






 , Mingjiang Liu 1,*
, Mingjiang Liu 1,*1 Department of Orthopaedics, The Affiliated Changsha Central Hospital, Hengyang Medical School, University of South China, 410004 Changsha, Hunan, China
2 Institute of Pediatrics, Laboratory of Pediatric Nephrology, Central South University, 410011 Changsha, Hunan, China
†These authors contributed equally.
Abstract
Background: Delayed wound healing, a common problem in
patients with diabetes mellitus (DM), is associated with impaired keratinocyte
autophagy. Epigallocatechin gallate (EGCG), a catechin, has been proven to
promote diabetic wound healing. This study aims to explore the therapeutic
mechanism of EGCG on diabetic wound healing. Methods: High glucose
(HG)-induced keratinocytes and streptozotocin (STZ)-induced DM rats were prepared
and intervened with EGCG to examine its therapeutic effects in in vivo
and in vitro settings. The AMPK inhibitor, Compound C, was
utilized to determine whether EGCG exerted its therapeutic effects through the
AMPK/ULK1 pathway. Results: In vitro, EGCG improved
HG-induced autophagy impairment in keratinocytes by increasing LC3II/LC3I,
Becline1, and ATG5 levels and decreasing p62 level. Mechanically, EGCG activated
the AMPK/ULK1 pathway, thereby promoting keratinocyte autophagy through the
phosphorylation of AMPK and ULK1. Notably, EGCG promoted the proliferation,
migration, synthesis and release of C-C motif chemokine ligand 2 (CCL2) in
HG-treated keratinocytes. Furthermore, EGCG indirectly promoted the activation of
fibroblasts, as evidenced by increased alpha-smooth muscle actin (
Keywords
- diabetic cutaneous ulcers (DCU)
- epigallocatechin gallate (EGCG)
- AMPK/ULK1 pathway
- keratinocytes
- autophagy
Diabetic cutaneous ulcers (DCU) are one of the complications of diabetes mellitus (DM) [1]. Glucose metabolism disorder, neuropathy, blood circulation disorder, and local infection are considered important factors causing the onset of DCU [2]. Clinically, DCU treatment includes debridement and dressing changes, infrared radiation, injection or oral administration of hypoglycemic drugs, and even amputation surgery in severe cases [3]. The long duration of treatment, high recurrence rate, and disability rate of DCU bring great physical and mental pain and economic burden to patients and families [4]. Promoting early wound healing in DCU and reducing its disability rate and treatment cost remain challenging. Therefore, it is urgent to further explore the pathogenesis of DCU, identify potential therapeutic targets, and develop new therapeutic methods.
Epigallocatechin gallate
(EGCG), a main active component in green tea, possesses several biological
activities such as anti-inflammatory [5], antioxidant [6], hypoglycemic [7],
antibacterial [8], and anticancer [9]. EGCG has displayed remarkable potential in
wound healing, as shown through in vitro and in
vivo studies [10]. In a
study of skin inflammation, EGCG can reduce the basal release and upregulation of
vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) in normal
human keratinocytes stimulated by tumor necrosis factor-
Skin wound healing is a complex process that requires collaboration between cells located in different layers of the skin [14]. During the proliferation stage of wound healing, cells from the epidermis and dermis proliferate and migrate to the wound bed to close the wound [15]. Keratinocytes in the epidermis are essential in wound healing. In DCU, keratinocytes exhibit reduced proliferation and impaired migration [16]. Autophagy, an important cellular process, plays a key role in maintaining cell homeostasis. Autophagy disorder contributes to the occurrence of skin diseases and delayed wound healing [17]. High glucose (HG) hinders keratinocyte autophagy by reducing autophagy-related protein 5 (ATG5), p62, and microtubule-associated protein 1 light chain 3 (LC3II) expressions and inhibiting keratinocyte migration, thereby delaying DCU wound healing [18]. Fibroblasts have been identified as key players in skin repair, responsible for the regeneration of granulation tissue as well as re-epithelialization [19, 20]. During wound healing, keratinocytes induce C-C motif chemokine 2 (CCL2) expression through autophagy, and the abundance of CCL2 is required to promote keratinocyte proliferation and migration and fibroblast activation [21]. Thus, the restoration of keratinocyte autophagy may be a promising method to promote wound healing in DCU.
AMP-activated protein kinase (AMPK) is an energy sensor capable of regulating a variety of metabolic and physiological processes [22]. However, AMPK activity is impaired in DM and its complications [23]. A previous study revealed that drugs that activate and regulate AMPK can enhance glucose uptake by cells and inhibit its intracellular production, offering a potential therapeutic effect on DM and its complications [24]. Additionally, the AMPK/Unc-51-like kinase-1 (ULK1) pathway is an important pathway involved in regulating autophagy. Inhibition of this pathway can reduce autophagosome production and downregulate LC3 and Beclin1 expressions [25, 26]. AMPK regulates autophagy via the direct phosphorylation of ULK1 [27]. These findings suggest that enhancing AMPK/ULK1-mediated autophagy may help to reduce the risk of DM and its complications [28, 29]. The study has found that EGCG can induce autophagy by enhancing AMPK activity, and ULK1 is crucial for EGCG-induced autophagy [30]. However, the role of EGCG in DCU by regulating AMPK/ULK1-mediated autophagy has not been reported.
Therefore, HG-induced cell models and streptozotocin (STZ)-induced DM rat models were constructed and intervened with EGCG to determine whether EGCG could restore keratinocyte autophagy, promote the activation of keratinocytes and fibroblasts, as well as accelerate wound healing in DCU by activating the AMPK/ULK1 pathway.
Keratinocytes (HaCaT, AW-CNH203, Abiowell, Changsha, China) and fibroblasts
(HFF-1, AW-CCH222, Abiowell, Changsha, China) were cultured in Dulbecco’s
modified Eagle’s medium (DMEM, D5796, Sigma,
Saint. Louis, MO, USA) and RPMI-1640 medium (2144322, Biological Industries,
Kibbutz Beit-Haemek, Israel). These medium were supplemented with 10% of fetal
bovine serum (FBS, 10099141, Gibco, Carlsbad, CA, USA) and 1%
of Penicillin/Streptomycin (AWI0070a, Abiowell, Changsha, China). They were
placed in an incubator (DH-160I, SANTN, Shanghai, China) containing 5% CO
For HaCaT cells, grouping 1 included 0 µM EGCG, 6.25 µM EGCG, 12.5
µM EGCG, 25 µM EGCG, 50 µM EGCG, and 100 µM EGCG.
Grouping 2 included Control, HG, HG + 6.25 µM EGCG, HG + 12.5 µM
EGCG, HG + 25 µM EGCG, and HG + 50 µM EGCG. The HaCaT cells in the
Control group were cultured normally. The HaCaT cells in the other groups were
treated with HG (50 mM glucose) for 24 h and EGCG (2503504, Shanghai Yuanye
Bio-Technology Co., Ltd., Shanghai, China) at different concentrations for 24 h
[31]. Grouping 3 included Control, HG, HG + EGCG, and HG + EGCG + Compound C.
Except for the Control group, HaCaT cells in other groups were treated with HG
for 24 h. HaCaT cells in the HG + EGCG group were treated with 50 µM EGCG
for 24 h after HG induction. HaCaT cells in the HG + EGCG + Compound C group were
treated with 50 µM Compound C (D426448,
Aladdin, Shanghai, China) and 50 µM
EGCG for 24 h after HG induction [32]. The information about the processing of
different groups was shown in Table 1. To induce the synthesis and release of
CCL2, cells in each group were treated with 100 ng/mL TNF-
| Group name | HG induction | EGCG | Compound C | |
| Grouping 1 | 0 µM EGCG | - | - | - |
| 6.25 µM EGCG | - | 6.25 µM | - | |
| 12.5 µM EGCG | - | 12.5 µM | - | |
| 25 µM EGCG | - | 25 µM | - | |
| 50 µM EGCG | - | 50 µM | - | |
| 100 µM EGCG | - | 100 µM | - | |
| Grouping 2 | Control | - | - | - |
| HG | + | - | - | |
| HG + 6.25 µM EGCG | + | 6.25 µM | - | |
| HG + 12.5 µM EGCG | + | 12.5 µM | - | |
| HG + 25 µM EGCG | + | 25 µM | - | |
| HG + 50 µM EGCG | + | 50 µM | - | |
| Grouping 3 | Control | - | - | - |
| HG | + | - | - | |
| HG + EGCG | + | 50 µM | - | |
| HG + EGCG + Compound C | + | 50 µM | 50 µM |
EGCG, Epigallocatechin gallate; HG, High glucose.
Male adult Sprague-Dawley rats (SPF, 7–8 weeks, 180–200 g) were bought from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). After adaptive feeding, rats were randomly divided into Normal, DM, DM + EGCG, and DM + EGCG + Compound C groups with 10 rats in each group. The DM rat model was established according to a previously described method [33]. Rats were intraperitoneally injected with STZ (80 mg/kg) (AWH0492a, Abiowell, Changsha, China) dissolved in citrate buffer solution (0.1 M, pH 4.5) to induce type I DM [34], with the same dose of citrate buffer solution being the control in the Normal group. Follow-up experiments were performed when the blood glucose level reached 16.7 mM. After the rats were anesthetized with intraperitoneal injection of 1% pentobarbital (30 mg/kg), a 15 mm diameter wound was created on the back using the full-thickness skin defect method. Rats in Normal and DM groups were treated daily with 1% carboxymethyl cellulose (C501052, Aladdin, Shanghai, China). Rats in the DM + EGCG group were treated with 1 mg/mL EGCG daily [35]. Rats in the DM + EGCG + Compound C group were given 1 mg/kg Compound C near the wound at 30 min before EGCG intervention [36]. The information about the processing of different groups was shown in Table 2. On days 0, 3, 7, and 14, the wound area was measured and photographed, and the wound healing rate was calculated according to the methods described in the literature [35]. On day 14, rats were sacrificed by cervical dislocation, and neonatal epithelial tissues were taken for follow-up analysis.
| Group name | STZ induction | EGCG | Compound C | |
| Grouping 1 | Normal | - | - | - |
| DM | 80 mg/kg | - | - | |
| DM + EGCG | 80 mg/kg | 1 mg/mL | - | |
| DM + EGCG + Compound C | 80 mg/kg | 1 mg/mL | 1 mg/kg |
Notes: EGCG, Epigallocatechin gallate; DM, diabetes mellitus; STZ, streptozotocin.
HaCaT cells in the logarithmic growth phase were digested with trypsin (AWC0232,
Abiowell, Changsha, China) to prepare a cell suspension. Cells were inoculated
into 96-well plates at a density of 1
Total proteins were extracted from HaCaT cells and neonatal epithelial tissues
using RIPA lysate (AWB0136, Abiowell, Changsha, China). The proteins were
separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
and transferred to nitrocellulose (NC) membranes. NC membranes were blocked with
5% skimmed milk (AWB0004, Abiowell, Changsha, China) and incubated with the
following primary antibodies respectively, including p62 (1:10,000, 66184-1-Ig,
Proteintech, Chicago, IL, USA), Beclin1
(1:2000, 11306-1-AP, Proteintech, Chicago, IL, USA), ATG5 (1:5000, ab108327,
Abcam, Cambridge, UK), LC3 (2 µg/mL, ab48394, Abcam, Cambridge, UK),
phosphorylated-AMPK (p-AMPK, 1:5000, ab92701, Abcam, Cambridge, UK), AMPK
(1:5000, 10929-2-AP, Proteintech, Chicago, IL, USA), p-ULK1 (1:5000,
29006-1-AP, Proteintech, Chicago, IL, USA), ULK1 (1:20,000, ab133747, Abcam,
Cambridge, UK), CCL2 (1:2000, 66272-1-Ig, Proteintech, Chicago, IL, USA),
transforming growth factor-
Cell slides were washed with phosphate-buffered saline (PBS), fixed with 4%
paraformaldehyde, permeabilized with 0.3% Triton, and blocked with 5% bovine
serum albumin (BSA). For tissue slices, antigen repair was performed after
dewaxing to water. The tissue slices were washed with PBS, and sequentially
placed in sodium borohydride solution, 75% ethanol solution, and Sudan black dye
solution. After washing with PBS, the tissue slices were blocked with 5% BSA.
The cell slides were incubated respectively with primary antibodies of LC3
(1:100, 14600-1-AP, Proteintech, Chicago, IL, USA), p62 (1:50, 18420-1-AP,
Proteintech, Chicago, IL, USA), alpha-smooth
muscle actin (
HaCaT cells (1
The scratch assay was applied to detect the migration ability of
HaCaT cells in each group. HaCaT cells from different groups
were digested to make a cell suspension. The 5
The HaCaT cell cultures in different groups were centrifuged at 1000 g at 4 °C for 15 min, and the supernatant was used for detection. The content of CCL2 released to supernatant was assessed according to the instruction of the CCL2 ELISA kit (CSB-E04655h, CUSABIO, Wuhan, China).
The epithelial tissue slices were dewaxed in xylene and dehydrated with gradient ethanol (75–100%). They were stained with hematoxylin (AWI0001a, Abiowell, Changsha, China), and then returned to blue in PBS. Next, the slices were stained with eosin (AWI0029a, Abiowell, Changsha, China), and dehydrated with gradient alcohol (95–100%). The slices were cleared in xylene and then sealed with neutral gum (AWI0238a, Abiowell, Changsha, China) for observation with a microscope.
After being dewaxed to water, the epithelial tissue slices were immersed in
citrate buffer (0.01 M, pH 6.0) (AWI0206a, Abiowell, Changsha, China), and boiled
for antigen retrieval. Subsequently, the endogenous enzymes were inactivated with
1% periodic acid. The slices were incubated with the primary antibody of
The epithelial tissue slices were dewaxed to water and stained with hematoxylin. They were washed with tap water and distilled water in turn. Next, the slices were soaked in PBS to make the nucleus return blue and stained with an acid fuchsin stain solution. Then, the slices were reacted with a phosphomolybdic acid differentiation solution, stained with aniline blue counterstain, and rinsed with absolute ethanol. The slices were blow-dried, cleared in xylene, and then sealed with neutral gum before observation with a microscope.
Data were analyzed by
GraphPad
Prism 8.0.1 (GraphPad Software Inc., San Diego, CA, USA) and expressed as mean
To screen suitable EGCG concentration, keratinocytes were treated with a series of EGCG concentrations (0, 6.25, 12.5, 25, 50, and 100 µM) for 24 h, and the cell viability was measured with the CCK-8 assay. No significant effect on cell viability was observed with EGCG at 0, 6.25, 12.5, 25, and 50 µM. However, However, keratinocyte viability was significantly decreased with 100 µM EGCG. Thus, 50 µM EGCG was selected for subsequent experiments (Fig. 1A). To explore the effect of EGCG on autophagy impairment, keratinocytes were first induced with HG (50 mM glucose) for 24 h, and then treated with EGCG for another 24 h, after which the levels of autophagy-related proteins were analyzed. After HG induction, autophagy was impaired in keratinocytes, showing decreased LC3II/LC3I, Beclin1, and ATG5 levels, and increased p62 level. However, after EGCG treatment, LC3II/LC3I, Beclin1, and ATG5 levels raised whereas p62 level declined in HG-treated keratinocytes, and the extent of these changes was EGCG concentration-dependent (Fig. 1B). Collectively, the above results demonstrated that EGCG improved HG-induced autophagy impairment in keratinocytes.
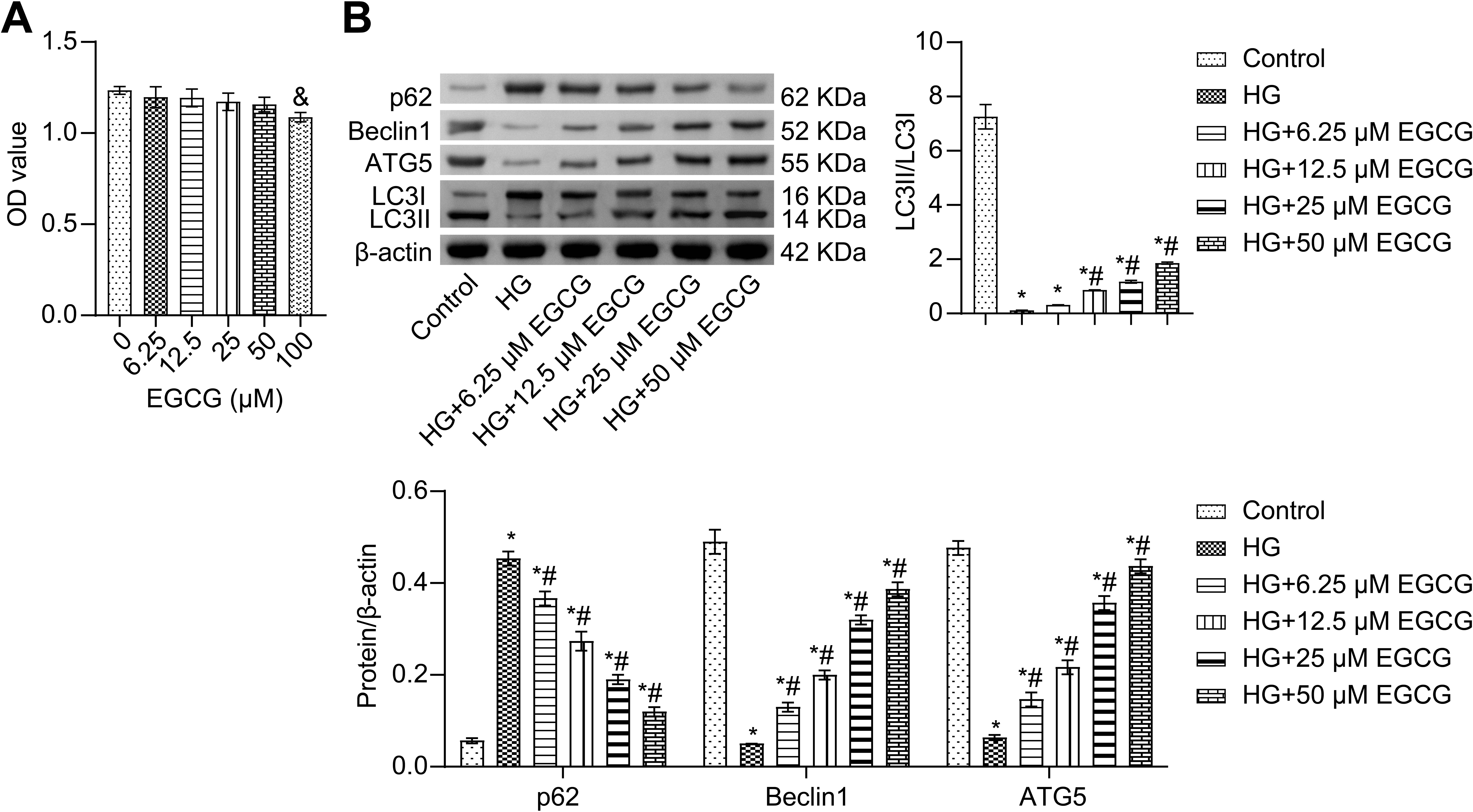 Fig. 1.
Fig. 1.EGCG induced autophagy in HG-treated keratinocytes. (A) The
viability of keratinocytes treated with different concentrations of EGCG was
evaluated by the CCK-8 assay. (B) The levels of p62, Beclin1, ATG5, LC3I, and
LC3II in keratinocytes were analyzed by Western blot. &p
To clarify the regulation of the AMPK/ULK1 pathway in autophagy induction by EGCG in HG-treated keratinocytes, an AMPK inhibitor (Compound C) was utilized to intervene, and proteins related to autophagy and the pathway were analyzed. HG induction led to a reduction in LC3, Beclin1, and ATG5 levels while a rise in p62 level in keratinocytes. Compared with the HG group, LC3, Beclin1, and ATG5 levels were raised and p62 level was declined in keratinocytes in the HG + EGCG group. However, the use of Compound C significantly reversed the autophagy induction of EGCG (Fig. 2A,B). Further analysis demonstrated that HG induction decreased the phosphorylation of AMPK and ULK1 in keratinocytes. Compared with the HG group, phosphorylation of AMPK and ULK1 in keratinocytes increased in the HG + EGCG group. However, the use of Compound C significantly reversed the effect of EGCG on AMPK/ULK1 pathway activation (Fig. 2C). These results suggested that EGCG enhanced autophagy in HG-treated keratinocytes by activating the AMPK/ULK1 pathway.
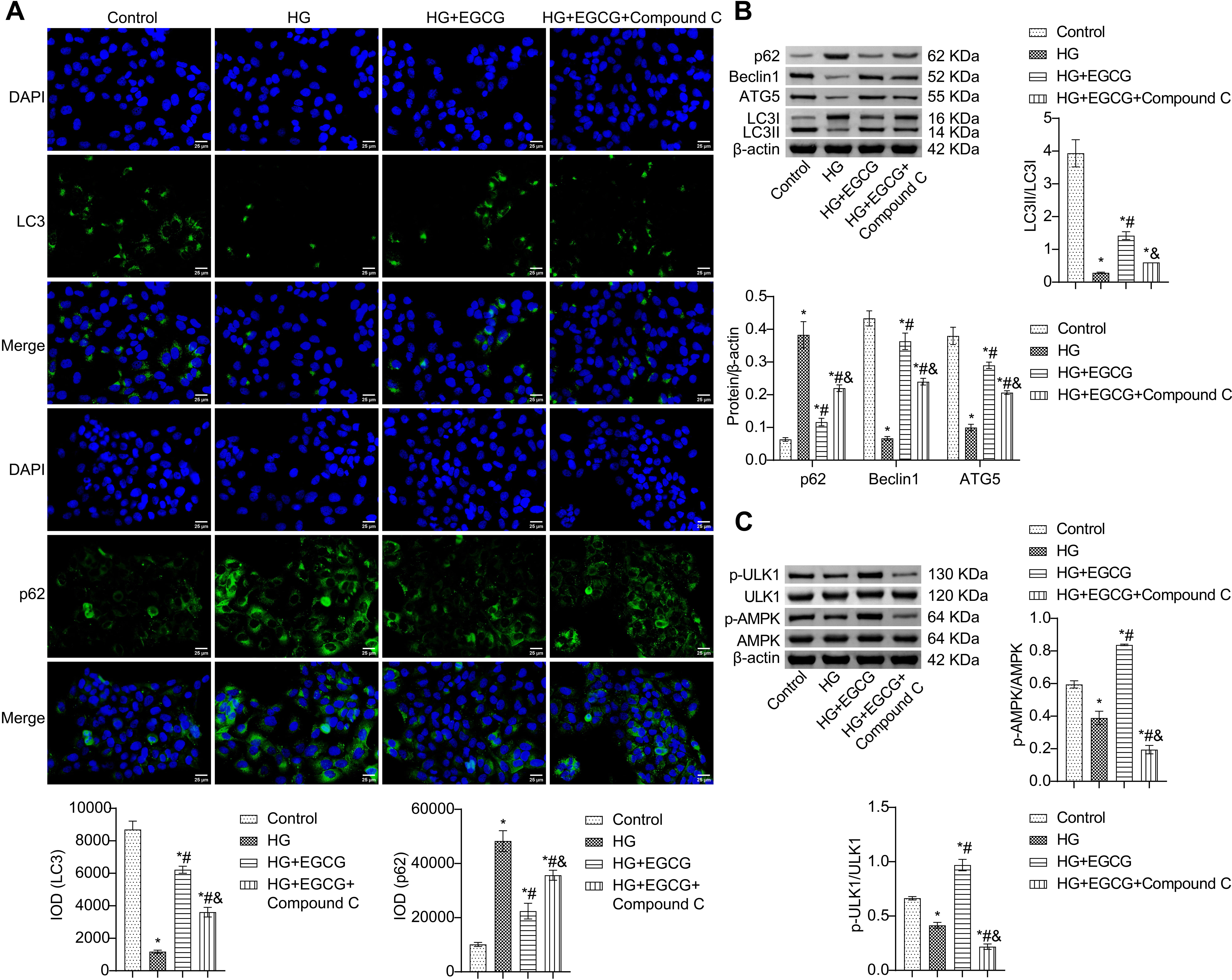 Fig. 2.
Fig. 2.Activation of the AMPK/ULK1 pathway could promote autophagy
induction of EGCG to HG-treated keratinocytes. (A) The levels of LC3 and p62 in
keratinocytes were detected by IF staining. (B,C) The levels of
p62, Beclin1, ATG5, LC3I, LC3II, p-ULK1, ULK1, p-AMPK, and AMPK in keratinocytes
were determined by Western blot. *p
Based on the above results, we further conducted EdU staining, scratch assay, and Western blot analyses on the proliferation and migration of keratinocytes. The proliferation and migration of keratinocytes were significantly inhibited following HG induction. Compared with the HG group, the proliferation, and migration of keratinocytes were significantly raised in the HG + EGCG group. However, the use of Compound C significantly reversed the promoting effects EGCG on the proliferation and migration of keratinocytes (Fig. 3A,B). Through the detection of chemokine CCL2, we observed a decrease in the synthesis of CCL2 in keratinocytes, as well as a decline in the content of CCL2 released into the culture supernatant after HG induction. Compared with the HG group, the synthesis and release of CCL2 in keratinocytes were raised in the HG + EGCG group. Notably, the use of Compound C significantly reversed the promoting effects of EGCG on the synthesis and release of CCL2 (Fig. 3C,D). These results displayed that EGCG promoted the proliferation and migration of HG-treated keratinocytes by enhancing autophagy.
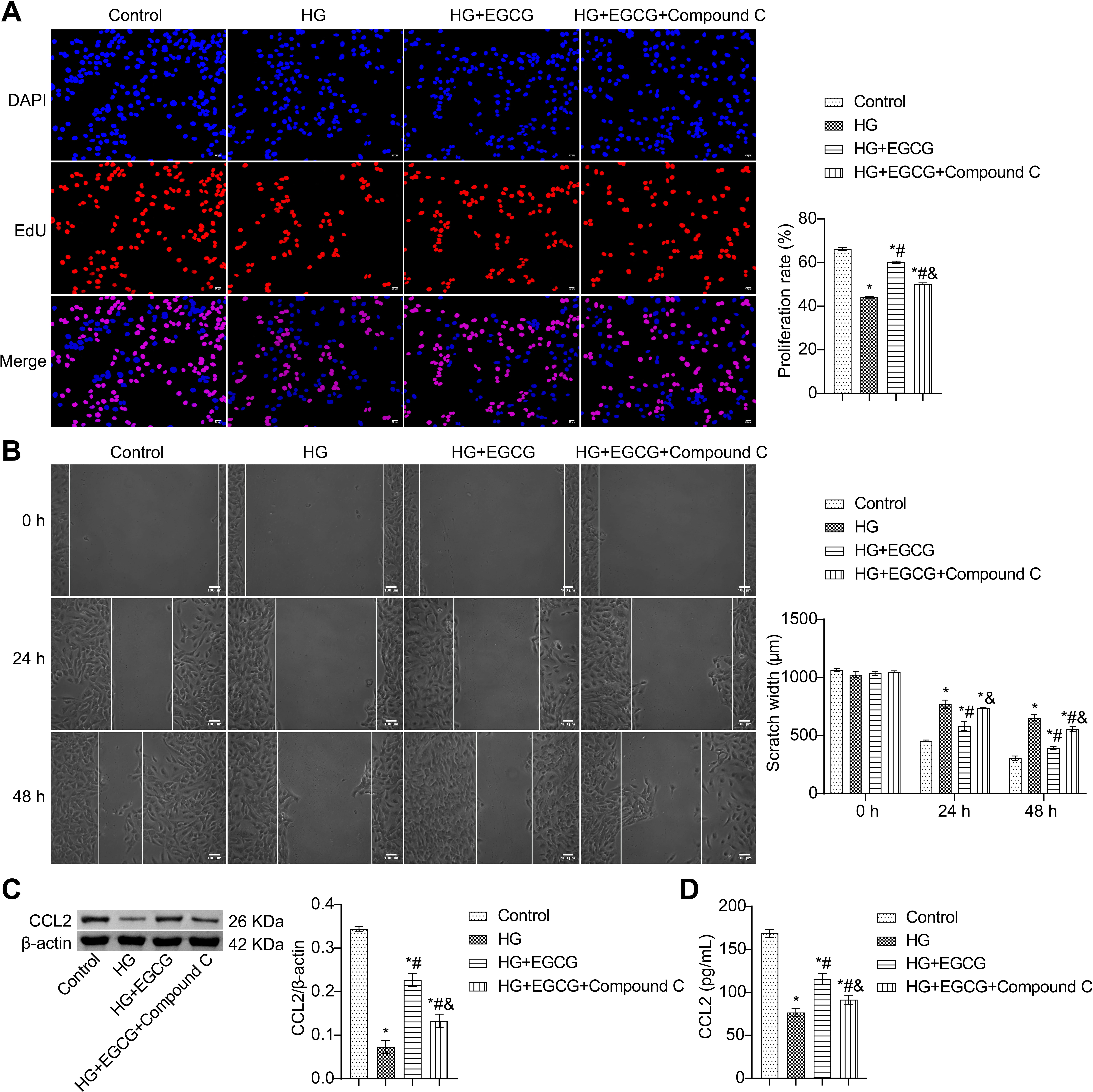 Fig. 3.
Fig. 3.EGCG promoted the proliferation and migration of HG-treated
keratinocytes by enhancing autophagy. (A) EdU staining of keratinocyte
proliferation. (B) Scratch assay of keratinocyte migration. (C) The synthesis of
CCL2 in keratinocytes was analyzed by Western blot. (D)
The content of CCL2 in the keratinocyte culture
supernatant was determined by ELISA. *p
To explore the impact of EGCG-induced keratinocyte autophagy on fibroblasts, the
culture supernatant of keratinocytes from different treatment groups was added to
the fibroblasts and incubated for 48 h. Fibroblast markers, including
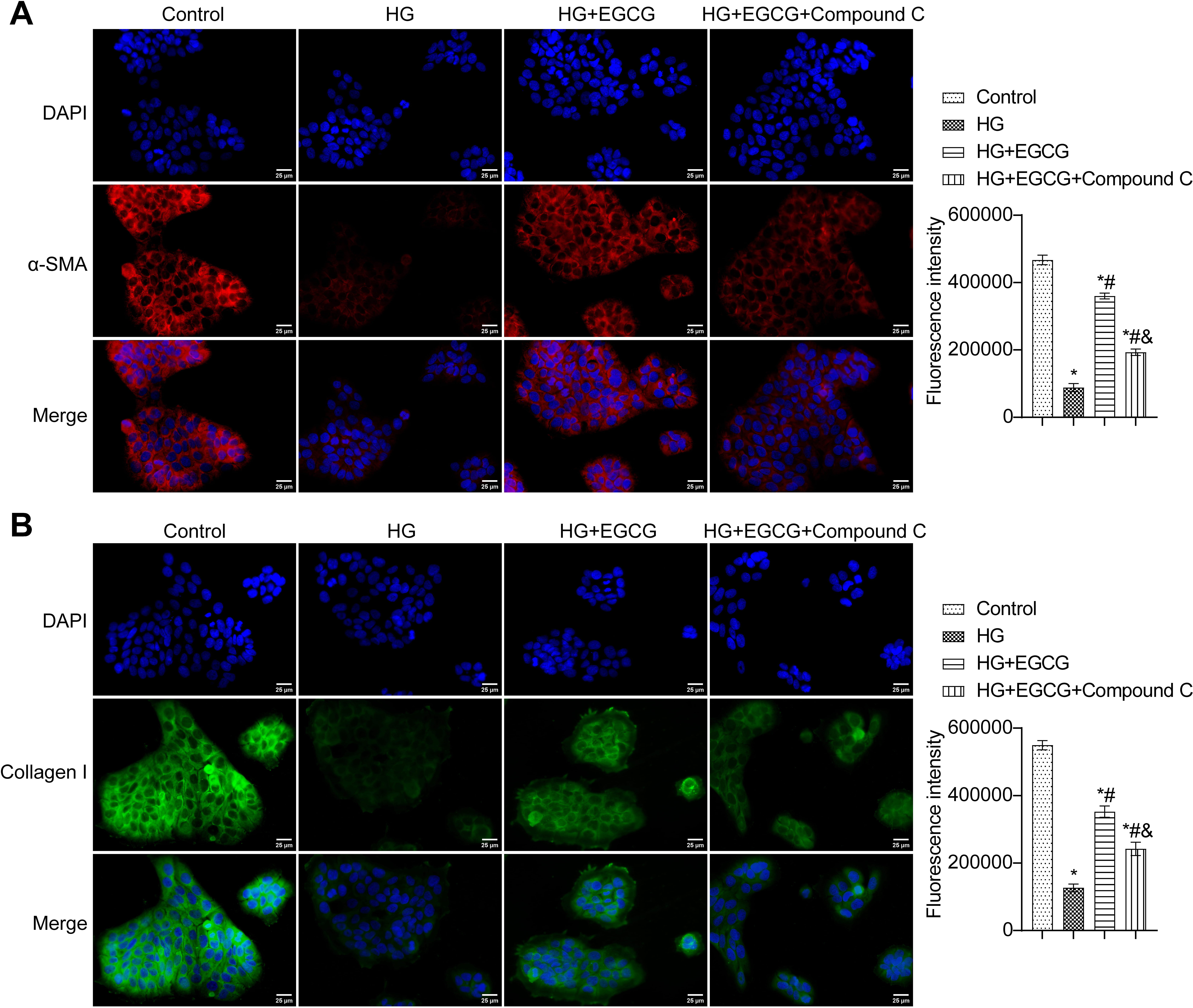 Fig. 4.
Fig. 4.Autophagy induction of EGCG on keratinocytes promoted fibroblast
activation. (A,B) The levels of
To explore the effect of EGCG on DCU, wounds on the back of DM rats were intervened with EGCG and Compound C, and the wound healing process was monitored on days 0–14. In the Control group, the wound area was gradually decreased with time and was fully epithelialized by day 14. By contrast, the wound in the DM group healed slowly and was not completely healed by day 14. However, wound healing in the DM + EGCG group was significantly improved, with the wound appearing nearly fully healed by day 14. Notably, the use of Compound C significantly reversed the beneficial effect of EGCG on wound healing (Fig. 5A,B). HE staining of the wounds on day 14 showed that the granulation tissue was reduced, and the re-epithelialization was weakened, accompanied by many inflammatory infiltrates in the DM group compared with the Control group. After EGCG intervention, inflammatory infiltration decreased, granulation tissue increased, and re-epithelialization enhanced. Unfortunately, the treatment with Compound C significantly reversed the pro-epithelialization effect of EGCG (Fig. 5C). Further analysis showed that compared with the Control group, the levels of LC3II/LC3I, Beclin1, ATG5, and the phosphorylation of AMPK and ULK1 declined in the DM group, whereas the level of p62 was increased. After EGCG intervention, the levels of LC3II/LC3I, Beclin1, ATG5, and phosphorylation of AMPK and ULK1 were increased, whereas the level of p62 was decreased. Again, the use of Compound C significantly reversed the promoting effects of EGCG on epidermal autophagy and AMPK/ULK1 pathway activation (Fig. 5D–F). These results illustrated that EGCG enhanced epidermal autophagy through the AMPK/ULK1 pathway to promote diabetic wound healing.
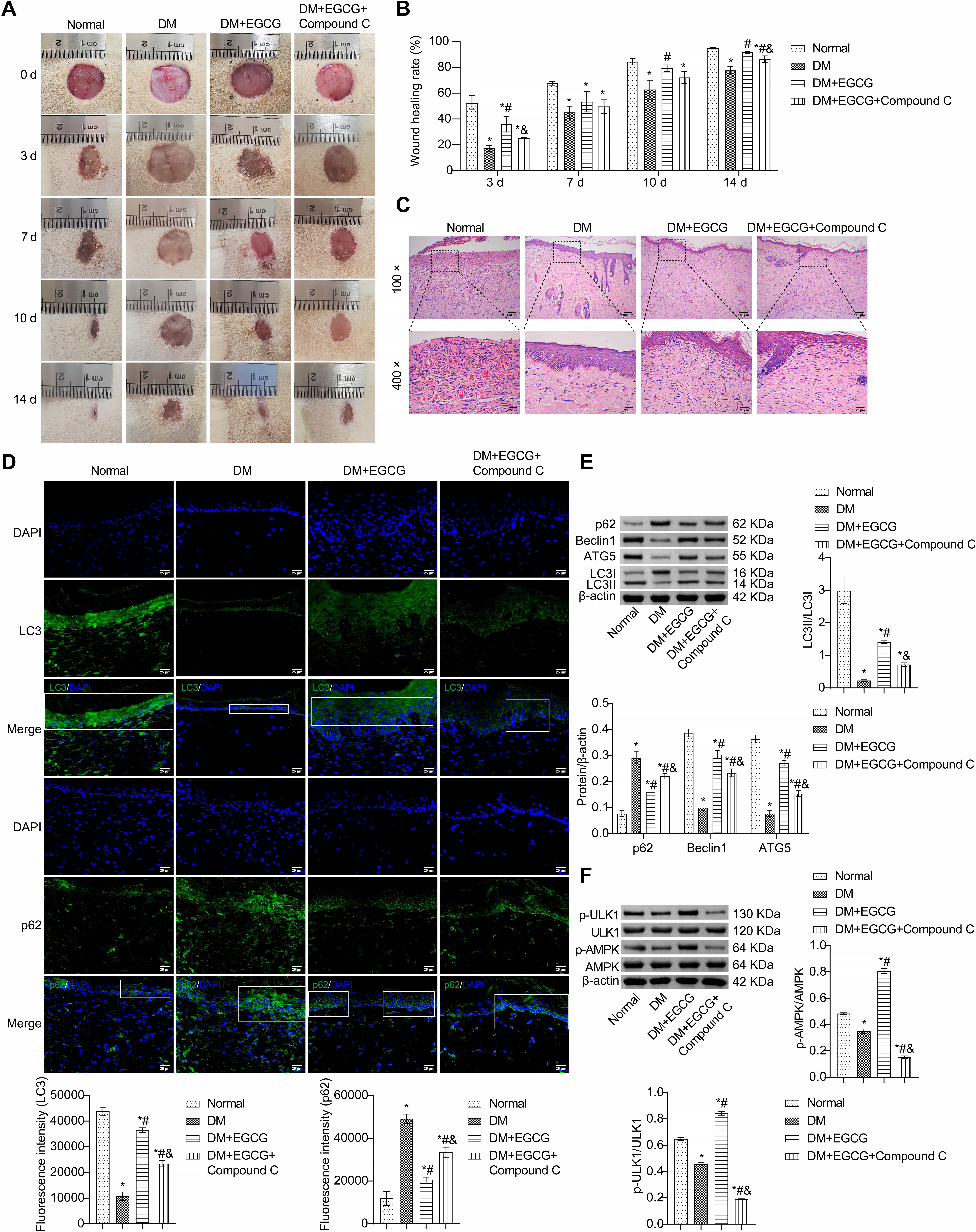 Fig. 5.
Fig. 5.EGCG enhanced epidermal autophagy through the AMPK/ULK1 pathway
to promote diabetic wound healing. (A) Wound images. (B) Wound healing rate. (C)
Wound epithelialization was observed on day 14 by HE staining. (D) The levels of
LC3 and p62 in wounds on day 14 were analyzed by IF staining. (E,F)
The levels of p62, Beclin1, ATG5, LC3I, LC3II, p-ULK1, ULK1,
p-AMPK, and AMPK in wounds on day 14 were analyzed by Western blot. *p
We examined keratinocyte proliferation and differentiation and fibroblast
activation-related proteins in the wound on day
14. IF staining showed a decrease in the
number of ATG5/KRT14- and ATG5/KRT10-positive
cells in the DM group compared with the Normal group. After EGCG intervention,
the number of ATG5/KRT14- and ATG5/KRT10-positive cells was increased. However,
the use of Compound C significantly reversed the effects of EGCG (Fig. 6A,B).
Masson staining displayed that collagen deposition was reduced and collagen
fibers were disordered in the DM group compared with the Normal group. After EGCG
intervention, collagen deposition recovered and the arrangement of collagen
fibers tended to be orderly. However, the use of Compound C significantly
reversed the promotion of EGCG on collagen deposition (Fig. 6C). Further study
displayed that the levels of
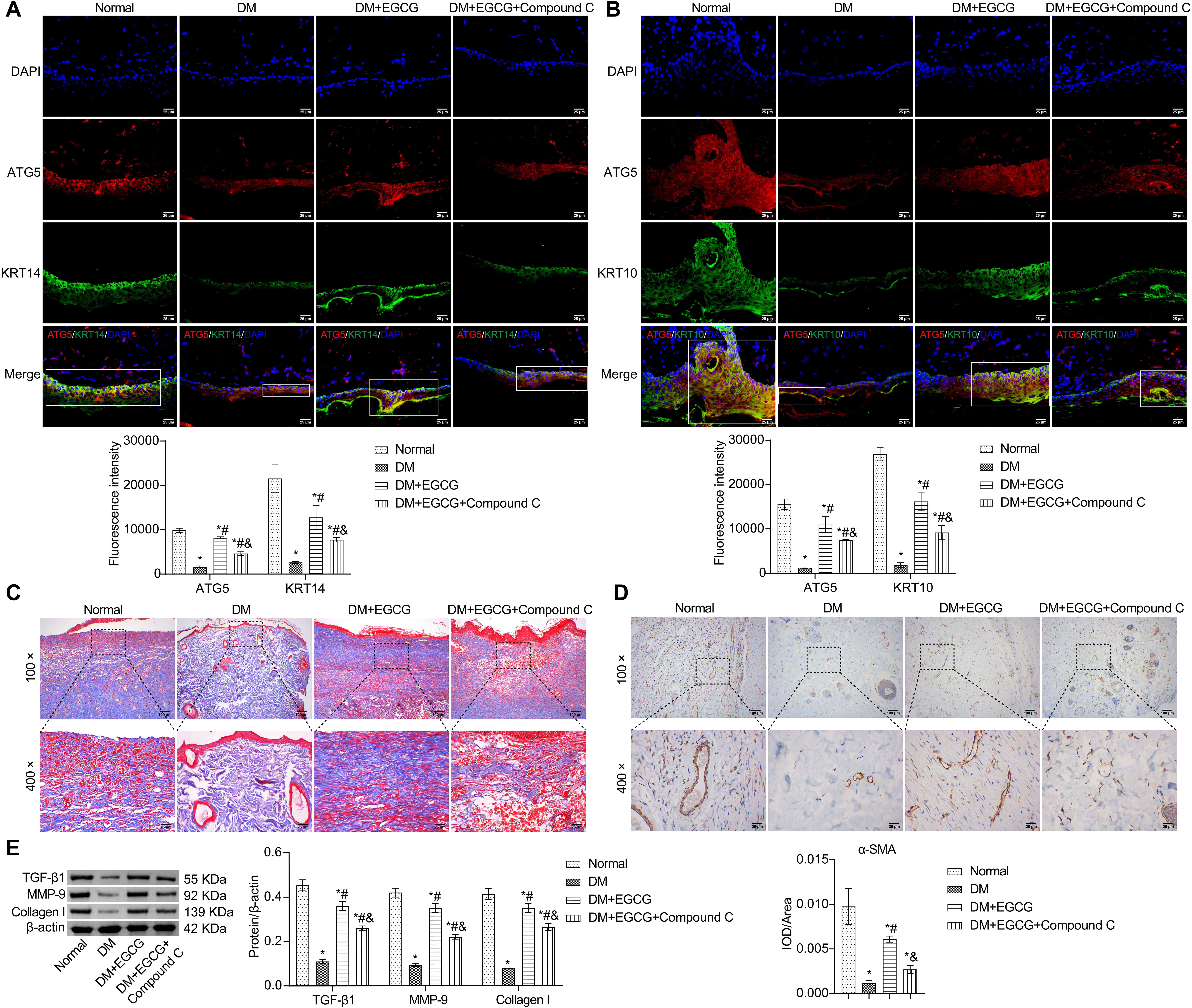 Fig. 6.
Fig. 6.EGCG promoted keratinocyte proliferation and differentiation and
fibroblast activation by enhancing epidermal autophagy. (A,B) The levels of
ATG5, KRT10, and KRT14 in wounds on day 14 were analyzed by IF staining. (C)
Collagen deposition in wounds on day 14 was assessed by Masson staining. (D) The
level of
DM is a common public health problem in modern society. Treatment for DCU, a major complication of DM, is still under investigation. EGCG, as a component of green tea, possesses various properties [37, 38, 39]. In addition, EGCG has beneficial effects on skin wound healing [10]. However, the precise mechanism by which EGCG promotes wound healing in DCU remains unknown. Here, our findings suggested that EGCG could promote DCU wound healing by restoring keratinocyte autophagy through the AMPK/ULK1 pathway.
Autophagy, one of the modes of cell death, is believed to exert a key regulatory role in the recovery from diseases [40]. Recently, the role of autophagy in wound healing has attracted attention [41]. Physiological dysfunction of epidermal keratinocytes plays an important role in delayed diabetic wound healing, including impaired autophagy, proliferation, and migration [42]. Damage to keratinocytes and other skin cells from an HG environment is a major cause of poor diabetic wound healing [43]. Here, keratinocyte autophagy was significantly reduced after HG induction. Unsurprisingly, the addition of EGCG upregulated LC3II/LC3I, Beclinl, and ATG5 levels and downregulated p62 level in HG-induced keratinocytes. In related studies, the increased expression of Beclinl, an autophagy-related gene, contributed to burn wound healing [44]. Beclin1/LC3-mediated autophagy may be beneficial to maintain the survival of injured cells [45]. In addition, increasing ATG5/ATG7 levels can promote the functional recovery of keratinocytes under oxidative stress [46]. A previous study showed that p62 knockdown enhances keratinocyte motor function [47]. Cell proliferation and migration are important processes in normal wound healing. The recovery of keratinocyte proliferation and migration further verified the ameliorative effect of EGCG on HG injury. CCL2 has the function of promoting angiogenesis and immunomodulatory, and is an important chemokine to accelerate wound healing [48]. The addition of EGCG significantly promoted the synthesis and release of CCL2 in HG-induced keratinocytes. These results suggested that EGCG might promote diabetic wound healing by improving the autophagy injury of keratinocytes and thereby promoting their proliferation and migration.
Among the various types of skin cells, keratinocytes and fibroblasts are the
main cells involved in the process of wound healing [49]. Fibroblasts are immune
regulatory factors in wound healing that play a major role in the construction
and remodeling of extracellular matrix in DCU treatment [50]. MMP-9 is thought to
be involved in keratinocyte migration, granulation tissue remodeling, and
epithelialization during wound healing [51]. Mice deficient in MMP-9 exhibited
disturbed collagen fibrogenesis and delayed epithelialization [52]. Collagen
synthesis and deposition are key factors in wound closure and are related to the
expression of TGF-
Autophagy initiation is mediated by the ULK1 complex, which is regulated by AMPK [56]. In addition, the downregulation of ULK1 has been associated with autophagy injury in DM and its complications [57]. The AMPK signaling pathway plays a major role in autophagy induction, and the upregulation of p-AMPK expression can induce glucose metabolism [58]. The use of EGCG significantly promoted the phosphorylation of AMPK and ULK1 in HG-induced keratinocytes and neonatal epithelial tissues in DM rats. Therefore, we speculated that the role of EGCG is related to the AMPK/ULK1 pathway. To further verify the regulatory role of the AMPK/ULK1 pathway in EGCG promoting diabetic wound healing, Compound C, a selective AMPK inhibitor, was selected for intervention in vitro and in vivo. As a result, autophagy, proliferation, and migration of keratinocytes and activation of fibroblasts were inhibited, and collagen synthesis and deposition were reduced, thus delaying epithelialization and wound healing. These results displayed that EGCG accelerated diabetic wound healing through activation of the AMPK/ULK1 pathway.
Despite the promising findings of this study, there are several limitations that need to be addressed in future research. Firstly, all experiments were performed using in vitro and in vivo models, and the findings may not necessarily reflect the complexities of diabetic wound healing in humans. Further clinical trials are needed to validate the therapeutic potential of EGCG in diabetic wound healing. Secondly, while the study focused on the role of EGCG in keratinocyte autophagy, the potential effects on other pathways involved in wound healing, such as oxidative stress and inflammation, were not investigated. Future studies should explore the comprehensive mechanisms underlying the therapeutic effects of EGCG in diabetic wound healing. Finally, the study only examined the effects of EGCG on the AMPK/ULK1 pathway. Other pathways that regulate autophagy may also be involved in the therapeutic effects of EGCG, and further studies are needed to elucidate these mechanisms.
To conclude, EGCG promoted diabetic wound healing, which might be achieved by restoring keratinocyte autophagy through the AMPK/ULK1 pathway to promote the activation of keratinocytes and fibroblasts. This study will provide value for the study of the pathogenesis and treatment of DCU.
DCU, Diabetic cutaneous ulcers; DM, diabetes mellitus; EGCG, Epigallocatechin
gallate; TNF-
All raw data can be provided upon request.
LJ and ML designed the research study. CT, YF, TC, ZZ, and XH performed the research. CT and YF analyzed the data. CT and YF wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This research has been approved by the Institutional Animal Ethics Committee of South China University (No. LSZ2023D110H).
Not applicable.
This research was supported by Natural Science Foundation of Changsha (No. kq2208441 and kq2208446), Key Project in Changsha Central Hospital (No. YNKY202107), and Hunan Clinical Medical Technology Innovation Guidance Project (No. 2020SK53304).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.