























 , Lulu Qin 1,*, Rufeng Lei 1
, Lulu Qin 1,*, Rufeng Lei 11 School of Information Engineering, Jingdezhen Ceramic University, 333403 Jingdezhen, Jiangxi, China
Abstract
Background: 5-methylcytosine (m5C) is a key post-transcriptional modification that plays a critical role in RNA metabolism. Owing to the large increase in identified m5C modification sites in organisms, their epigenetic roles are becoming increasingly unknown. Therefore, it is crucial to precisely identify m5C modification sites to gain more insight into cellular processes and other mechanisms related to biological functions. Although researchers have proposed some traditional computational methods and machine learning algorithms, some limitations still remain. In this study, we propose a more powerful and reliable deep-learning model, im5C-DSCGA, to identify novel RNA m5C modification sites in humans. Methods: Our proposed im5C-DSCGA model uses three feature encoding methods initially—one-hot, nucleotide chemical property (NCP), and nucleotide density (ND)—to extract the original features in RNA sequences and ensure splicing; next, the original features are fed into the improved densely connected convolutional network (DenseNet) and Convolutional Block Attention Module (CBAM) mechanisms to extract the advanced local features; then, the bidirectional gated recurrent unit (BGRU) method is used to capture the long-term dependencies from advanced local features and extract global features using Self-Attention; Finally, ensemble learning is used and full connectivity is used to classify and predict the m5C site. Results: Unsurprisingly, the deep-learning-based im5C-DSCGA model performed well in terms of sensitivity (Sn), specificity (SP), accuracy (Acc), Matthew’s correlation coefficient (MCC), and area under the curve (AUC), generating values of 81.0%, 90.8%, 85.9%, 72.1%, and 92.6%, respectively, in the independent test dataset following the use of three feature encoding methods. Conclusions: We critically evaluated the performance of im5C-DSCGA using five-fold cross-validation and independent testing and compared it to existing methods. The MCC metric reached 72.1% when using the independent test, which is 3.0% higher than the current state-of-the-art prediction method Deepm5C model. The results show that the im5C-DSCGA model achieves more accurate and stable performances and is an effective tool for predicting m5C modification sites. To the authors’ knowledge, this is the first time that the improved DenseNet, BGRU, CBAM Attention mechanism, and Self-Attention mechanism have been combined to predict novel m5C sites in human RNA.
Keywords
- RNA
- 5-methylcytosine site identification
- DenseNet
- BGRU
- improved CBAM attention
- self-attention
- deep learning
- ensemble learning
Post-transcriptional modifications are an essential area in bioinformatics research, with more than 170 RNA modifications having been identified [1]. RNA undergoes a variety of post-transcriptional chemical modifications, including N1-methyladenosine (m1A), N7-methylguanosine (m7G), N4-methylcytosine (m4C), 5-methylcytosine (m5C), 5-hydroxymethylcytosine (hm5C), and N6-methyladenosine (m6A) [2]. Among them, 5-methylcytosine (m5C) is one of the most common modifications involved in various cellular processes. Additionally, 5-methylcytosine (m5C) is a widespread mRNA modification that occurs in the untranslated region of mRNA transcripts [3, 4]. It is essential for many biological functions, including tRNA recognition, RNA metabolism, and stress response. Studies have shown that m5C modification sites play an important regulatory role in many aspects of gene expression, including ribosomal reorganization, translation, and RNA output [5]. Furthermore, m5C modification sites have been associated with the development of many cancers and diseases, such as lung cancer, liver cancer, breast cancer, autosomal recessive mental retardation, amyotrophic lateral sclerosis, and Parkinson’s disease [6, 7, 8, 9]. Therefore, the accurate identification of m5C modification sites in RNAs is significant for revealing the epigenetic regulation of related diseases and understanding the mechanisms and functions of such modifications.
In recent years, RNA modifications have received increasing attention and many computational methods have been developed to predict m5C modification sites in RNA. Some high-throughput sequencing techniques, such as oxidative bisulfite sequencing [10], bisulfite sequencing [11], m5C-RIP-seq [12, 13], Aza-IP-seq, and miCLIP-seq [14, 15], have been frequently used in the past to identify m5C modification sites in RNA. However, these methods are both costly and time-consuming. Therefore, a series of excellent models based on machine learning algorithms have been applied to m5C-modified sites, such as m5Cpred-SVM [16], m5Cpred-XS [17], iRNA5hmC [18], and Staem5 [19]. However, machine learning algorithms are only suitable for small-scale datasets and may not perform as well on larger data volumes. Deep-learning algorithms can automatically process large-scale datasets and can better extract the original features of the sequence, which improves the performance of the model. For example, Ali et al. [20] developed the iRhm5CNN model, which is an efficient and reliable computational prediction model for identifying RNA 5hmC sites. They extracted features from RNA sequences using one-hot encoding and achieved a better performance by using the convolutional neural network structure in deep learning. Therefore, there is a need to identify or develop a novel and effective deep-learning method to identify m5C modification sites in human RNA.
The traditional convolutional neural network (CNN) [21, 22] is more prone to gradient disappearance problems when the network depth is deeper, while the ResNet [23] can train deeper CNN models to achieve a higher accuracy (Acc). The ResNet has a large number of parameters during the learning process, while the densely connected convolutional network (DenseNet) [24] enhances feature propagation to greatly reduce the number of parameters and alleviate the gradient disappearance problem. These advantages allow the use of DenseNet to achieve better performance than ResNet with fewer parameters and computational costs. For example, Wang et al. [25] designed a predictor named MDCAN-Lys in 2020, which used DenseNet to identify lysine acetylation sites, while its use obtained excellent experimental results on an independent test dataset. Subsequently, Jia et al. [26] proposed a predictor for DeepDN_iGlu using the DenseNet and attention mechanism to predict lysine glutarylation sites. Therefore, our proposed im5C-DSCGA model introduces an improved DenseNet to extract more advanced local features in RNA sequences.
The traditional recurrent neural network (RNN) [27] is prone to problems of gradient disappearance or gradient explosion during the learning process, making it difficult to capture the dependencies between each base in a long RNA sequence. Thus, our proposed im5C-DSCGA model introduces a bidirectional gated recurrent unit (BGRU) [28] to capture the long-term dependencies between m5C features. Notably, the Attention mechanism in deep learning is also often applied to bioinformatics. Therefore, our proposed im5C-DSCGA model introduces the improved Convolutional Block Attention Module (CBAM) Attention [25, 29] module and the Self-Attention [30] module to capture the prominent key features and global features in RNA sequences.
In 2021, A. EI et al. [31] published a review on m5C modification site prediction models. The review clearly introduces the m5C modification site prediction models that are currently in common use and describes the evaluation metrics for the different models. In conclusion, the review provides researchers with a comprehensive understanding of m5C modification site prediction models and provides valuable guidance and insights for future research and application. Table 1 shows the performance of some m5C modification site prediction tools, in which NS refers to the number of samples and WS represents the word size.
| Species | Predictor | ML algorithm | Features | NS | WS | Sn | Sp | Acc |
| H. sapiens | RNAm5CPred | SVM | KNF, KSNPFs, and PseDNC | 240 | 41 bp | 90.83% | 94.17% | 92.50% |
| H. sapiens | iRNA-PseColl | SVM | PseKNC | 240 | 41 bp | 75.83% | 79.17% | 77.50% |
| H. sapiens | M5C-HPCR | Ensemble of SVM | PseDNC and HPCR | 240 | 41 bp | 90.83% | 95.00% | 92.92% |
| H. sapiens | IRNAm5C_NB | NB, RF, SVM, and AdaBoost | BPB, K-mer, ENAC, EIIP, and PseEIIP | 240 | 41 bp | 82.81% | 81.11% | 82.20% |
| H. sapiens | iRNAm5C-PseDNC | RF | PseDNC | 1900 | 41 bp | 69.86% | 99.86% | 92.37% |
| M. musculus | ||||||||
| A. thaliana | PEA-m5C | RF | Binary Encoding, k-mer, and PseDNC | 158 | 43 bp | 86.00% | 90.00% | 88.00% |
| A. thaliana | m5C-PseDNC | SVM | PSNP, KSPSDP, CPD, and PseDNC | 12,578 | 41 bp | 68.10% | 75.50% | 71.80% |
| H. sapiens | 538 | 41 bp | 85.50% | 80.00% | 82.80% | |||
| M. musculus | 11,126 | 41 bp | 75.75% | 72.80% | 74.30% | |||
| A. thaliana | m5CPred-SVM | SVM | PSNP, 4NF, 5SNPF, PseDNC, and 5SPSDP | 2000 | 41 bp | 75.40% | 79.90% | 77.50% |
| H. sapiens | 2000 | 41 bp | 79.90% | 74.90% | 71.40% | |||
| M. musculus | 138 | 41 bp | 75.50% | 76.10% | 75.80% | |||
| A. thaliana | iRNA-m5C_SVM | SVM | KNFC, MNBE, and NV | 10,578 | 41 bp | 79.40% | 80.90% | 80.15% |
| D. melanogaster | iRNA5hmC | SVM | K-mer | 1324 | 41 bp | 67.67% | 63.29% | 65.48% |
| D. melanogaster | iRNA5hmC-PS | LR | Ps-Mono (G-gap) DiMer | 1192 | 41 bbp | 80.00% | 79.50% | 78.30% |
| D. melanogaster | iRhm5CNN | CNN | One hot and NCP | 1324 | 41 bp | 82.00% | 80.00% | 81.00% |
ML, machine learning; NS, number of samples; WS, word size; Sn, sensitivity; Sp, specificity; Acc, accuracy; SVM, Support Vector Machine; NB, Naïve Bayes; RF, random forest; LR, Logistic Regression; CNN, Convolutional Neural Networks; KNF, K-mer Nucleotide Frequency; KSNPFs, K-spaced Nucleotide Pair Frequency; PseDNC, Pseudo dinu-cleotide composition; PseKNC, pseudo K-tuple nucleotide composition; HPCR, heuristic nucleotide physicochemical property reduction; BPB, Bi-profile Bayes; ENAC, Enhanced Nucleic Acid Composition; EIIP, Electron-lon Interaction Pseudopotentials; PseEIIP, Pseudo Electron-lon Interaction Pseudopotentials; PSNP, Position-specific nucleotide propensity; KSPSDP, K-spaced position-specific dinucleotide propensity; CPD, Chemical property density; 4NF, 4-nucleotide frequency; 5SNPF, 5-spaced nucleotide pair frequency; 5SPSDP, 5-spaced position-specific dinucleotide propensity; KNFC, K-tuple nucleotide frequency component; MNBE, mono-nucleotide binary encoding; NV, natural vector; NCP, Nucleotide Chemical Property.
Traditional medical experimental methods in bioinformatics are costly and time-consuming. Therefore, it is crucial to develop computational techniques and derive some excellent predictors. We propose a predictor for identifying m5C modification sites in the context of deep learning. Moreover, our predictor only needs to input an RNA sequence to predict whether this RNA sequence is an m5C modification site or not, which can provide biologists with a more convenient tool to help them better understand the role of m5C modification sites in human RNA in relation to gene expression. In this study, we designed a hybrid network structure based on a combination of improved DenseNet, BGRU, CBAM Attention, and Self-Attention, called the im5C-DSCGA model, to predict m5C modification sites in human RNA. Details on the im5C-DSCGA model and the network structure of each module are provided in Section 2.
In this work, we proposed the identification of 5-methylcytosine sites in human RNA using a deep-learning-based approach. Hereafter, we present the work in this section in four parts: benchmark dataset, model architecture, feature extraction, and evaluation metrics.
The selection of the dataset is a critical part of model construction, and Hasan M M et al. [32] were used for the benchmark data in this study. They used humans as subjects to study the distribution of m5C modification sites in RNA and collected nucleotide sequences of 41 bp in length. To obtain a high-quality dataset, they used CD-HIT [33] software to remove DNA sequences with more than 90% similarity. Notably, to assess the robustness of the model, we used the same strategy as in the recent study, whereby 20% (11,630 m5Cs and 11,630 non-m5Cs) were randomly selected from the original dataset and treated as independent datasets. However, the remaining 80% (46,559 m5Cs and 46,559 non-m5Cs) was used as the training dataset to develop the prediction model. Details of the benchmark dataset are shown in Table 2.
| Original dataset | Positive sample | Negative sample |
| Total | 58,159 | 58,159 |
| Training dataset | 46,529 | 46,529 |
| Testing dataset | 11,630 | 11,630 |
For the model architecture, our discussion will be in two parts. First, we will summarize the overall architecture of the im5C-DSCGA model, and then, offer a detailed description of the structure for each module.
In this study, we concluded the prediction methods for identifying m5C modification sites on the same dataset and the current advancements in m5C modification site prediction. Although the Deepm5C model [32] has made quite a lot of progress, there are still some deficiencies to overcome. Therefore, we designed a novel deep-learning model called im5C-DSCGA to identify m5C modification sites in human RNA.
Fig. 1 summarizes the design of the prediction and evaluation processes for the im5C-DSCGA model. This process consisted of four parts, respectively: feature encoding, im5C-DSCGA model framework, ensemble learning module, and performance evaluation. In the feature encoding part, we used three encoding methods, namely, one-hot encoding, nucleotide chemical property (NCP), and nucleotide density (ND). In the model framework part, for a given RNA sequence, the network framework consists of six modules, which are the input module, improved DenseNet module, improved CBAM Attention module, BGRU module, Self-Attention module, and output module. The input module is used to feed the original RNA sequences into the subsequent DenseNet module after three kinds of features are encoded. Then, using the improved DenseNet module, the network can extract more advanced features than the residual networks and ordinary convolutional neural networks. The improved CBAM Attention module is used to extract more critical and prominent features by multiplying the Spatial Attention module and the Channel Attention module at the corresponding positions of their respective feature matrices. The BGRU module also used the output feature vector above as an input. The BGRU module is designed to obtain long-term dependencies between high-level features more efficiently than gated recurrent unit (GRU) and ordinary recurrent neural networks. The Self-Attention mechanism module is used to evaluate the importance of RNA sequence features. The output module uses a fully connected neural network to receive these high-level features as the input and calculates probability values between 0 and 1 using the softmax activation function. In the Ensemble learning model section, we used the homogeneous ensemble [34] method, which ultimately uses soft voting for classification. Here, the average of the three probability values was taken to obtain the final prediction probability. If the probability value is greater than 0.5, a m5C modification site is identified; conversely, the opposite is true. In the performance evaluation section, we show the evaluation of the im5C-DSCGA model by cross-validation and independent tests.
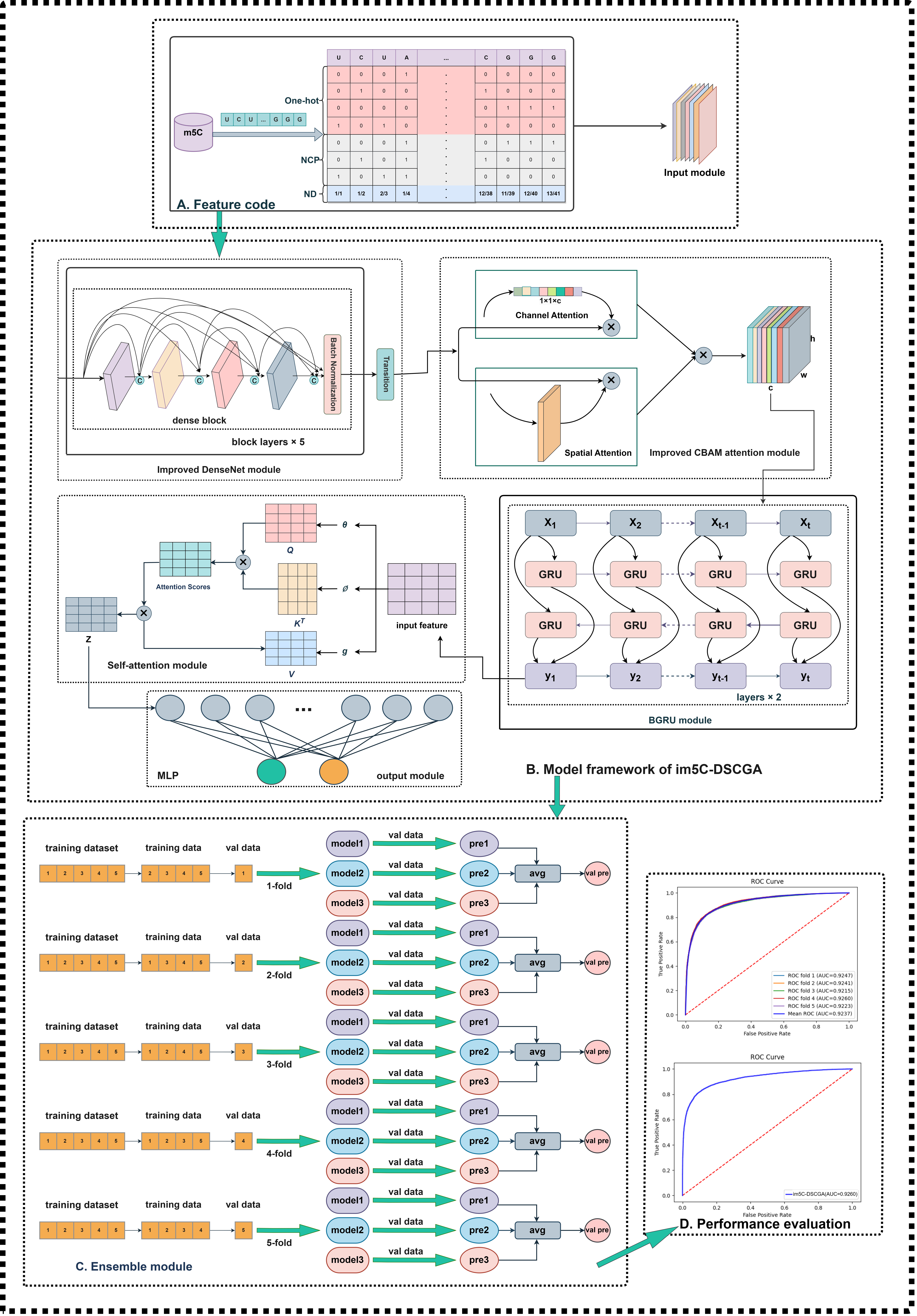 Fig. 1.
Fig. 1.The im5C-DSCGA model structure. (A) Feature encoding. RNA
sequences were feature encoded using one-hot, NCP,
and nucleotide density (ND) to obtain an 8
The ResNet [23] is an improved convolutional neural network that solves the degradation problem, which can occur in the CNNs, as shown in Fig. 2. It is shown that the overall performance of the CNN is largely affected by the number of network layers. Specifically, the more layers the neural network has, the more complex feature extractions the network can perform, and theoretically improved results can be achieved. Nevertheless, the accuracy saturates or even decreases when the depth reaches a certain level. This is known as the degradation problem and makes it increasingly difficult to train deeper neural networks. However, the residual network uses shortcut connections to solve the problem of model degradation in deep networks. The shortcut connections are added between the other two layers compared to the traditional neural networks, and the deeper layers are brought into play by residual learning. As the number of network layers increases, the residual convolutional neural network can obtain better learning results.
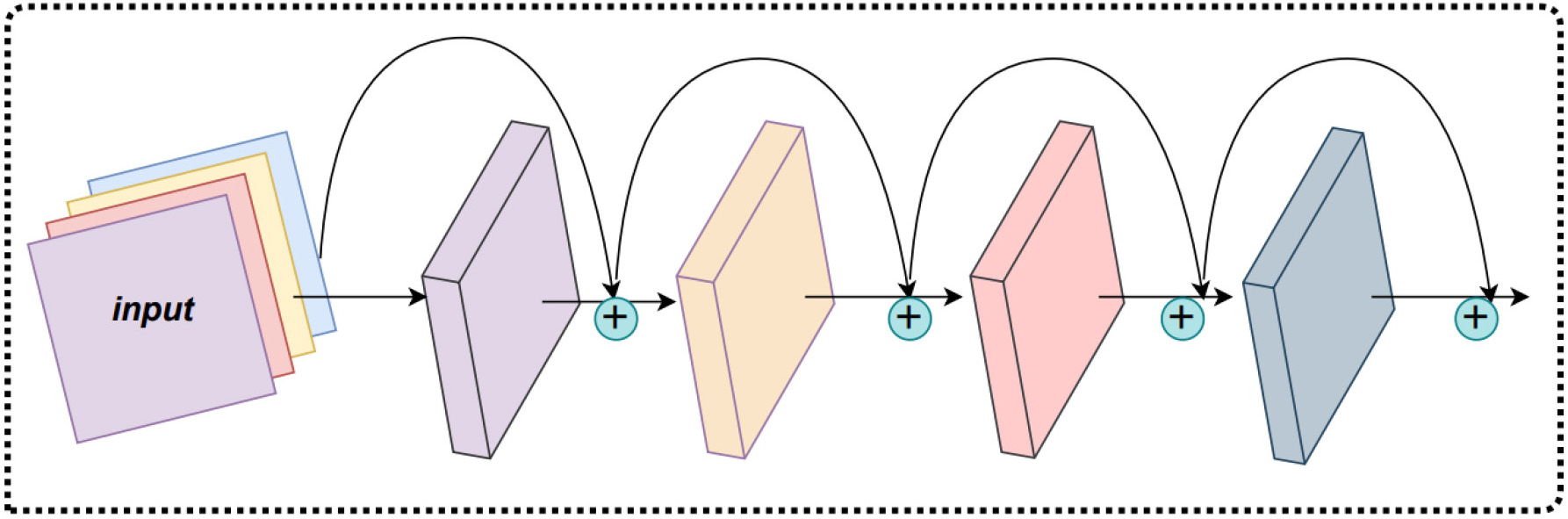 Fig. 2.
Fig. 2.The residual neural network (ResNet) structure.
The residual neural network is composed of a series of basic blocks called
residual blocks and learns the required mappings and uses a special
short-circuiting mechanism to connect them. The residual is the difference
between the observed value and the estimated value. Suppose within a certain
layer that the solved mapping (optimal function) is noted as
where the function
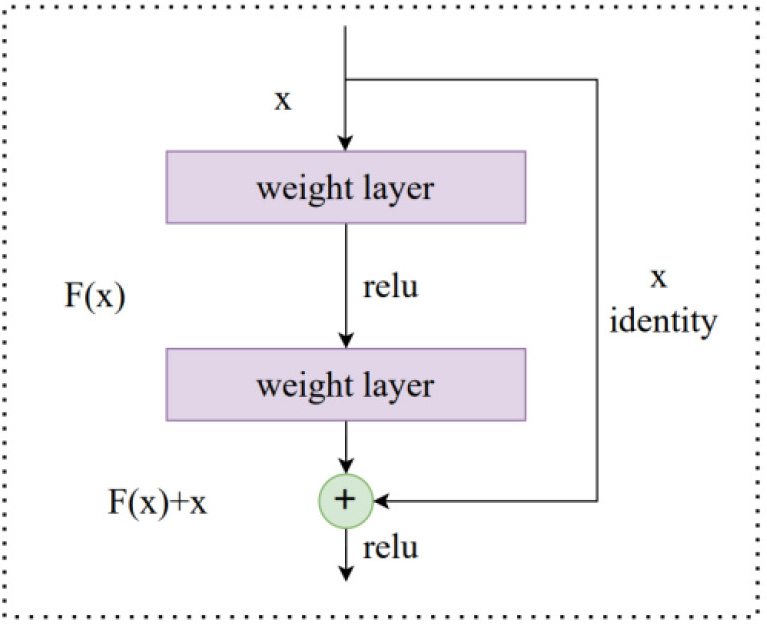 Fig. 3.
Fig. 3.The ResNet shortcut connection.
Residual Block
A ResNet is composed of a series of basic blocks
called residual blocks. For the residual block, we used a three-layer bottleneck
structure, which can be seen in Fig. 4. It reduces the size of the feature map by
1
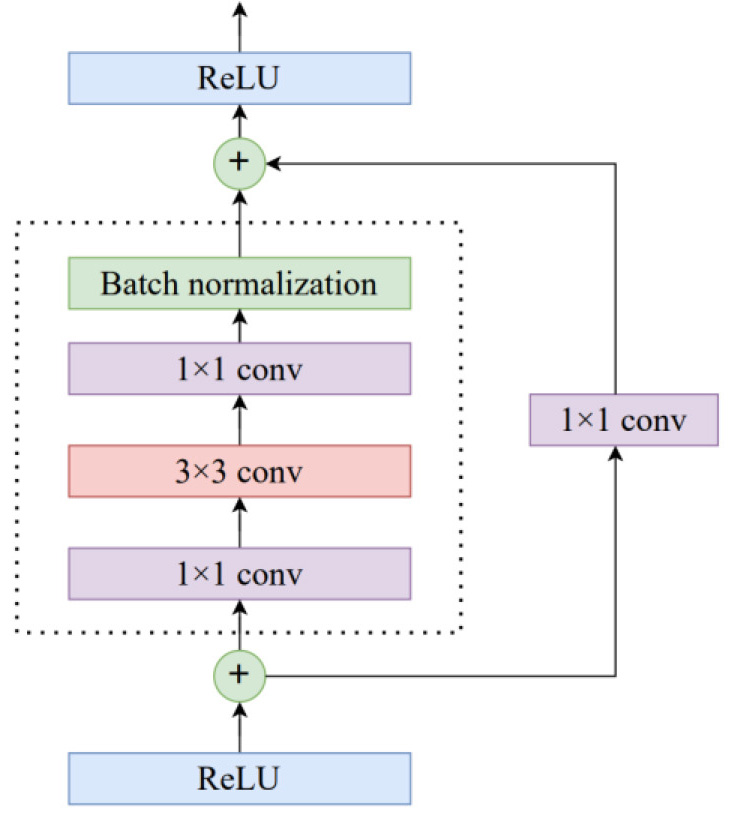 Fig. 4.
Fig. 4.The residual block structure.
Traditional CNNs may suffer from inadequate feature extraction, which can lead to network degradation. DenseNet are an improved convolutional neural network that is based on the ResNet. A convolutional layer, the dense block layer, and the transition layer form its network architecture. The RNA sequences are encoded by three feature encoding methods and the feature matrix is first passed through a convolutional layer, then, the dense block layer, and finally the transition layer. In this study, we improved the original network structure of DenseNet. In detail, we removed one layer of the convolutional layer and the feature matrix that RNA sequences were encoded by and three feature encoding methods were directly input into the dense block layer, and a batch normalization layer was added between the dense block layer and the transition layer to, finally, obtain the high-level features of the RNA sequences.
The improved DenseNet extracts the original feature information for RNA sequences at a deeper level and enhances the robustness of the im5C-DSCGA model, resulting in better generalization ability. Fig. 5 shows the network structure of the improved DenseNet.
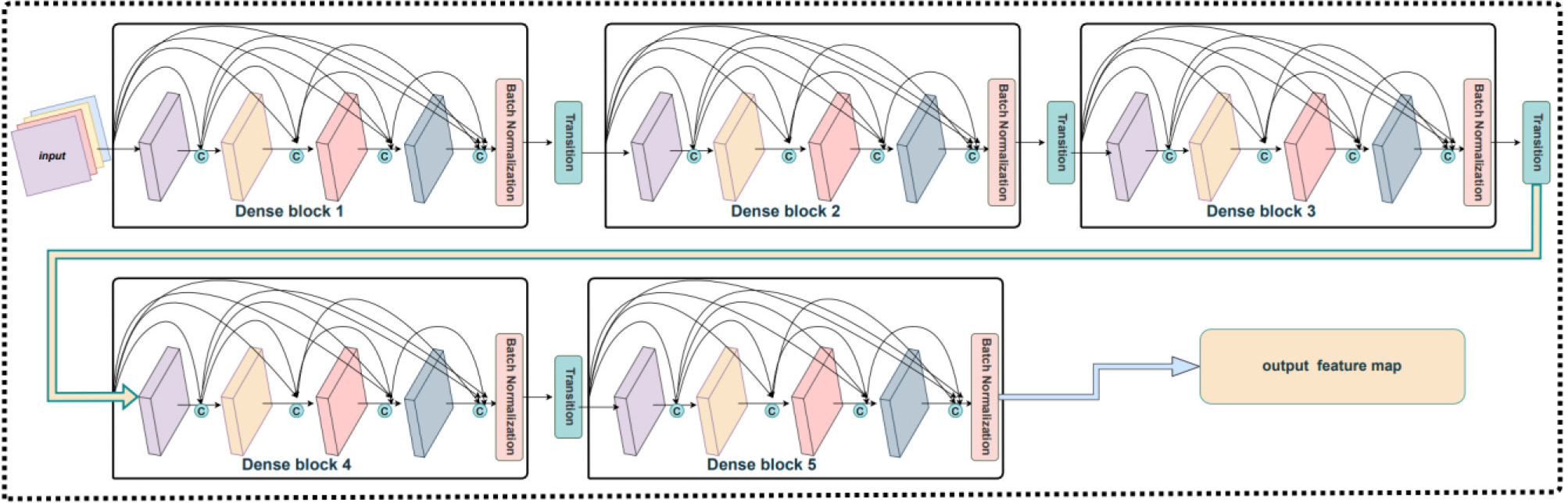 Fig. 5.
Fig. 5.The improved DenseNet structure.
2.2.3.1 Dense block
The DenseNet uses a dense block structure, which is a dense connection mechanism. It is used specifically to concatenate the outputs of all the previous convolutional layers together as the input of the next convolutional layer, to achieve feature reuse. The dense block structure improves the efficiency of the model and also enhances its expressiveness.
Fig. 6 presents the network structure of the dense block, which consists of the
L-layer network structure with a nonlinear transformation function. The nonlinear
transformation function consists of a 3
where
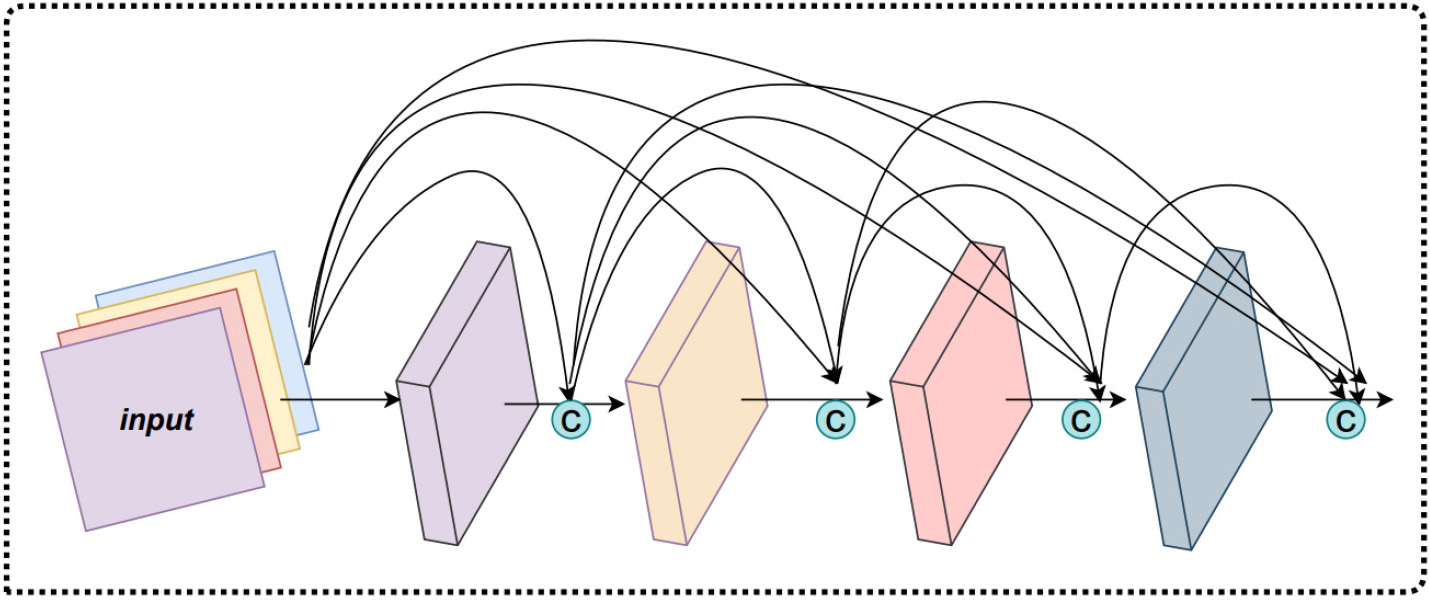 Fig. 6.
Fig. 6.The dense block structure.
2.2.3.2 Transition
The main role of the transition layer is to connect the two adjacent dense
blocks and reduce the size of the output feature map. The transition layer
consists of a 1
We add a BN layer before the 1
where
Since considering the various importance of the different features, we introduced an improved CBAM Attention [25] module after the DenseNet module to weight the feature mapping, thereby allowing the further improvement of the prediction ability by the im5C-DSCGA model. The CBAM Attention is a simple and effective attention mechanism in feedforward convolutional neural networks, which includes two modules, a Channel Attention module, and a Spatial Attention module, as shown in Fig. 7.
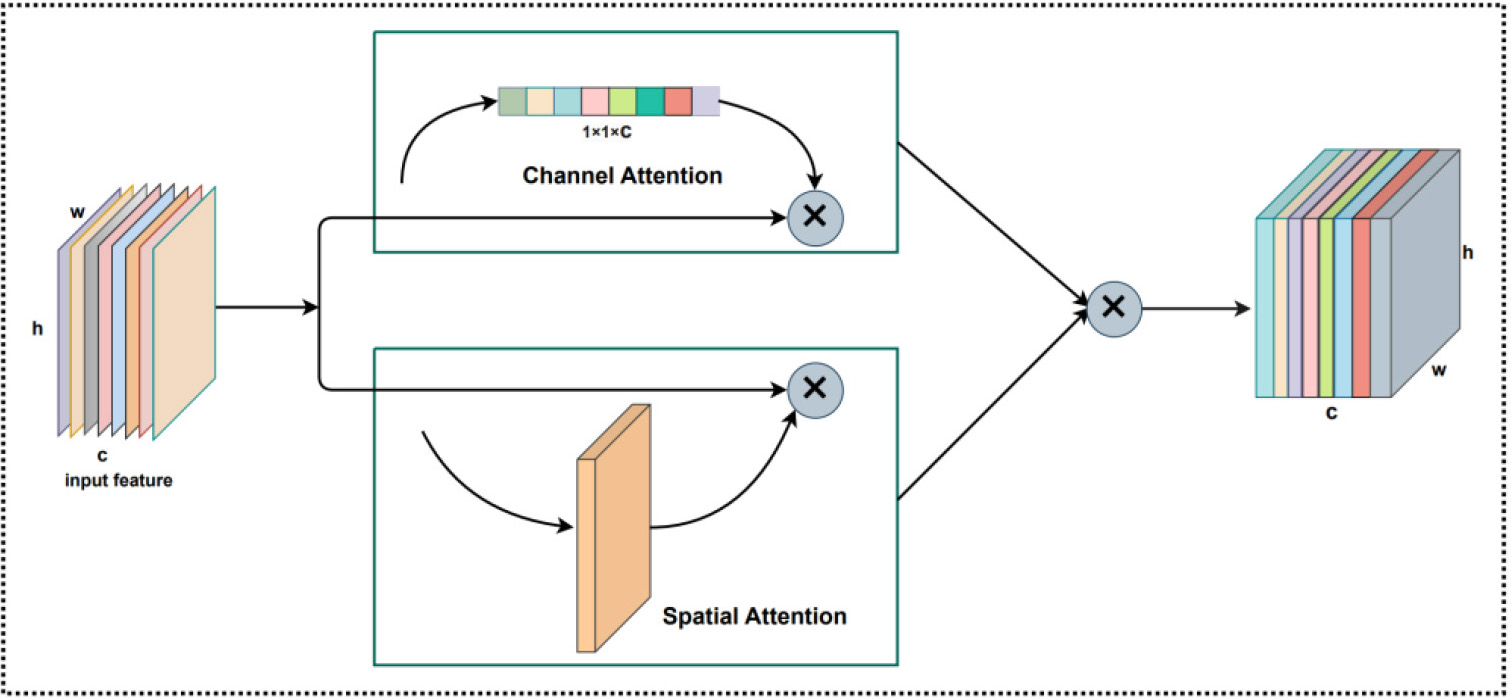 Fig. 7.
Fig. 7.The improved CBAM Attention module structure.
The original CBAM Attention first evaluates the original features using the Channel Attention module, after which it feeds the feature map produced by the Channel Attention module back to the Spatial Attention module, which then outputs the feature map. However, this serial connection has the drawback of calculating in a particular way, which might result in improper weight calculation in the Spatial Attention module and loss of channel weight information in the finished feature map. Therefore, we improved the CBAM Attention with the idea that the output features from the DenseNet module are fed into the Channel Attention and Spatial Attention modules, multiplying the feature maps of these two outputs by each other at the corresponding positions. By using this approach, we can increase the expressiveness of the features and maximize their retention for each attention module after evaluation.
2.2.4.1 Channel Attention
Considering that the channels in the feature map are different, it has varying degrees of importance. Therefore, we used Channel Attention to compute various weights for each channel, and the structure is shown in Fig. 8. Channel Attention compresses the feature map in the spatial dimension to obtain a one-dimensional vector before manipulating it.
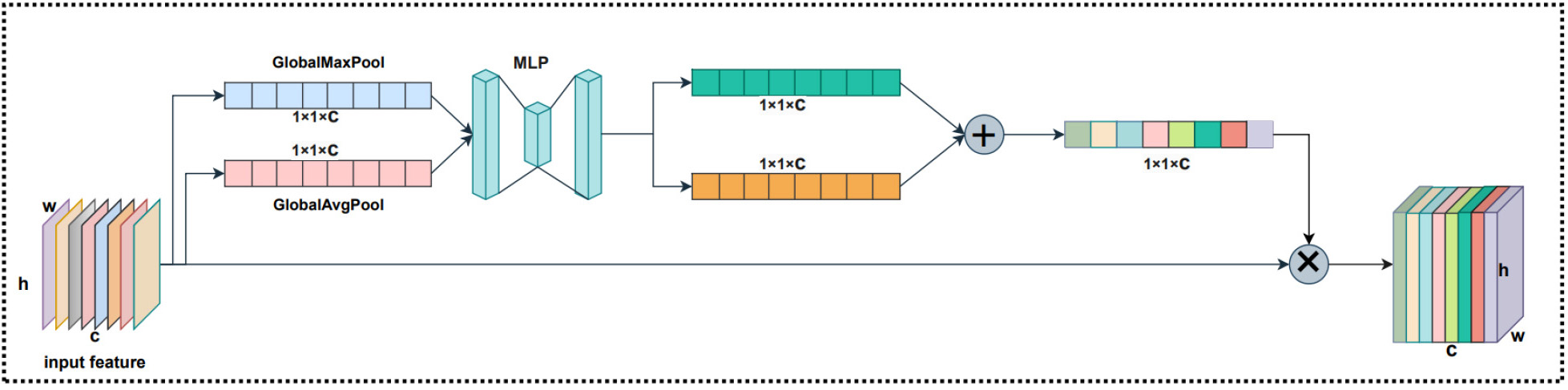 Fig. 8.
Fig. 8.The Channel Attention structure.
When compressing in the spatial dimension, the Spatial Attention considers both the global average pooling and the global max pooling. For Channel Attention, global max pooling is used to extract the maximum value of the feature map for each channel, while global average pooling is used to extract the average value of the feature map for each channel. Channel Attention is first aggregated by two parallel average pooling and max pooling to aggregate the spatial information of the feature mapping and sent to a shared fully connected neural network (MLP). Secondly, the weights of the Channel Attention module are obtained using the sigmoid activation function after the two results output by the MLP are added element by element. Finally, to obtain the feature map of the Channel Attention module weights, these weights are multiplied by the input feature map. It is possible to express the Channel Attention as:
where pooling is global max pooling and global average pooling,
2.2.4.2 Spatial Attention
Considering that the receptive domains are different, the degree of their influence on the feature map is also different. Therefore, to calculate the weights between the receptive domains, we employed Spatial Attention. The structure for Spatial Attention is illustrated in Fig. 9. Spatial Attention is compressed for channels, and global max pooling and global average pooling are performed in the channel dimension, respectively. For Spatial Attention, the operation of global max pooling is to extract the maximum value at each position on the channel, while the operation of global average pooling is to extract the average value at each position on the channel. Spatial Attention is firstly processed by two parallel global max pooling and global average pooling operations for feature mapping, and the two obtained feature maps are performed in CONCAT based on the channels. Secondly, after a convolution operation, it can be down-dimensioned to 1 channel before the Spatial Attention feature map is generated by the sigmoid function. The final generated features are obtained by multiplying this feature map by the Spatial Attention input feature map. The Spatial Attention can be expressed as:
where pooling is global max pooling and global average pooling and
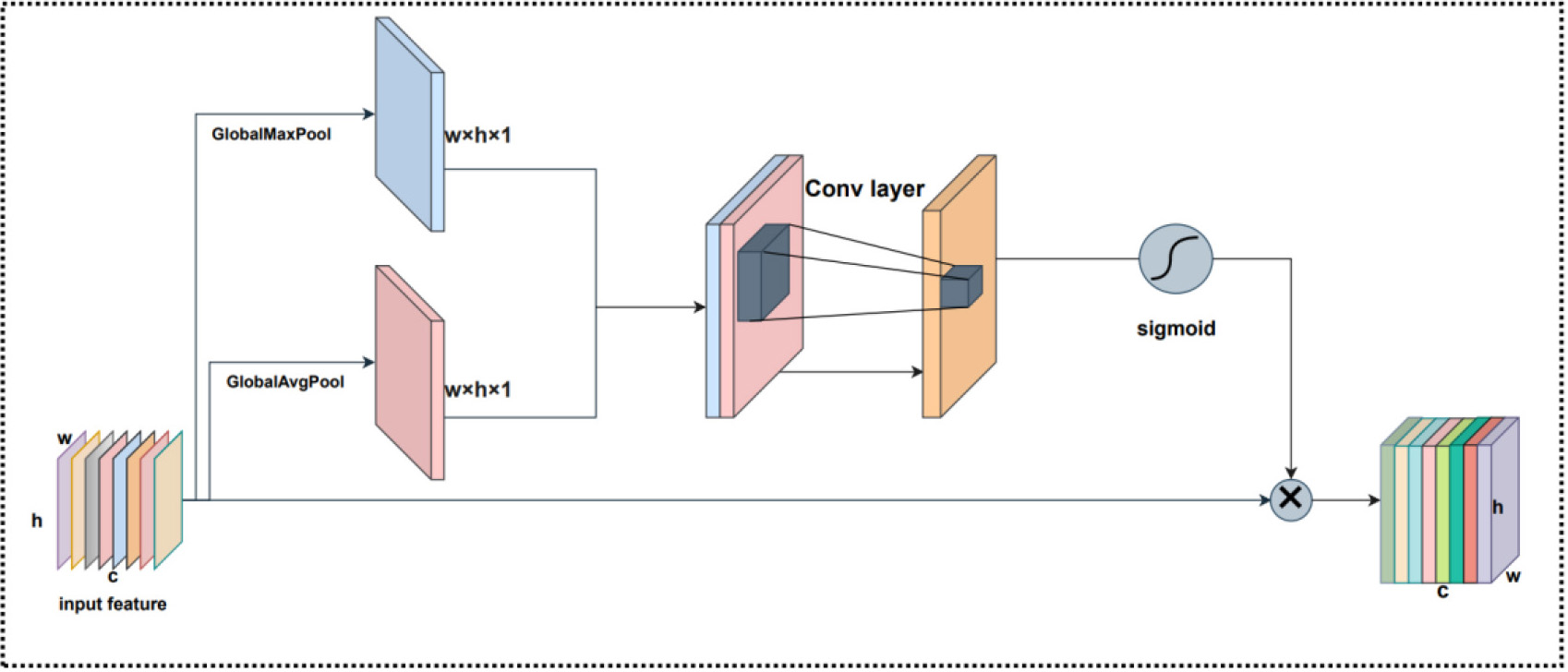 Fig. 9.
Fig. 9.The Spatial Attention structure.
The bidirectional gated recurrent unit (BGRU) [28] is a recurrent neural network (RNN), which is a variant of the traditional RNN. It is capable of learning long-term dependencies between sequences in RNA sequence prediction tasks and mitigating gradient disappearance or explosion phenomena. Fig. 10 illustrates the BGRU network structure. The BGRU consists of a forward GRU and a reverse GRU. The network of the GRU is simpler compared to the long short-term memory network (LSTM) [35], which synthesizes the forgetting gate and the input gate into a new gate called the update gate. The update gate controls the amount of data that previous memory information continues to be retained until the current moment. Although there is one less gate, there are fewer parameters and GRU can save a lot of time in case of a large training dataset. For a GRU, it can be expressed as:
where
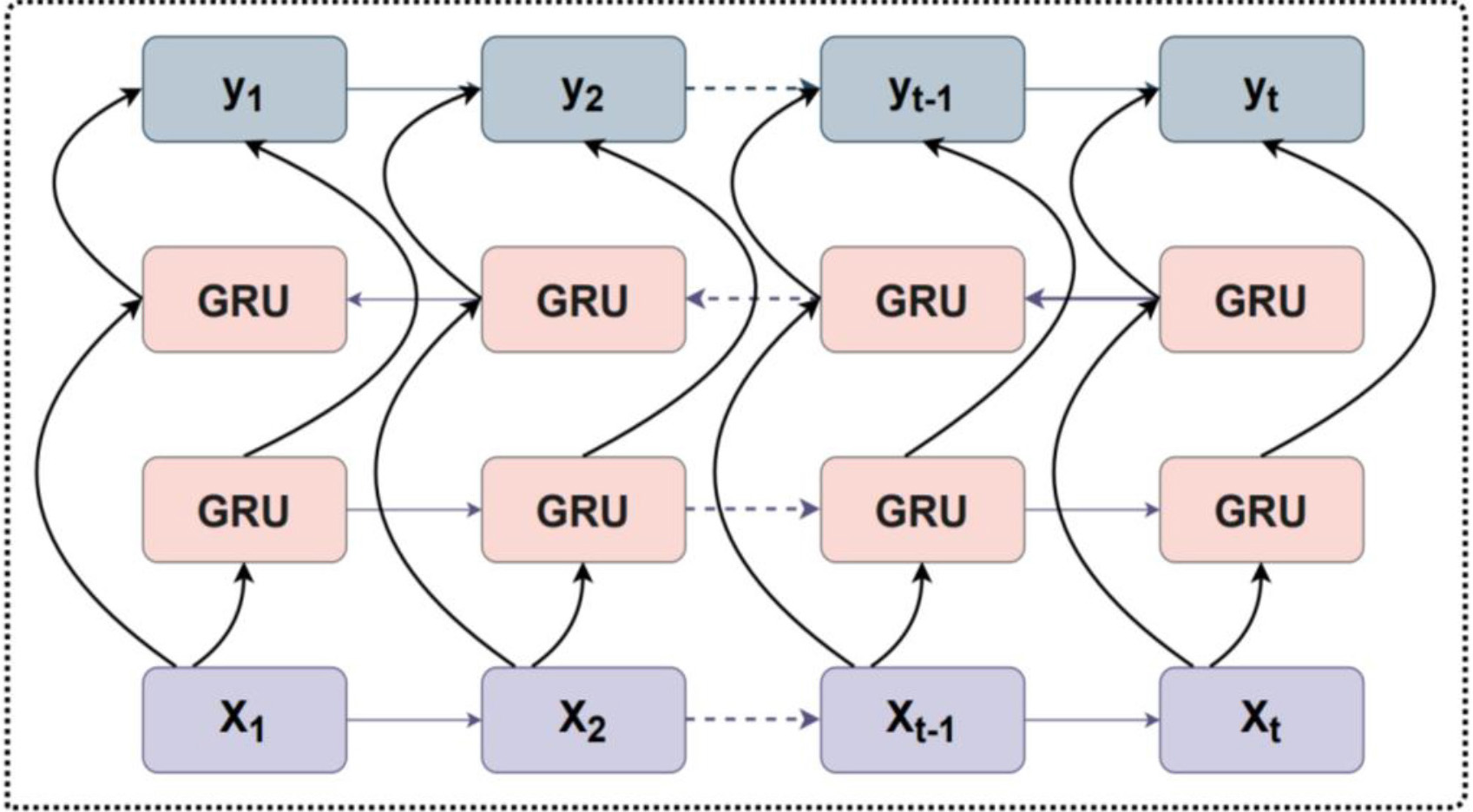 Fig. 10.
Fig. 10.The bidirectional gated recurrent unit (BGRU) structure.
The Self-Attention [30] mechanism is a widely used mechanism in deep learning, and the specific structure is shown in Fig. 11. It is actually a weighting method, whereby a certain part of the input sequence is given a higher weight than other parts. It can be understood since it wants the machine to notice the correlation between the different parts in the whole input features so that it can better capture the information in the input features.
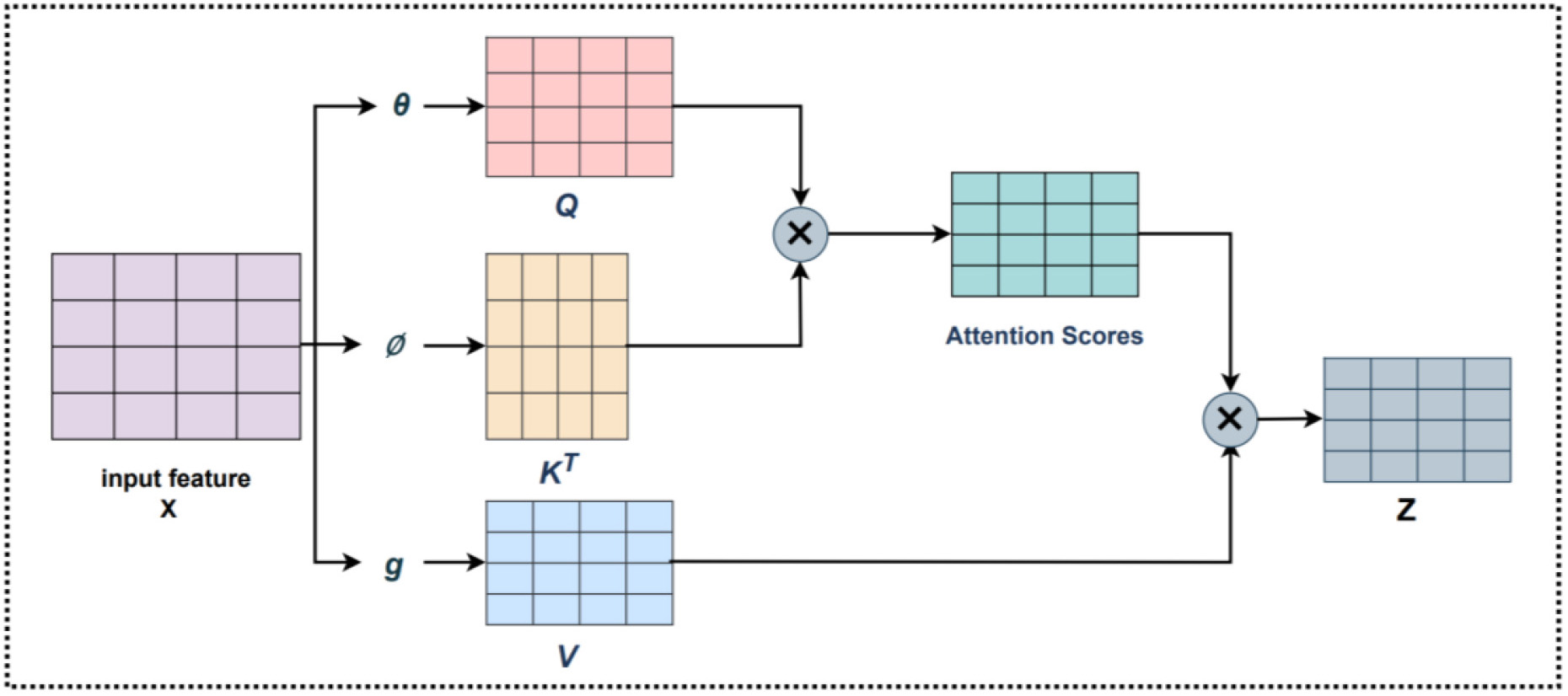 Fig. 11.
Fig. 11.The Self-Attention structure.
The Self-Attention mechanism converts the input data into three vectors
where
It is obtained by querying the correlation of the vector with the corresponding vector to calculate the weight of each value vector. The calculation method is shown as:
where
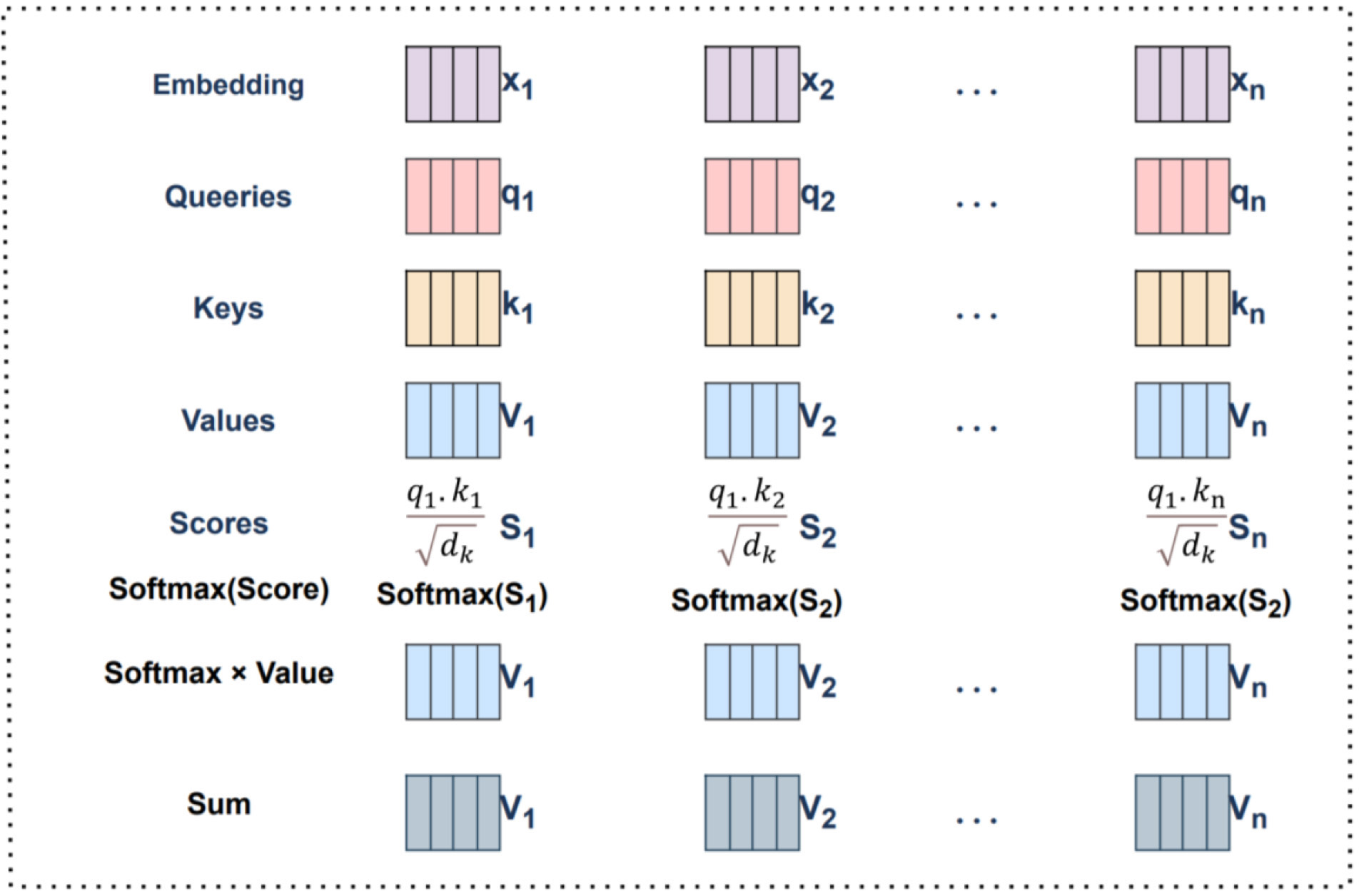 Fig. 12.
Fig. 12.Self-Attention mechanism calculation using vectors.
In machine learning, the independent test datasets are taken and fed into several identical or different models, to compute several predictions before averaging them. This ensemble learning strategy is named model averaging, the benefit of which is that various models typically do not result in the same mistakes on independent test datasets, thereby providing a highly effective method of lowering generalization errors. In this study, the ensemble learning [34] module refers to the use of the same feature encoding and model framework methods for the same training dataset. It precisely utilizes the idea of model averaging that was introduced above. In this study, we employed five-fold cross-validation, whereby we divided the training dataset into five parts, four of which were used for training and one for validation. The final prediction results are obtained by a soft voting method. For the training dataset, we used three identical network frameworks, which had been trained three times in each fold, to obtain three models. We put the validation dataset into three models in each fold to get three probability values. The validation results for each fold are obtained by adding and averaging these three probability values. If the probability value is greater than 0.5, the m5C modification site is identified; otherwise, the opposite is true. For the independent test dataset, we used the same method as the training dataset. The specific structure of the ensemble learning module is shown in Fig. 1.
Feature encoding is an essential step in building a predictive model. It converts the letters in a biological sequence into numerical information that can be recognized by a computer. To investigate the impact of multiple features on experiments, we used three feature encoding methods in this work. We used one-hot encoding, nucleotide chemical property encoding (NCP), and nucleotide density encoding (ND) to identify m5C modification sites in human RNA, which will be described in detail in the following sections.
One-hot encoding [36] is a simple and effective feature encoding method that has
been widely used in bioinformatics. It represents the four bases of adenine (A),
cytosine (C), guanine (G), and uracil (U) in the nucleotide chain of an RNA
molecule as a binary vector of zeros and ones. Each base in the m5C sequence in
human RNA is converted into a four-dimensional feature vector with the four bases
A, C, G, and U represented by the codes (1, 0, 0, 0), (0, 1, 0, 0), (0, 0, 1, 0),
and (0, 0, 0, 1), respectively. For example, a human RNA m5C sequence of
UCUAU…GCGGG can be represented as shown in Fig. 2. The length of the human RNA
m5C sequence in this work is 41 bp, meaning that each sequence is transformed
into a 4
Recently, the nucleotide chemical property (NCP) [37] encoding method has been applied to many studies in bioinformatics. This encoding method is based on three chemical properties and is a relatively simple encoding scheme. Each nucleotide has a different chemical property. Therefore, we can encode RNA sequences depending on the structure of the loop, chemical structure, and hydrogen bond interactions.
Analyzed from the perspective of the functional groups contained in nucleotides,
both A and C contain amino groups, and both G and U contain ketone groups; from
the perspective of ring structures, A and G contain two ring structures, and G
and C have only one ring structure; analyzed from the perspective of base
complementary pairing, A and U when paired are linked by two hydrogen bonds,
whereas G and C are linked by three hydrogen bonds. Each base in the human RNA
m5C sequence was converted into a three-dimensional feature vector, with the four
bases A, C, G, and U encoded by (1, 1, 1), (0, 1, 0), (1, 0, 0), and (0, 0, 1),
respectively. In this study, the length of the human RNA m5C sequence was 41 bp,
meaning each sequence was transformed into a 3
The nucleotide density (ND) [38] encoding method is also one of the RNA
sequences encoding methods and is often used in combination with the NCP encoding
method. Its main principle is to take one or several bases in an RNA sequence
sample as an element and calculate the frequency of this element occurring in the
sample where it is located. Suppose the RNA sequence samples are composed of
take the calculation of single nucleotide density as an example, where
where
here, we take a 41 bp long m5C sequence “UCUAU…GCGGGG” in RNA as the example,
“A” is at positions 4, 12, …, 31, and 33, with densities
¼1/4, 2/12, …, 3/31, and 4/33, respectively. “C” is at
positions 2, 6, …, 36, and 38, with densities of 1/2, 2/6, …, 11/36, and
12/38, respectively. “G” is in positions 8, 18, …, 40, and 41, with densities
of 1/8, 2/18, …, 12/40, and 13/41, respectively. “U” is in positions 1, 3,
…, 27, and 28, with densities of 1/1, 2/3, …, 11/27, and 12/28, respectively.
Each base in the human RNA m5C sequence is converted into a one-dimensional
feature vector. In this study, the length of the human RNA m5C sequence is 41 bp,
thereby meaning that using this method, each sequence is transformed into a 1
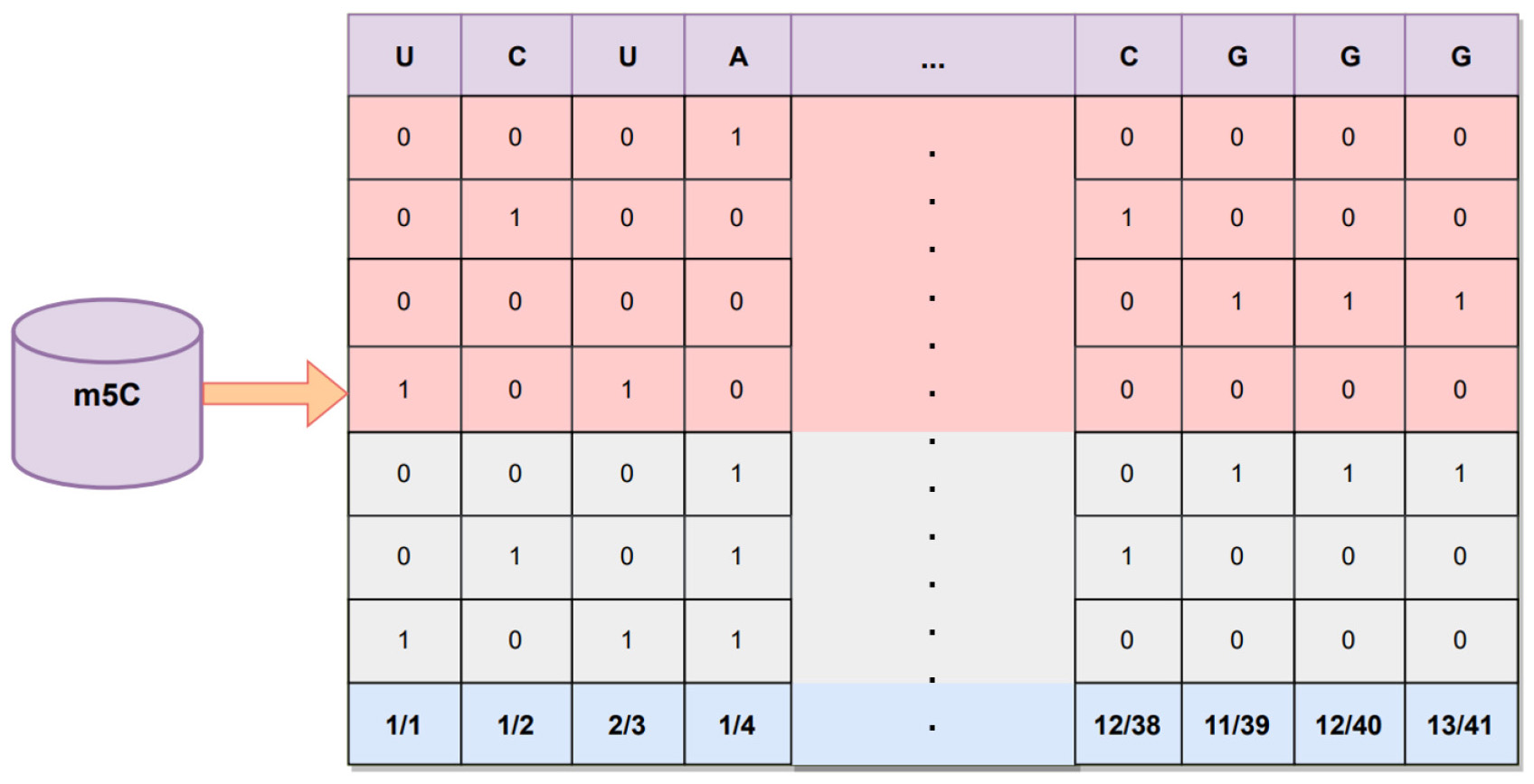 Fig. 13.
Fig. 13.One-hot, NCP, and ND encoding.
In this study, we chose four evaluation metrics to assess the im5C-DSCGA model, namely, sensitivity (Sn), specificity (SP), accuracy (Acc), and Matthew’s correlation coefficient (MCC), defined as in Eqn. 15.
where TP, TN, FP, and FN denote true positives, true negatives, false positives, and false negatives, respectively. Sn and SP denote the proportion of positive and negative samples, respectively, which are correctly predicted. Acc represents the proportion of the whole sample predicted correctly, and MCC can accurately assess the performance of the model. Notably, in this study, we used the MCC metric to evaluate the global model performance.
Furthermore, we also added the receiver operating characteristic curve (ROC) [39] and calculated the area under the ROC curve (AUC) to evaluate the overall performance. The value of AUC ranges from (0,1), and its value is positively correlated with the prediction performance, and the closer the AUC value is to 1 the more effective the model is.
To facilitate comparisons with existing models, we used datasets from existing models to train the im5C-DSCGA model. In the experiments, an NVIDIA GeForce RTX 3080 Ti GPU was used to train the neural network for the im5C-DSCGA model. In the model training, the optimizer used Adam to prevent the loss function from falling into local optimal points. Meanwhile, we used a cross-entropy loss function to propagate the gradients and used regularization, dropout, and early stop strategies to avoid overfitting. In addition, the optimal hyperparameters were determined by comparison experiments. All parameter settings and model training were based on Python 3.8 (Elemental Security, Dallas, TX, USA, https://www.python.org/) and Keras 2.8.0 (Google, Mountain View, CA, USA, https://keras.io/) for implementation into the im5C-DSCGA model. Table 3 shows all hyperparameters in the im5C-DSCGA model.
| Parameters | Number |
| Dense block | 5 |
| Convolution layer number of a dense block | 4 |
| Convolution kernel size | 96 |
| BGRU layer neurons | 500 |
| Self-Attention layer neurons | 500 |
| Dropout ratio | 0.5 |
| First dense layer neurons | 240 |
| Second dense layer neurons | 40 |
| Last dense layer neurons | 2 |
BGRU, bidirectional gated recurrent unit.
In order to specifically evaluate the performance of the im5C-DSCGA model and demonstrate the improvements in the model, we will discuss three aspects of the model, the variants, feature analysis, and structural analysis.
For the model variants, we designed four models, the RSCm5C model, RGAm5C model, DSCm5C model, and DGAm5C model. The first model variant was the RSCm5C model, which we designed without the BGRU module and Self-Attention module, while we also changed the DenseNet module to the ResNet module to compare it to the original im5C-DSCGA model, which can effectively respond to the effect of global features and more advanced local features in the prediction effect of the model.
The second model variant called the RGAm5C model, was designed as a CBAM module without improvements, while we again changed the DenseNet module to use the ResNet module to compare it to the original im5C-DSCGA model, which can effectively respond to the effect of important features and more advanced local features in the prediction effect of the model. The third model variant, named the DSCm5C model, eliminates the BGRU module and the Self-Attention module based on the original model structure, which can effectively respond to the influence of global features in the prediction effect of the model. The last model variant, called the DGAm5C model, eliminates the improved CBAM Attention module based on the original model structure, which can effectively respond to the effect of important features on the prediction effect of the model. All four model variants are classified by fully connected neural networks.
The structural design of these four model variants is compared to the original im5C-DSCGA model, thus, demonstrating the superiority of our model structure. Fig. 14 shows a detailed description of the four variant model structures. All model variants are trained on the benchmark dataset, and all use the same hyperparameter settings of our proposed im5C-DSCGA model.
 Fig. 14.
Fig. 14.Brief illustration of four variant models. (A) The RSCm5C model. (B) The RGAm5C model. (C) The DSCm5C model. (D) The DGAm5C model.
To further evaluate the performance of the im5C-DSCGA model, we compared it to four variants of the model based on deep learning, including RSCm5C, RGAm5C, DSCm5C, and DGAm5C. Through the experimental comparisons, we discovered that DenseNet can extract more advanced local features better than ResNet in the deep-learning framework. Moreover, the attention mechanism is effective in capturing some key features and more prominent important features. Fig. 15 demonstrates the performance of im5C-DSCGA and the four model variants on the training dataset with five-fold cross-validation. Fig. 16 presents the performance of im5C-DSCGA and the four model variants on the independent test dataset.
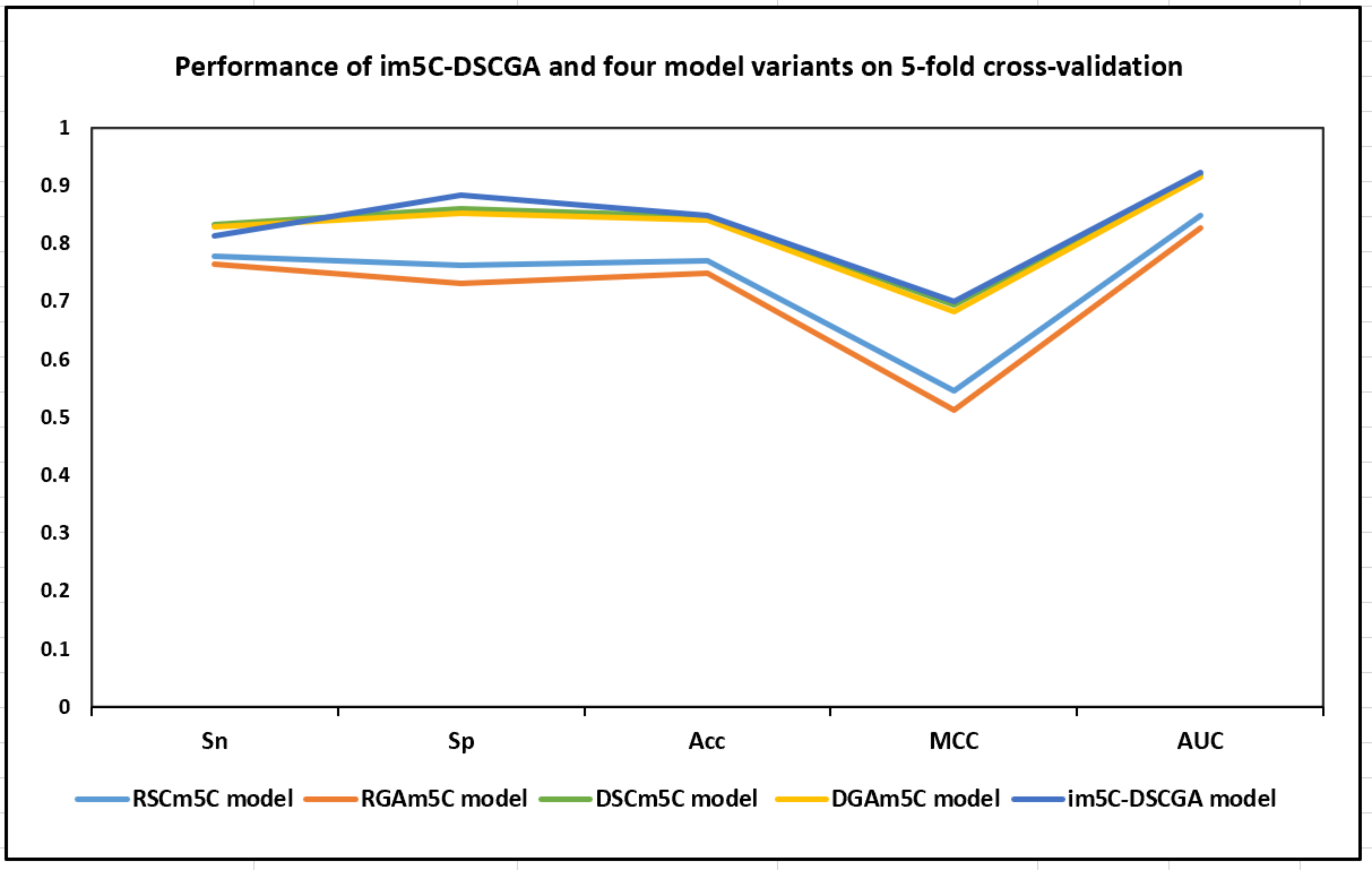 Fig. 15.
Fig. 15.Performance of im5C-DSCGA and four model variants on five-fold cross-validation.
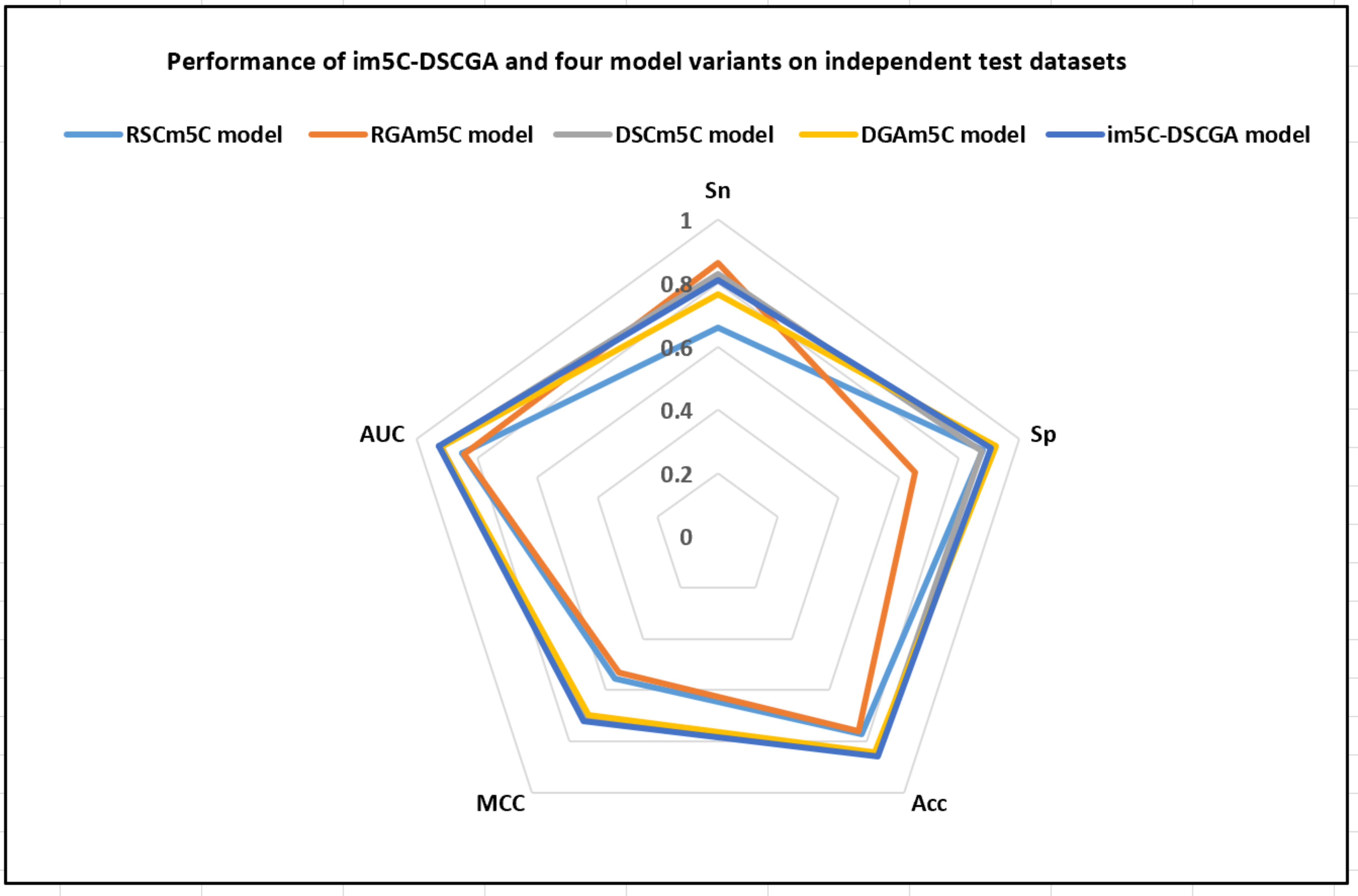 Fig. 16.
Fig. 16.Performance of im5C-DSCGA and four model variants on independent test datasets.
As can be visualized in Fig. 15, the four metrics SP, Acc, MCC, and AUC for the im5C-DSCGA model significantly outperformed the four model variants in the five-fold cross-validation on the training dataset. The SP was higher by 12.13%, 15.16%, 2.46%, and 3.22%, respectively. The Acc was higher by 7.82%, 10.03%, 0.25%, and 0.85%, respectively. The MCC was higher by 15.54%, 18.70%, 0.68%, and 1.74%, respectively. The AUC was higher by 7.45%, 9.50%, 0.32%, and 0.70%, respectively. Similarly, in Fig. 16, it is clear that the Acc, MCC, and AUC of the im5C-DSCGA model outperformed all four model variants in the independent tests. The Acc was higher by 8.87%, 10.02%, 0.66%, and 1.49%, respectively. The MCC was higher by 16.71%, 19.22%, 1.61%, and 2.46%, respectively. The AUC was higher by 7.79%, 8.45%, 0.53%, and 0.74%, respectively. Therefore, we chose the im5C-DSCGA model as the model for this research. In addition, Figs. 17,18 also show the ROC plots of the im5C-DSCGA model and the four model variants on the training dataset and the independent test dataset, respectively.
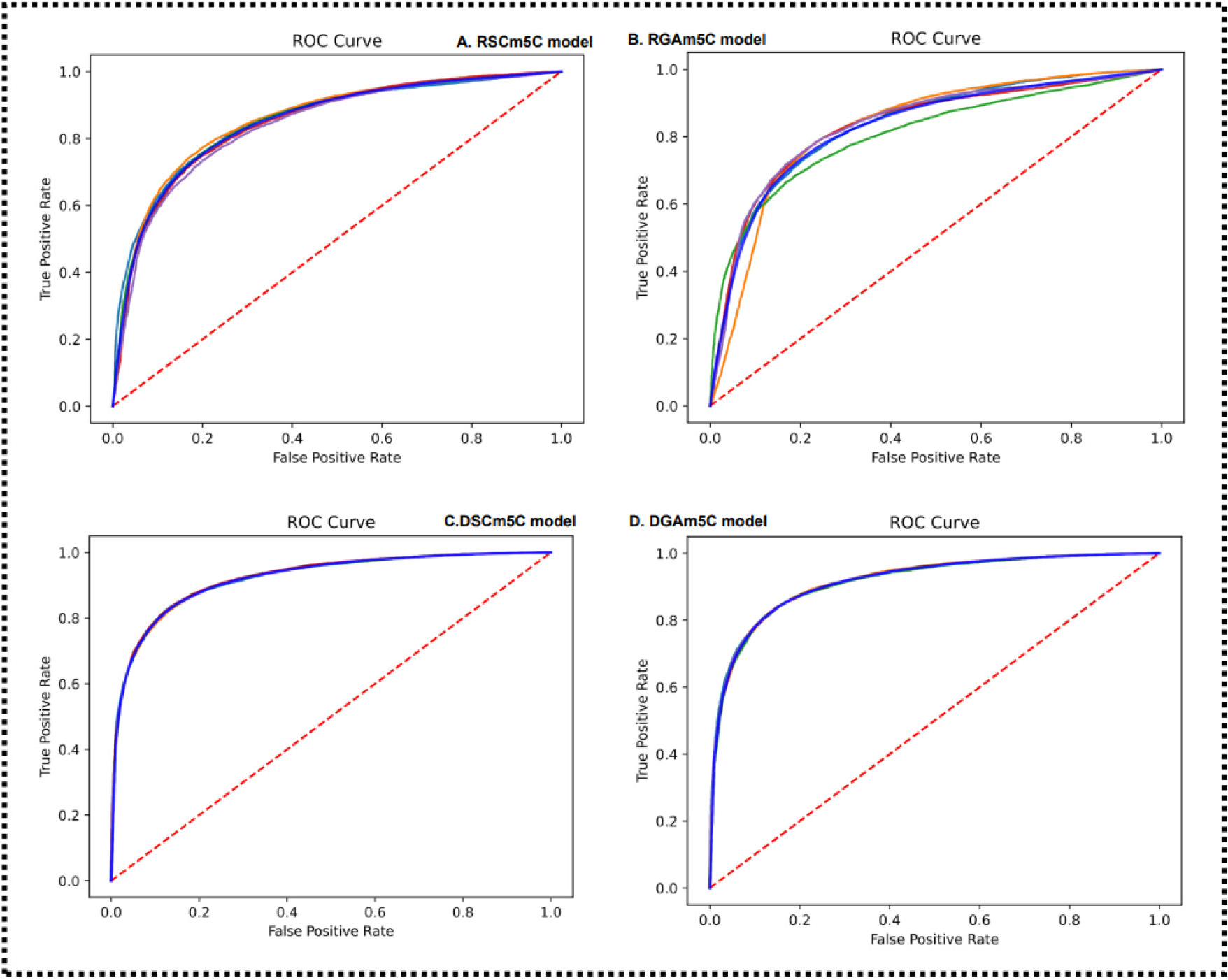 Fig. 17.
Fig. 17.Performance and four model variants on five-fold cross-validation.
 Fig. 18.
Fig. 18.Performance of four model variants on independent test datasets.
Since the number of dense blocks and the number of convolutional layers in each dense block in the DenseNet are important factors affecting the performance of our model, this study evaluated the performance using different numbers of dense blocks and a different number of convolutional layers in the dense blocks. Fig. 19 presents the performance of different numbers of dense blocks and the number of convolutional layers in the different dense blocks. It can be clearly seen that when four convolutional layers are used to build one dense block and five dense blocks are stacked together, the SP, Acc, and MCC metrics of the model are greater than the performance using other combinations of cases. Therefore, in this study, we chose to build one dense block using four convolutional layers and constructed the DenseNet with five dense blocks.
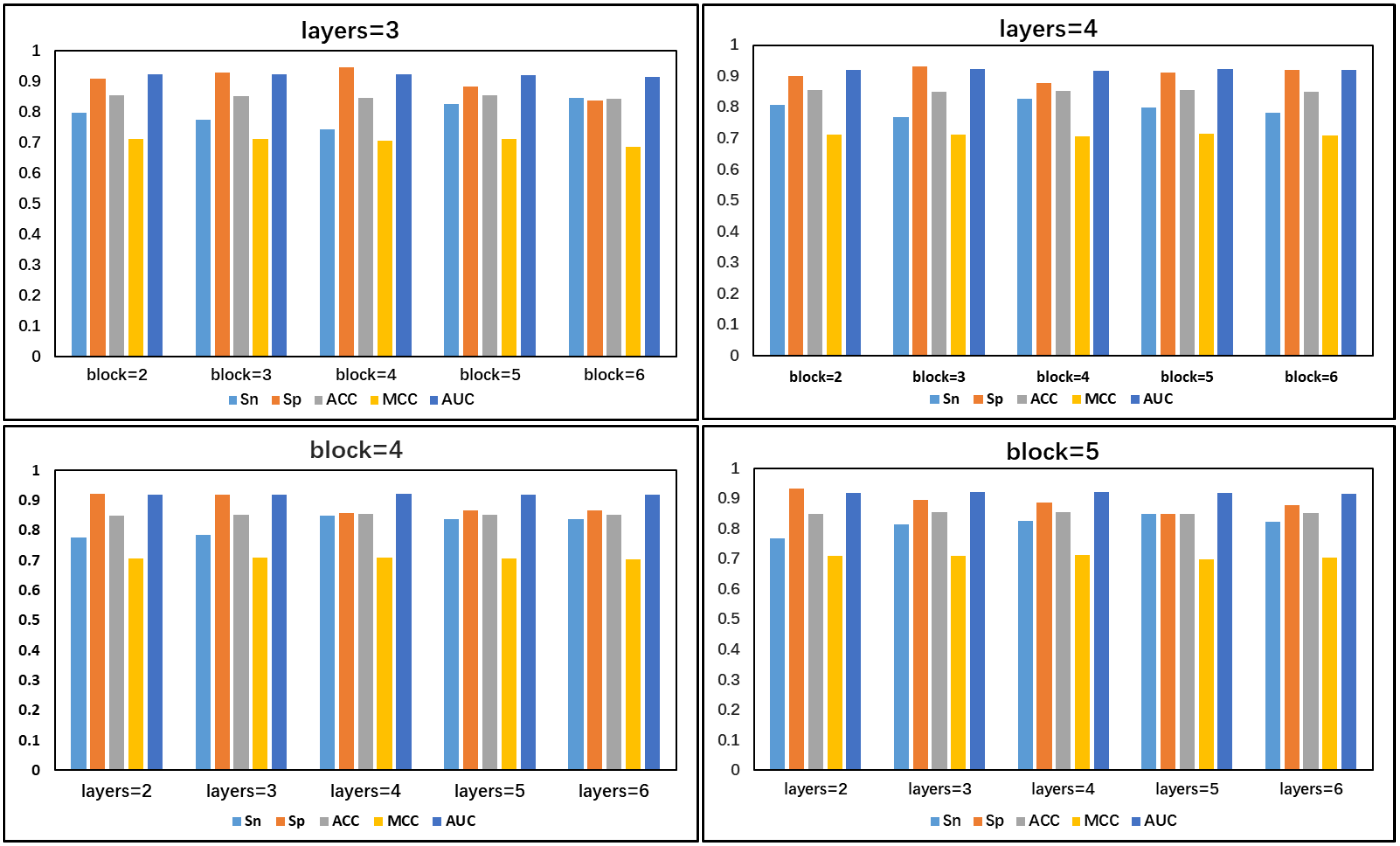 Fig. 19.
Fig. 19.Comparison of the number of dense blocks and dense block convolution layers.
To demonstrate the superiority of the deep-learning algorithms, we compared the three most representative algorithms on the five-fold cross-validation and independent test dataset using random forest (RF), logistic regression (LR), and AdaBoost. Here, we input the feature coding into each of the three machine-learning algorithms and presented the experimental results in Tables 4,5. The Sn, SP, ACC, and AUC of the three machine learning algorithms were all around 0.5, although the MCC was very low. This indicates that the deep-learning models we constructed performed better than the machine-learning models.
| Predictor | Sn | SP | Acc | MCC | AUC |
| RF | 0.531 (0.18) | 0.490 (0.19) | 0.509 (0.02) | 0.018 (0.03) | 0.511 (0.02) |
| LR | 0.507 (0.02) | 0.522 (0.03) | 0.514 (0.01) | 0.028 (0.03) | 0.521 (0.02) |
| AdaBoost | 0.504 (0.02) | 0.517 (0.03) | 0.510 (0.02) | 0.021 (0.03) | 0.516 (0.02) |
| im5C-DSCGA | 0.814 (0.02) | 0.885 (0.02) | 0.850 (0.00) | 0.701 (0.00) | 0.923 (0.00) |
MCC, Matthew’s correlation coefficient; AUC, area under the curve; LR, logistic regression. For comparison purposes, bold represents best results, and decimals in parentheses represent errors.
| Predictor | Sn | SP | Acc | MCC | AUC |
| RF | 0.479 | 0.509 | 0.494 | –0.01 | 0.491 |
| LR | 0.533 | 0.449 | 0.491 | –0.02 | 0.484 |
| AdaBoost | 0.520 | 0.445 | 0.482 | –0.03 | 0.475 |
| im5C-DSCGA | 0.810 | 0.908 | 0.859 | 0.721 | 0.926 |
For comparison purposes, bold represents best results.
To further evaluate the performance of the im5C-DSCGA model, we compared it to the existing most advanced computational method, the Deepm5C model, to identify m5C sites in human RNA sequences. Here, to provide a fair performance comparison, we used the same five-fold cross-validation and independent tests as the Deepm5C model to evaluate performances. The im5C-DSCGA model outperformed the Deepm5C model, further illustrating the better generalization capability of our proposed im5C-DSCGA model.
Table 6 presents the performance of the im5C-DSCGA model compared to the existing prediction method Deepm5C model on the training dataset for the five-fold cross-validation. Table 7 shows the performance of the im5C-DSCGA model compared to the existing prediction method Deepm5C model on the independent test dataset. The SP and MCC of the im5C-DSCGA model outperformed Deepm5C with the training data and five-fold cross-validation. Similarly, the SP, Acc, and MCC of the im5C-DSCGA model outperformed the im5C-DSCGA model by 5.1%, 0.7%, and 3.0%, respectively, using independent tests. This result indicates the strong potential of using the im5C-DSCGA model in the RNA modification site prediction task.
| Predictor | Sn | SP | Acc | MCC | AUC |
| Deepm5C | 0.835 (0.06) | 0.875 (0.01) | 0.855 (0.07) | 0.697 (0.10) | 0.941 (0.05) |
| im5C-DSCGA | 0.814 (0.02) | 0.885 (0.02) | 0.850 (0.00) | 0.701 (0.00) | 0.923 (0.00) |
| Predictor | Sn | SP | Acc | MCC | AUC |
| Deepm5C | 0.846 | 0.857 | 0.852 | 0.691 | 0.938 |
| im5C-DSCGA | 0.810 | 0.908 | 0.859 | 0.721 | 0.926 |
>For comparison purposes, bold represents best results, and decimals in parentheses represent errors.
In addition, our im5C-DSCGA model was tested as a transfer learning model using 240 samples from H. sapiens, which were from the iRNA-PseColl model [40]. Our model scored 59.2%, 75.4%, and 53.8% on the SP, Acc, and MCC metrics, respectively, although there was a slight decrease, which may be due to the fact that the two benchmark datasets involve different tissues and cell types. It was 15.9% higher on the Sn metric at 91.7%. However, as a transfer learning model, ours still showed good results in predicting samples from H. sapiens. Additional information on the prediction methods of the m5C site can be explored from the review [31].
In the im5C-DSCGA model proposed in this work, Fig. 20 shows the performance of the model for five-fold cross-validation on the training dataset. It can be clearly seen that the performance of each fold of the five-fold cross-validation is relatively stable. In addition, Fig. 21 shows the ROC curve plots for the im5C-DSCGA model on the training dataset for the five-fold cross-validation and on the independent tests. This result further highlights the stability and reliability of the im5C-DSCGA model.
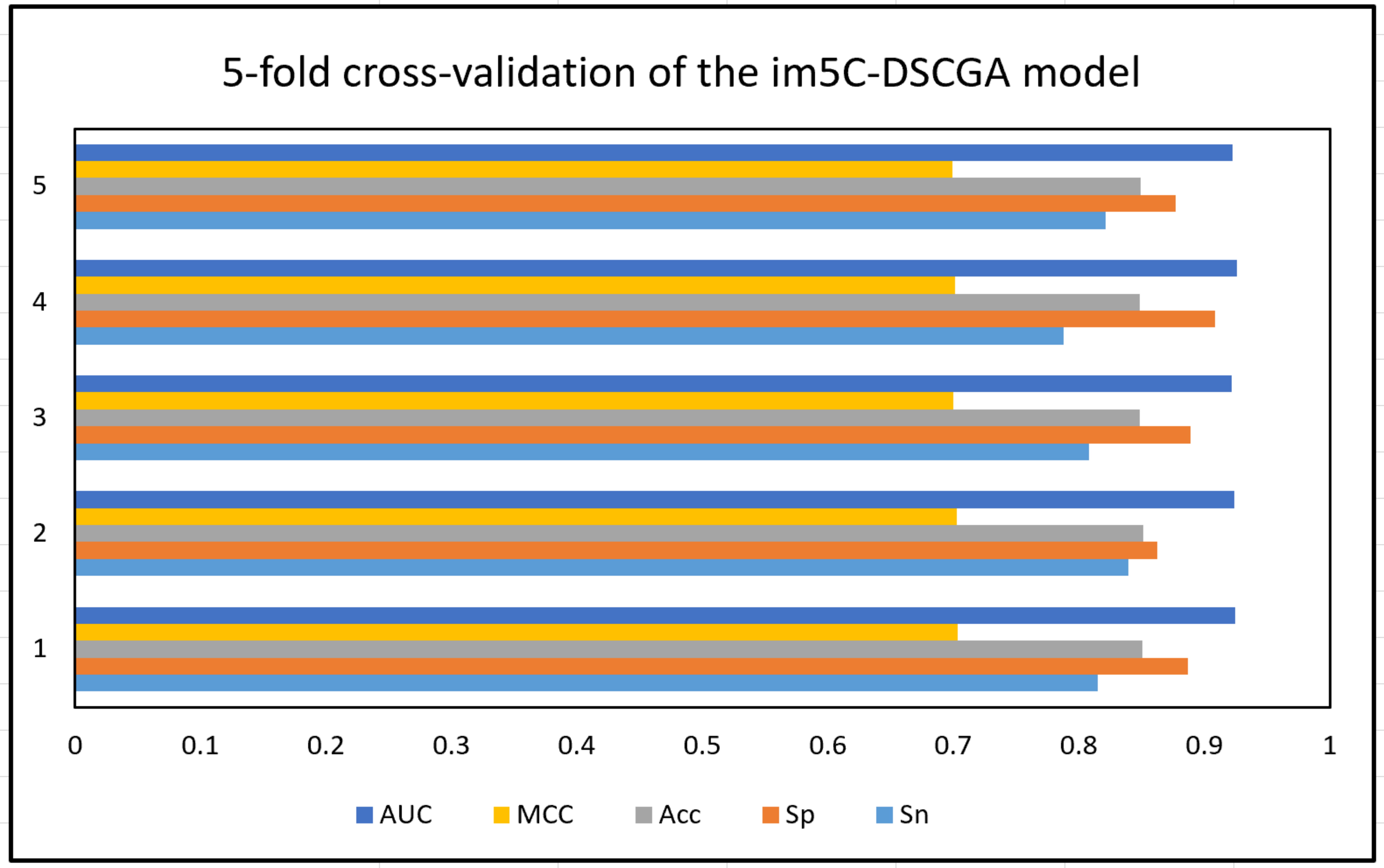 Fig. 20.
Fig. 20.Performance of im5C-DSCGA model on five-fold cross-validation.
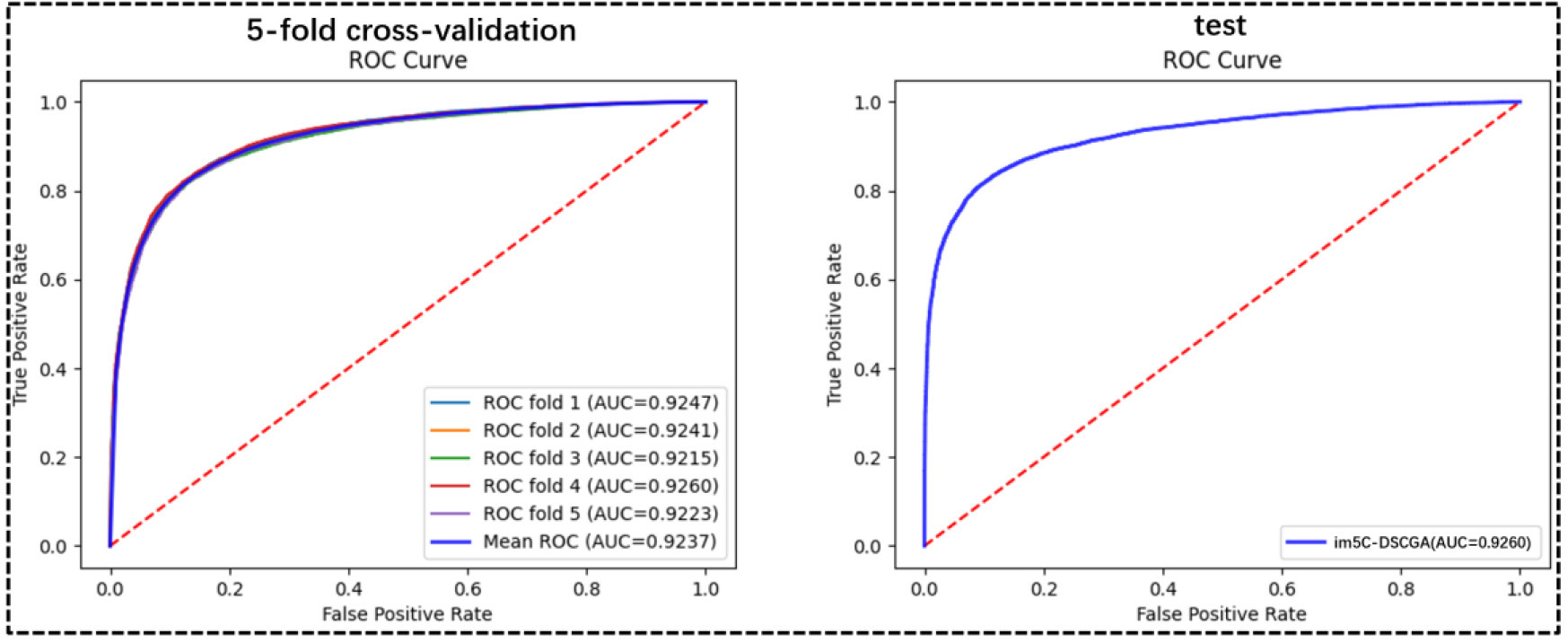 Fig. 21.
Fig. 21.Performance of im5C-DSCGA model.
Using the im5C-DSCGA network framework proposed in this work, we compared the
performance of five different feature encoding methods, including one-hot,
one-hot + NPF, one-hot + ND, NPF + ND, and one-hot + NPF + ND. They encode RNA
sequences into 4
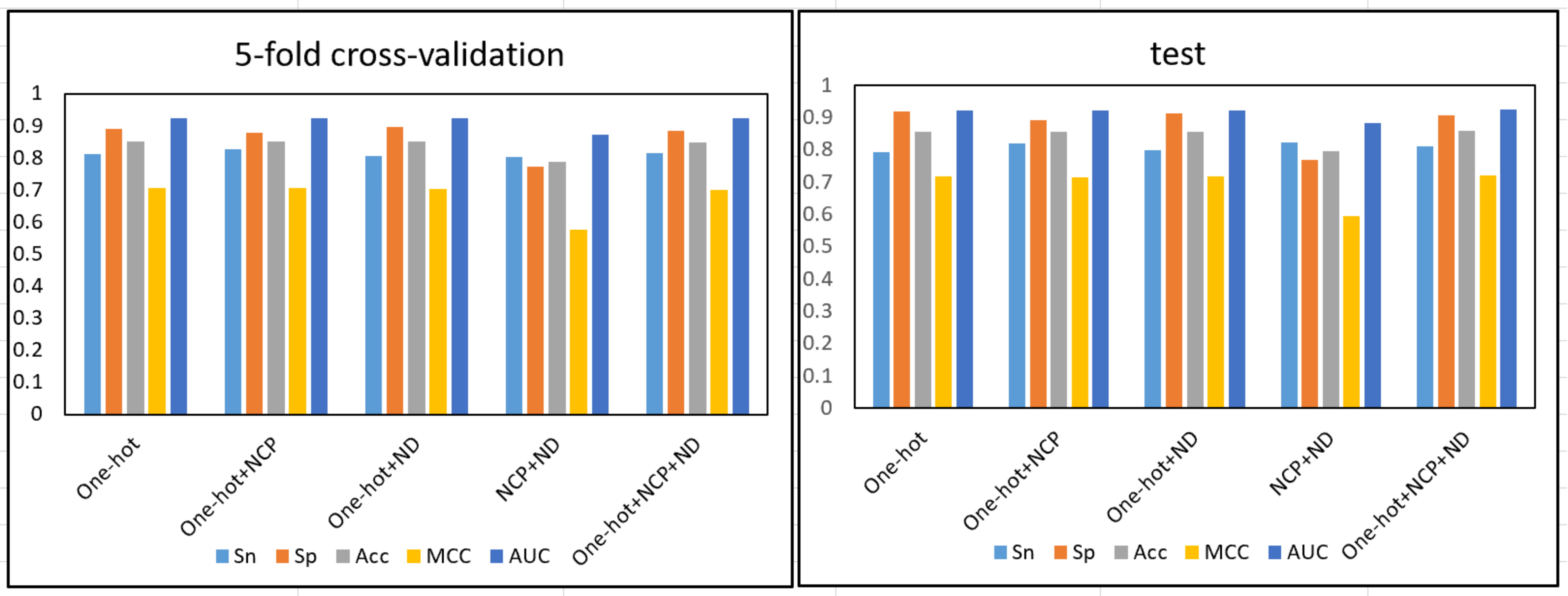 Fig. 22.
Fig. 22.Ablation experiment of feature encoding method.
It can be distinctly seen that the four feature encoding methods, one-hot, one-hot + NPF, one-hot + ND, and one-hot + NPF + ND, significantly outperform the NCP + ND feature encoding. However, the feature encoding for the one-hot + NPF + ND combination outperformed the feature encoding for the other combinations, in terms of MCC performance evaluation metrics. Therefore, we adopted the one-hot + NPF + ND feature encoding as the final feature encoding method for the im5C-DSCGA network framework.
In this study, we designed a novel deep learning-based model named im5C-DSCGA to accurately identify m5C modification sites in human RNA. The main innovation of the im5C-DSCGA model exists in the following three aspects. First, we used the improved DenseNet method and the CBAM Attention mechanism as advanced local feature extractors. Secondly, we used the BGRU method to capture the long-term dependencies of high-level local features and used Self-Attention to extract global features. Finally, we used the ensemble learning method to generate a better generalization ability in the im5C-DSCGA model. From the metrics of the experimental results, the deep learning im5C-DSCGA model proposed in this study obtains a satisfactory prediction result. The MCC metric reached 72.1% on the independent test, which is 3.0% higher than the current state-of-the-art Deepm5C model prediction method. Overall, the im5C-DSCGA model achieves a more accurate and stable performance than the Deepm5C model, which further proves the effectiveness of our model.
The completion of the im5C-DSCGA model will help researchers to better identify m5C modification sites in human RNA. Furthermore, we will extend this work in subsequent studies by trying to build a network framework using the BERT method and Transformer model alongside deep learning. In the future, we will consider building a network of servers, which can provide many conveniences. In addition, all datasets and source code for the im5C-DSCGA model are freely accessible at https://github.com/lulukoss/im5C-DSCGA.
It is simple to extract the dataset and source code for this work from https://github.com/lulukoss/im5C-DSCGA.
The experiments were created and designed by JJ and LQ; LQ has written the manuscript, and JJ and RL have revised it. Moreover, LQ and RL performed model creation, feature extraction, model testing, and performance evaluation. JJ supervised the research. The final version has been adopted by all authors, who also contributed to the research and writing of this study.
Not applicable.
Not applicable.
The authors are grateful for the constructive comments and suggestions made by the reviewers. This work was partially supported by the National Natural Science Foundation of China (Nos. 61761023, 62162032, and 31760315), the Natural Science Foundation of Jiangxi Province, China (Nos. 20202BABL202004 and 20202BAB202007), the Scientific Research Plan of the Department of Education of Jiangxi Province, China (GJJ190695). These funders had no role in the study design, data collection and analysis, decision to publish or preparation of manuscript.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.