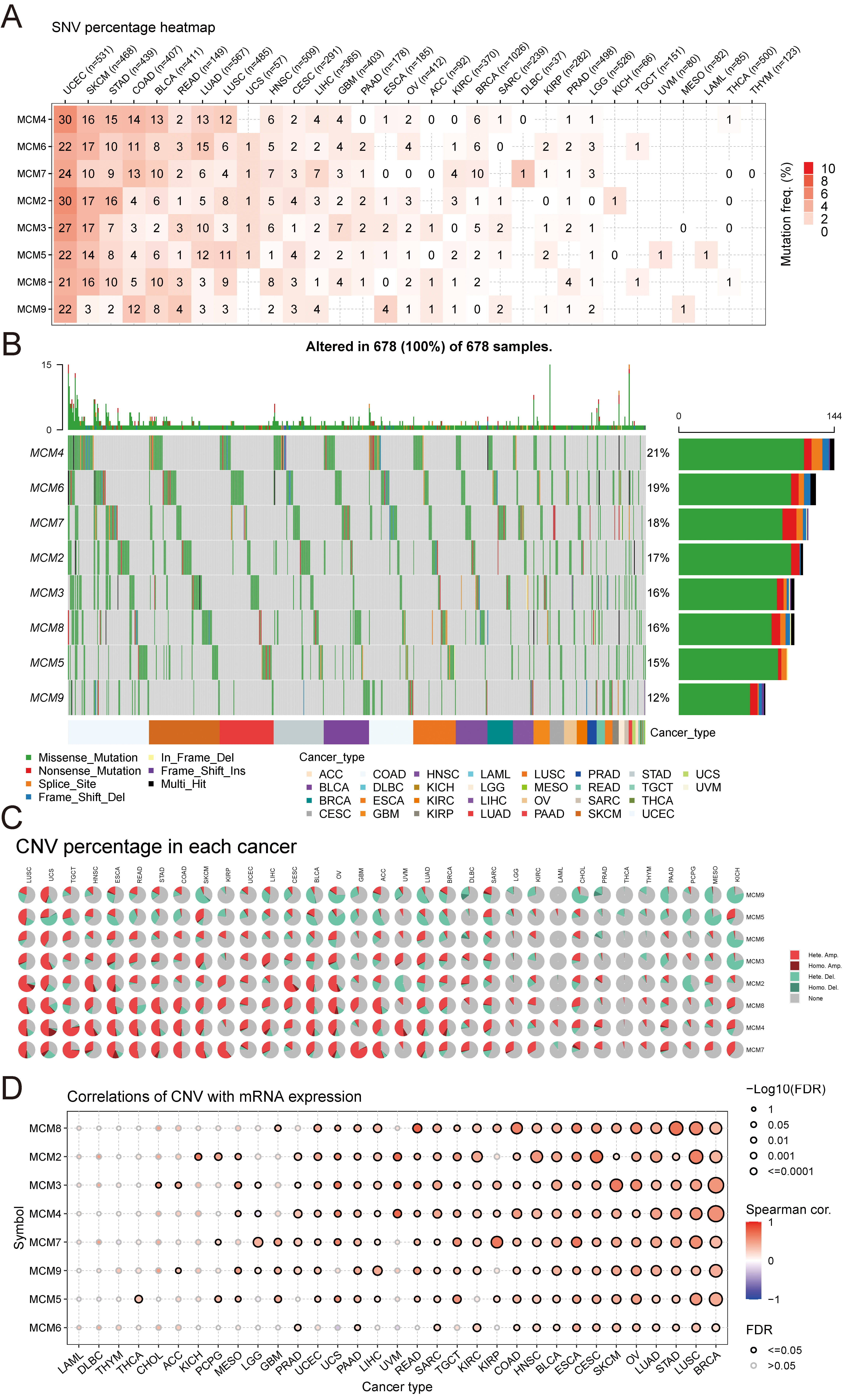 Fig. 2.
Fig. 2.Relationship between the MCM gene expressions and
genomic alterations. (A) Heatmap demonstrating the SNV frequency in
MCM2–9 genes in tumours. (B) Waterfall plot demonstrating SNV
distribution and its classification in MCM genes. (C) Pie chart
demonstrating the proportion of different types of copy number variations (CNVs)
in each gene across various cancer types. (D) Bubble plots demonstrating the
relationship between mRNA expression and CNV frequency. The colour of the bubbles
indicates the strength of correlations, with blue bubbles representing negative
correlations and red bubbles representing positive correlations. The bubble sizes
are proportional to the significance, based on the FDR values. Black borders
indicate an FDR of
The study found a strong negative correlation between the methylation levels and mRNA expression in most MCM family members. However, this correlation was not observed in MCM7 in CHOL and MCM3 in CESC, DLBC, KICH, KIRC, acute myeloid leukaemia (LAML), OV, PAAD, pheochromocytoma and paraganglioma (PCPG), PRAD, THYM, and UCS (Fig. 3A). Analysis of the differential methylation revealed that the methylation levels of the MCM genes were significantly lower in tumour tissues compared to the matched normal tissues: MCM2 in BLCA, BRCA, ESCA, HNSC, KIRC, KIRP, LIHC, LUAD, LUSC, PAAD, THCA, and UCEC; MCM3 in BLCA, BRCA, LIHC, LUSC, and PRAD; MCM4 in BRCA, ESCA, KIRC, LUAD, and THCA; MCM5 in BLCA, BRCA, COAD, KIRC, LIHC, LUAD, LUSC, PAAD, PRAD, and UCEC; MCM6 in BLCA, BRCA, HNSC, KIRC, LIHC, LUAD, LUSC, THCA, and UCEC; MCM7 in KIRP, LIHC, LUAD, PRAD, and THCA; MCM8 in BRCA and PRAD; MCM9 in BLCA, KIRP, LIHC, LUAD, LUSC, PRAD, and UCEC. However, the methylation levels of the following genes were higher in the tumour tissues than in the matched normal tissues: MCM3 in KIRC, KIRP, and THCA; MCM4 in KIRP; MCM5 in ESCA; MCM6 in PRAD; MCM7 in BRCA and LUSC; MCM8 in KIRC, LUSC, and THCA; MCM9 in BRCA, KIRC, and PAAD (Fig. 3B). Hypomethylation of MCM2–9 in various types of cancers was associated with an increased risk of mortality (Fig. 3C). The study found that hypermethylation of MCM2, MCM3, MCM7, and MCM8 was a significant risk factor for survival in certain types of cancers, including UVM, LGG, CESC, and ACC (Fig. 3D–H). Altogether, these results suggest that abnormal DNA methylation regulates abnormal expression of MCMs, thereby influencing tumour progression.
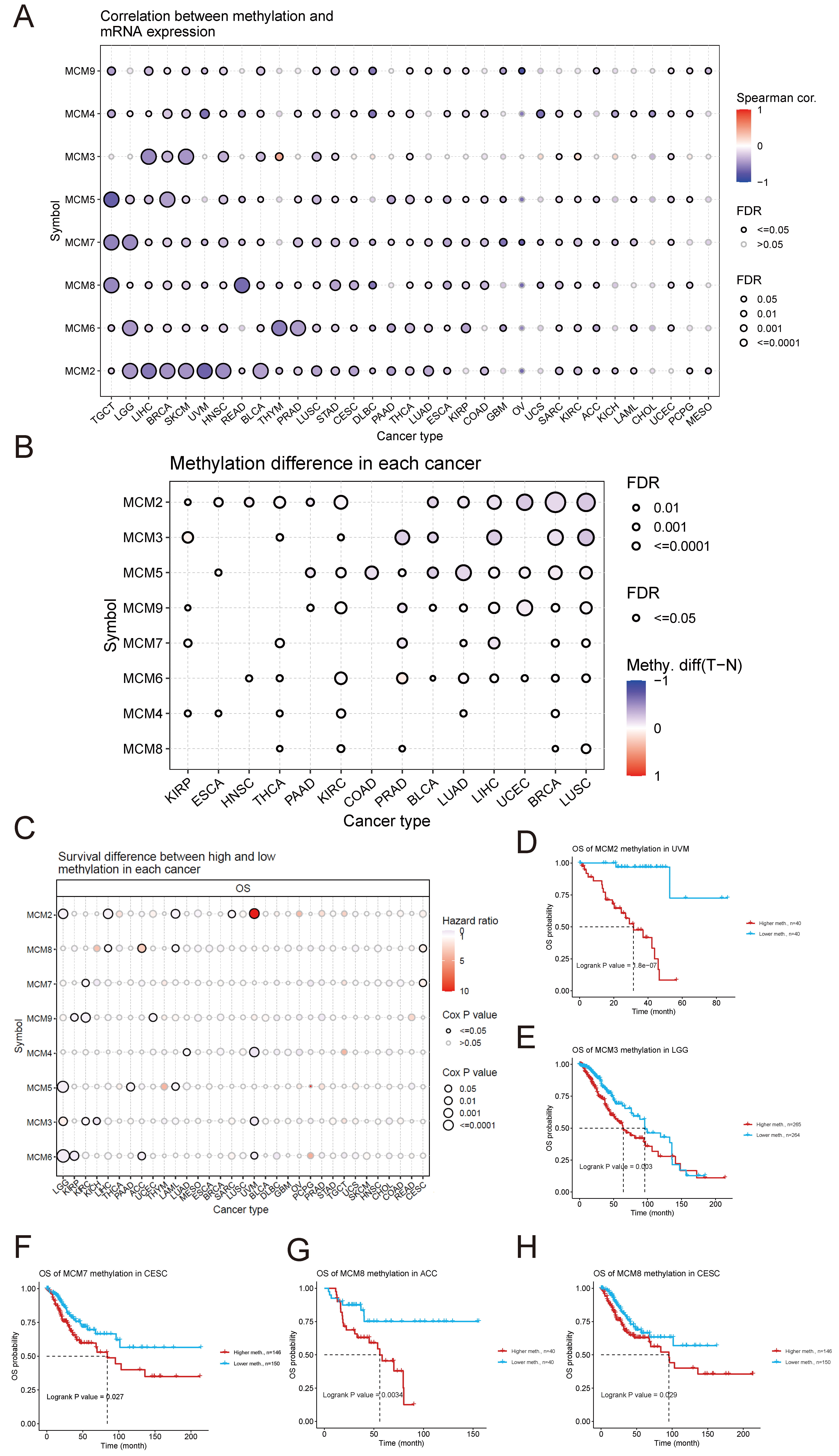 Fig. 3.
Fig. 3.Methylation alterations in MCM2–9 genes and overall
survival (OS). (A) Correlation between the mRNA expression and methylation
levels of MCM genes. Negative correlations are represented by blue
bubbles, whereas positive correlations are represented by red bubbles. The
intensity of the colour reflects the strength of the correlation, with darker
colours indicating a stronger correlation. The size of the bubbles is positively
correlated with significance, based on FDR values. Black borders indicate an FDR
of
The survival analysis results indicate that in most cancer types, a high expression of MCM2–9 was linked to a poor prognosis. However, a high expression of certain MCM genes was associated with a better prognosis in specific cancer types: MCM2 in CESC, STAD, and UVM; MCM3 in CESC, OV, STAD, and THCA; MCM4 in READ and THYM; MCM5 in CESC and THCA; MCM6 in CESC, READ, and THYM; MCM7 in DLBC and THCA; MCM9 in THYM (Fig. 4A–C). Various tumours were found to be influenced by the MCM2–9 genes, in terms of their pathological staging. Specifically, MCM4, MCM6, and MCM2 were associated with KIRP; MCM7, MCM5, MCM6, and MCM2 were associated with KIRC; MCM3 and MCM2 were associated with BRCA; MCM5 and MCM6 were associated with THCA; MCM7 was associated with KICH; MCM8 was associated with SKCM; MCM3 was associated with TGCT (Fig. 4D). According to the findings for KICH, a positive correlation existed between MCM2 expression and the pathological stage. This suggests that tumours with higher stages tend to exhibit higher levels of MCM2 expression (Fig. 4E). Furthermore, the MCM2–9 genes exhibited varying levels of expression in different clinical stages of tumours. Specifically, MCM4, MCM6, and MCM2 were differentially expressed in different clinical stages of KIRP (Fig. 4F). While the expression of MCM3 increased with the clinical stage in TGCT (Fig. 4G). This study suggests that the abnormal expression of MCM genes is closely linked to cancer prognosis.
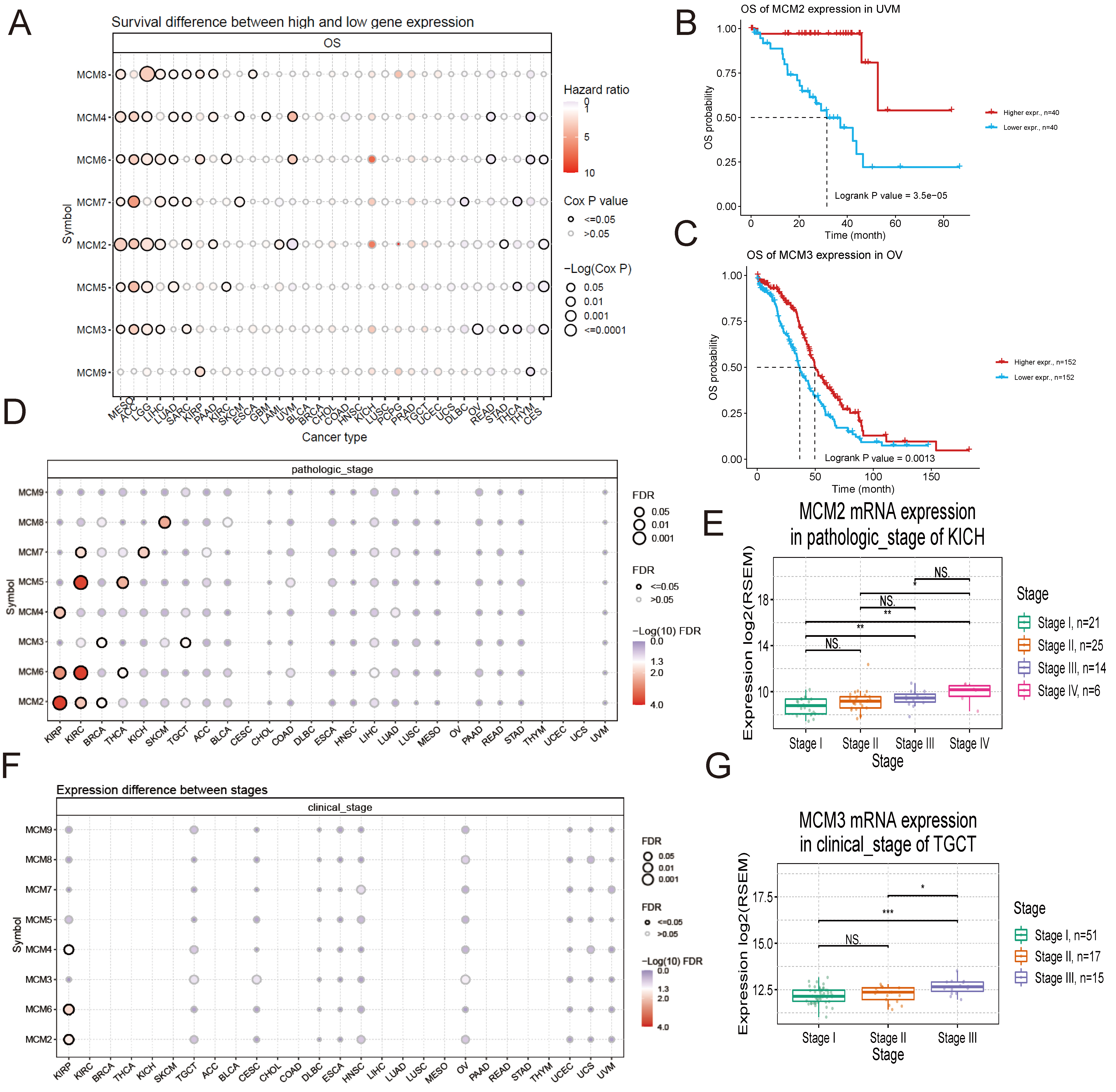 Fig. 4.
Fig. 4.Relationship between MCM gene expressions and overall
survival (OS) in pan-cancer. (A) Heatmap demonstrating the univariate Cox
proportional-hazards regression analysis –log
This study investigated the correlation between MCM gene expressions
and immune cell infiltration in the tumour microenvironment (TME). MCM2
expression significantly correlated with the immune, stromal, and
microenvironmental scores. A negative correlation was observed between the immune
scores and MCM2 expression in cancers, including GBM, UCEC, ESCA,
stomach and oesophageal carcinoma (STES), SARC, KIRP, LUSC, high-risk Wilms
tumour (WT), neuroblastoma (NB), TGCT, and PCPG (Fig. 5A). In this study, we
investigated the relationship between the infiltration levels of 24 immune cell
types and GSVA scores. Our findings suggest the existence of two distinct
clusters of cancer types based on this correlation (Fig. 5B). The two clusters
exhibited different patterns of immune cell infiltration. The MCM gene
expressions positively correlated with the infiltration levels of the
immunosuppressive cells in cancers, including the natural regulatory T (nTreg)
cells, induced regulatory T (iTreg) cells, exhausted, dendritic cells (DCs), and
macrophages. However, the expression of the MCMs was significantly negatively
correlated with the infiltration levels of the immune effector cells, including
the natural killer (NK) cells, CD8
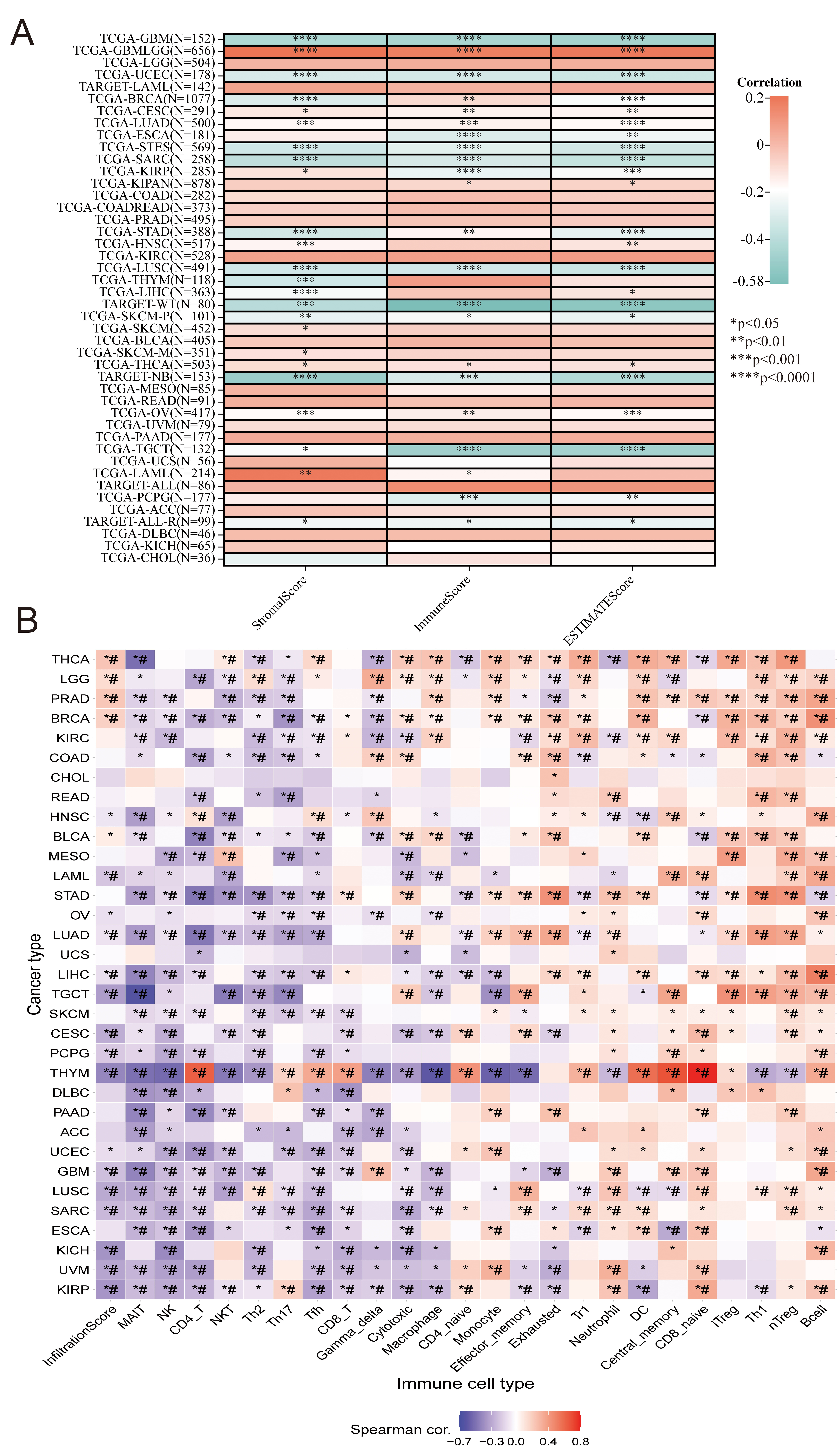 Fig. 5.
Fig. 5.Relationship between the MCM gene expressions and the
tumour microenvironment. (A) Heatmap demonstrating the correlation between
MCM2 expression and immune, stromal, and microenvironmental scores. (B)
Relationship between the MCM gene expressions and immune cell
infiltration. The heatmap summarises the significant correlations based on
p-values and FDRs from Spearman correlation analysis of GSVA scores and
immune cell infiltration. Blue represents the negative correlations, whereas red
represents positive correlations. Note: *, p
To investigate the impact of the MCM genes on drug efficacy, the study
analysed the correlation between gene expression and drug sensitivity using data
obtained from the GDSC and CTRP databases. The results indicated that the
increased expression of the MCM2–9 genes was linked to heightened
resistance to drugs, such as 17-AAG, RDEA119, trametinib, and selumetinib (Fig. 6A). Moreover, the study found that there was a negative correlation between the
expressions of MCM2–9 and drug sensitivity. This correlation was
validated by analysing the IC
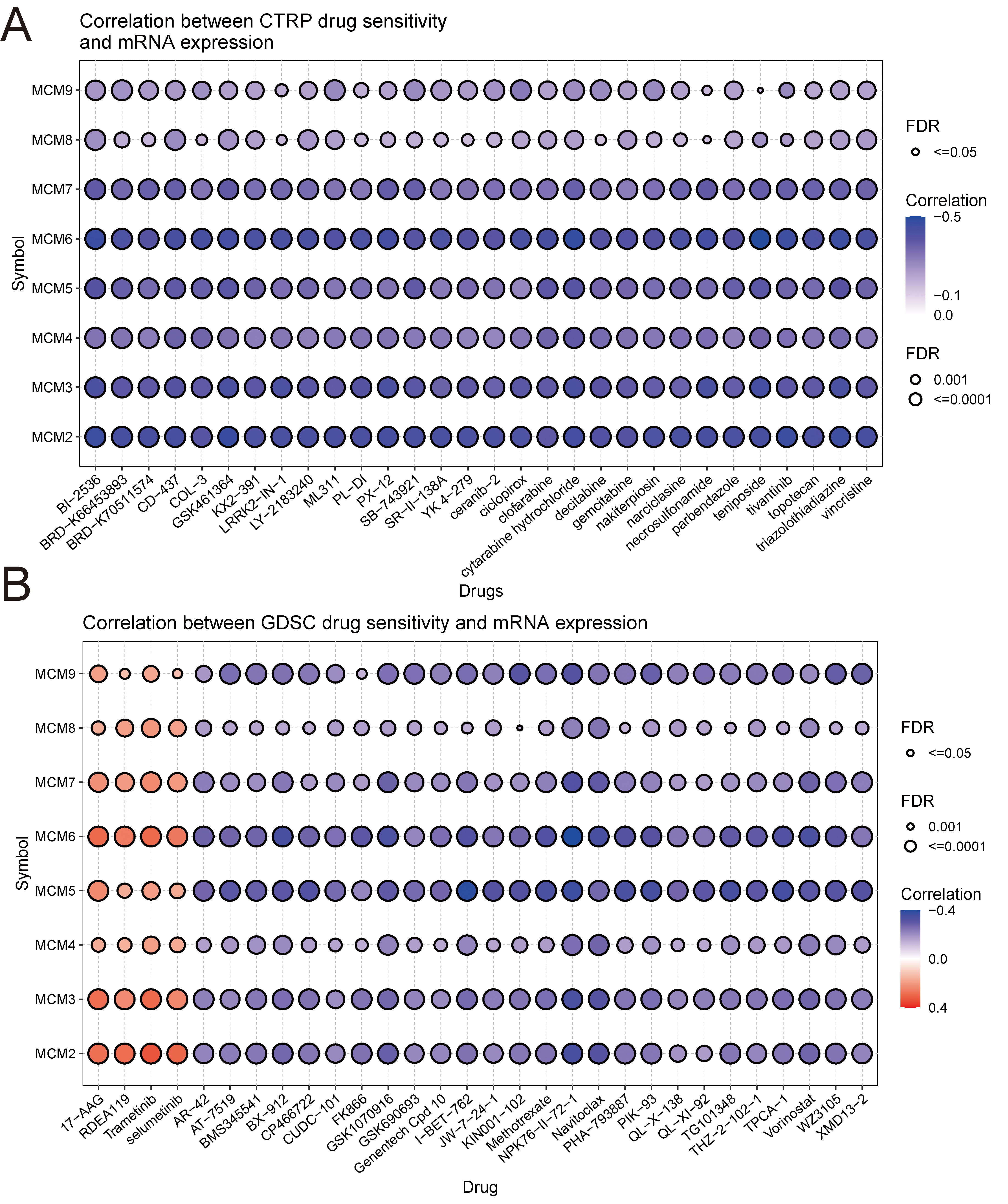 Fig. 6.
Fig. 6.Relationship between the MCM gene expressions and drug
sensitivity. (A) Correlation between the expressions of the MCM genes
and drug sensitivity based on Genome Therapy Response Portal (CTRP) data. (B)
Correlation between the expressions of the MCM genes and drug
sensitivity based on GDSC data. In the scatterplot, blue bubbles indicate
negative correlations and red bubbles indicate positive correlations, while the
bubble size positively correlates to significance based on FDR values. Darker
colours indicate stronger correlations. Additionally, black borders indicate FDR
values of
The miRNA–gene network analysis revealed that all eight MCM genes were regulated by multiple miRNAs (Fig. 7A). The expressions of the MCM genes were regulated by a complex network of miRNAs, which play an essential role in tumour development and progression. Pathway enrichment analysis revealed that the MCM2–9 genes were associated with several pathways, including the TSC/mTOR signalling, cell cycle-related, PI3K/AKT signalling, RTK signalling, RAS/MAPK signalling, and apoptosis-related pathways (Fig. 7B). The MCM genes had a positive impact on specific pathways in several cancers, whereby MCM7 and MCM5 were primarily involved in the activation of Androgen Receptor (AR) and Epithelial–Mesenchymal Transition (EMT), respectively (Fig. 7C,D). The GSVA demonstrated that the expressions of the MCM gene sets were linked to cell cycle regulation and apoptosis in cancer (Fig. 7E). This finding suggests that the MCMs have a significant role in controlling cancer-related pathways.
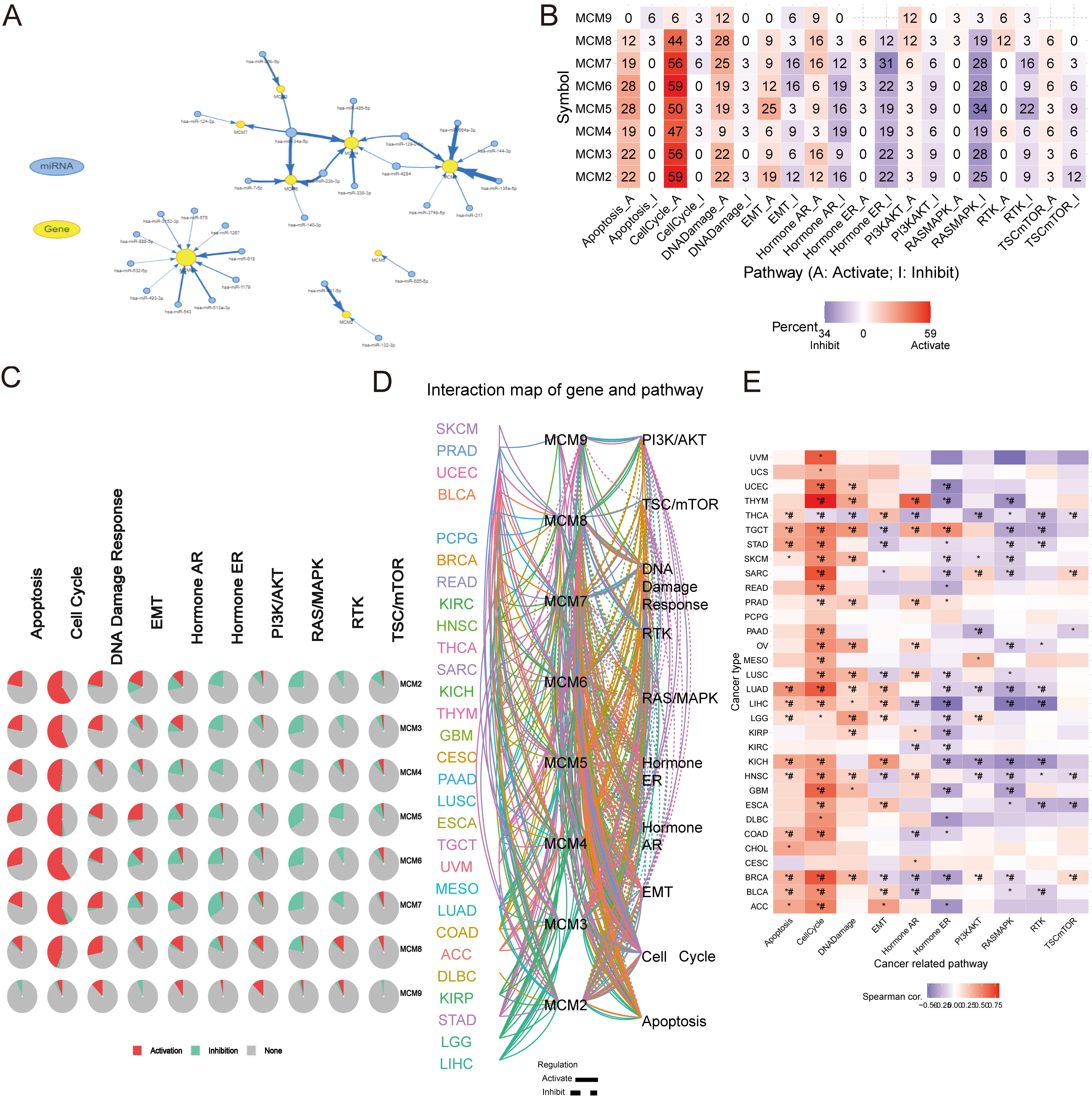 Fig. 7.
Fig. 7.Potential molecular mechanisms underlying altered MCM
gene expressions. (A) The miRNA regulatory network is representative of miRNAs
and target genes, whereby nodes represent miRNAs or target genes, edges represent
the miRNA-to-gene conversion regulation, and edge widths indicate the absolute
correlation coefficients. (B) The combined percentage of the effects of the MCM
complex on pathway activity. (C) Pie chart demonstrating the proportion of genes
contributing to the pathway activity across 32 cancer types. (D) A network
demonstrating the connection between genes and pathways using straight lines.
Activation is represented by solid lines, whereas inhibition is denoted by dashed
lines. The line colours are used to indicate different cancer types. (E)
Association between GSVA scores and cancer-related pathway activity. *,
p-value
Recently, DNA replication has emerged as a major research focus for investigating the occurrence and development of tumours. The MCM family comprises replicative DNA helicases that play an indispensable role in DNA replication and perform essential functions during all stages of the cell cycle [2]. Recent studies have demonstrated the significant roles MCM family members play in carcinogenesis. Abnormal expressions of the MCMs have been observed in various malignant tumours, such as cervical cancer, breast cancer, and human glioma. These findings suggest that the MCMs can be used to predict tumour development and prognosis [22, 23, 24]. This study investigated the correlation between MCM gene expressions and genomic alterations, TME features, prognosis, and drug sensitivity. It also identified the potential mechanisms through which the MCM family members contribute to tumour development. The results indicate that abnormal expressions of the MCMs could be a useful marker in early tumour diagnoses and in predicting treatment effectiveness.
The expressions of MCM2–9 were found to be elevated in most types of cancer. However, the expression of MCM3 was significantly downregulated in KICH and PRAD. The GSVA scores for the BLCA, BRCA, COAD, ESCA, HNSC, KICH, KIRC, KIRP, LIHC, LUAD, LUSC, STAD, and THCA tissues were significantly higher than for para-carcinoma tissues. Furthermore, survival analysis revealed that the high expressions of MCM2–9 were associated with a poorer prognosis in most tumour types, suggesting that the MCM2–9 genes play important roles in the pathological and clinical staging in the majority of tumours. These findings are consistent with those from previous studies, which suggested that abnormal MCM gene expressions were closely related to patient prognoses [25, 26, 27, 28, 29]. Breast cancer is a complex illness that can be classified into different subtypes, depending on gene expression patterns. An in-depth understanding of these subtypes may help to develop targeted therapies for each subtype, which can lead to more effective and individualised treatments [30, 31]. In this study, a strong correlation was observed between the expressions of MCM2–8 and the different BRCA subtypes. Specifically, the expressions of MCM2–8 were significantly different between luminal A and luminal B subtypes, indicating that the MCM2–8 genes are potential biomarkers for the stratification of patients with these two subtypes. In addition, the findings of this study suggest that the MCM genes are reliable predictors of prognosis in various types of cancer. Genomic instability is a major cause of cancer development. Although most cancer genomes undergo regular changes in chromosomal regions, the exact regulatory mechanisms that promote copy number alterations in specific regions of the genome remain unclear [32, 33]. Various mechanisms underlying the generation of CNVs have been proposed, including the stalling of the replication forks or the induction of double strand breaks during DNA replication. Another mechanism that leads to the generation of CNVs is the use of damaged intermediates as primers, which re-fuse specific segments of DNA back into the genome, ultimately, causing gene duplication or deletion [34]. In this study, frequent variations in the gene copy numbers were observed in the MCM complex. Our study revealed a significant positive correlation between the frequency of the copy number variations (CNVs) and the mRNA expression of the MCM members. Among the MCM genes, MCM4 and MCM6 had the highest mutation frequencies, with missense mutations being the primary type. These results indicate that alterations in copy number can have an impact on the expressions of the MCM genes, which could potentially lead to the development of tumours.
This study provides evidence that the expression of the MCM family members is significantly influenced by complex regulations at both the genomic and epigenomic levels. The methylation patterns of the MCM genes vary greatly among different types of cancer. However, with the exceptions of MCM7 in CHOL and MCM3 in CESC, DLBC, KICH, KIRC, LAML, OV, PAAD, PCPG, PRAD, THYM, and UCS, the expression of most MCM genes correlated negatively with their methylation levels. The study found that hypomethylation of MCM2–9 was generally associated with an increased risk of mortality in most types of cancer. However, in UVM and LGG, hypermethylation of MCM2 and MCM3, respectively, was found to be a risk factor. In CESC, hypermethylation of MCM7 was identified as a risk factor, while in ACC and CESC, hypermethylation of MCM8 was found to be a risk factor. Upregulated MCM2 expression has been associated with promoter methylation and clinical characteristics of patients [35]. Therefore, the aberrant expression of the MCM genes in tumours may be influenced by abnormal DNA methylation, which eventually affects the prognosis.
Targeting the TME has become a promising approach for cancer treatment in recent
years and our study found a significant correlation between MCM2
expression and immune, stromal, and microenvironmental scores. We also evaluated
the correlation between the infiltration levels of 24 different types of immune
cells and GSVA scores [36]. The MCM gene expressions correlated
positively with the infiltration levels of the immunosuppressive cells in ACC,
BLCA, BRCA, CESC, CHOL, COAD, DLBC, ESCA, HNSC, and KIRC. However, it correlated
negatively with the infiltration levels of NK cells, CD8
The increased expressions of the MCM genes can lead to apoptosis, hinder cell cycle progression, and trigger the DNA damage response in different types of cancer [15, 16, 37, 38]. In addition, the MCM genes affect tumour invasion and metastasis [39, 40]. The study highlights the crucial role the MCM2–9 genes play in regulating cancer-related pathways, such as PI3K/AKT signalling, RTK signalling, RAS/MAPK signalling, and apoptosis-related pathways. Drug sensitivity analysis revealed that the MCM gene expressions correlated with the resistance to 17-AAG, RDEA119, trametinib, and selumetinib. However, the mechanisms through which these drugs affect the expressions of the MCM genes and tumour development require further investigation.
Despite the significant advancements in cancer treatment, there are still several crucial matters that need to be resolved. Specifically, future studies should focus on elucidating the specific mechanisms underlying the high expression of MCM proteins in certain cancer types and investigating the associated genetic and epigenetic changes. In addition, the mechanisms through which the MCM family is regulated in malignant tumours warrant further investigation. To improve therapeutic strategies for cancer, it is crucial to identify potential upstream regulators of the MCM members and understand the mechanisms through which they affect them at the transcriptional and post-transcriptional levels. A comprehensive understanding of the MCM family can aid in the development of more effective treatments for cancer.
In this study, our focus was to investigate the expression and function of eight key MCMs within tumors and the TME. We explored genomic alterations and the miRNA network to uncover additional mechanisms that contribute to the dysregulation of MCMs in cancer. Our findings are consistent with previous research and also provide new insights for future investigations. Additionally, we discovered that these genes have significant effects on the TME and drug resistance, which can offer valuable insights for developing cancer treatment strategies. These findings may open up possibilities for alternative approaches in managing clinically refractory cancers.
ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; COADREAD, colon adenocarcinoma/rectum adenocarcinoma oesophageal carcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, oesophageal carcinoma; FPPP, FFPE Pilot Phase II; GBM, glioblastoma multiforme; GBMLGG, glioma; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIPAN, pan-kidney cohort (KICH + KIRC + KIRP); KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukaemia; LGG, brain low-grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OVO, Varian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; STAD, stomach adenocarcinoma; SKCM, skin cutaneous melanoma; STES, stomach and oesophageal carcinoma; TGCT, testicular germ cell tumours; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma; OS, osteosarcoma; ALL, acute lymphoblastic leukaemia; NB, neuroblastoma; WT, high-risk Wilms tumour.
All raw data can be provided upon request.
LW conceived the study, drafted the manuscript, and performed the analysis. XL analyzed and interpreted the data and ultimately reviewed the manuscript. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
Not applicable.
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.