
 , Yu-Dong Cai 2,*
, Yu-Dong Cai 2,*1 Department of Science and Technology, Binzhou Medical University Hospital, 256603 Binzhou, Shandong, China
2 School of Life Sciences, Shanghai University, 200444 Shanghai, China
3 Changping Laboratory, 102206 Beijing, China
4 Key Laboratory of Stem Cell Biology, Shanghai Jiao Tong University School of Medicine (SJTUSM) & Shanghai Institutes for Biological Sciences (SIBS), Chinese Academy of Sciences (CAS), 200031 Shanghai, China
5 College of Information Engineering, Shanghai Maritime University, 201306 Shanghai, China
6 Department of Computer Science, Guangdong AIB Polytechnic College, 510507 Guangzhou, Guangdong, China
7 Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 200031 Shanghai, China
8 CAS Key Laboratory of Tissue Microenvironment and Tumor, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 200031 Shanghai, China
†These authors contributed equally.
Abstract
Background: Different severities of coronavirus disease 2019 (COVID-19) cause different levels of respiratory symptoms and systemic inflammation. DNA methylation, a heritable epigenetic process, also shows differential changes in different severities of COVID-19. DNA methylation is involved in regulating the activity of various immune cells and influences immune pathways associated with viral infections. It may also be involved in regulating the expression of genes associated with the progression of COVID-19. Methods: In this study, a sophisticated machine-learning workflow was designed to analyze whole-blood DNA methylation data from COVID-19 patients with different severities versus healthy controls. We aimed to understand the role of DNA methylation in the development of COVID-19. The sample set contained 101 negative controls, 360 mildly infected individuals, and 113 severely infected individuals. Each sample involved 768,067 methylation sites. Three feature-ranking algorithms (least absolute shrinkage and selection operator (LASSO), light gradient-boosting machine (LightGBM), and Monte Carlo feature selection (MCFS)) were used to rank and filter out sites highly correlated with COVID-19. Based on the obtained ranking results, a high-performance classification model was constructed by combining the feature incremental approach with four classification algorithms (decision tree (DT), k-nearest neighbor (kNN), random forest (RF), and support vector machine (SVM)). Results: Some essential methylation sites and decision rules were obtained. Conclusions: The genes (IGSF6, CD38, and TLR2) of some essential methylation sites were confirmed to play important roles in the immune system.
Keywords
- COVID-19 severity
- DNA methylation
- machine learning
- rules
Coronavirus disease 2019 (COVID-19) is currently the most serious public health
problem and has caused millions of deaths worldwide. It can be classified into
mild, moderate, and severe infection categories according to the clinical
manifestations of the SARS-CoV-2 infection. These manifestations include
asymptomatic, fever or chills, cough, loss of taste and/or smell, muscle or body
aches, nausea or vomiting, and diarrhea. Studies have shown that severe COVID-19
is associated with host genetic variation related to host immune responses to
viral infection and inflammasome modulators. COVID-19 severity is closely
correlated with host factors [1]. Studies have revealed the interplay of genetic
and epigenetic alterations that control host responses. Epigenetic changes that
regulate chromatin structure have important implications for genome stability and
the maintenance of cellular homeostasis since they are related to the
pathophysiology of viral infections. DNA methylation is a heritable epigenetic
process, whereby methyl groups are added to the C-5 position of the DNA cytosine
loop by DNA methyltransferases. DNA methylation plays a key role in gene
imprinting, X inactivation, silencing of repeat elements, and transposon
expression. It also participates in cell development and aging [2]. DNA cytosine
methylation at the 5
An epigenome-wide association study of novel coronavirus infections revealed
important DNA methylation regulation processes associated with COVID-19
progression [12]. Specific differences in CpG methylation were found between
patients with severe and mild diseases. The differential methylation is primarily
related to the activation of the interferon signaling pathway and the
overactivation of B and T lymphocytes [13, 14, 15]. Transcriptomic studies have
confirmed that these pathways are associated with COVID-19 severity, while it was
shown that regulation of these pathways is mediated by epigenetic changes at the
promoter level of the relevant genes [16]. Moreover, epigenetic dysregulation
exists in the CD209 signaling pathway, phagocytosis pathway, and AKT
signaling pathway in COVID-19 patients with specific blood cell types.
CD209 is primarily expressed in B lymphocytes and dendritic cells (DCs),
and it interacts with CD209L in endothelial cells of SARS-CoV-2 target
tissues, which may contribute to virus invasion [17]. Therefore, hypermethylation
of the CD209 signaling pathway may be related to the protective effect
during SARS-CoV-2 infection. Interestingly, EDC3 hypermethylation in
severe cases may mediate the overexpression of the angiotensin-converting enzyme
(ACE) 2 protein in COVID-19 patients, thereby worsening the
infection [18]. The hypomethylation signatures associated with COVID-19 severity
include interferon-related signatures and lymphocyte activation signatures.
Interferon-related features are associated with systemic autoimmune disease in
mild and severe cases [19]. Further analysis revealed that the enrichment of
transcription-factor binding sites, which regulate the levels of cytokines (e.g.,
interleukin (IL)-6, IL-1
High-throughput sequencing data provides extensive molecular information relevant to patients with COVID-19. Our team has previously used machine learning to screen 49 key methylation sites associated with COVID-19. The degree of methylation at these sites was correlated to the age of the patient [25]. Accordingly, we aimed to further explore the COVID-19 mechanism based on whole-blood genome-wide DNA methylation profiling from 101 negative controls, 360 patients with mild infections, and 113 individuals with severe infections.
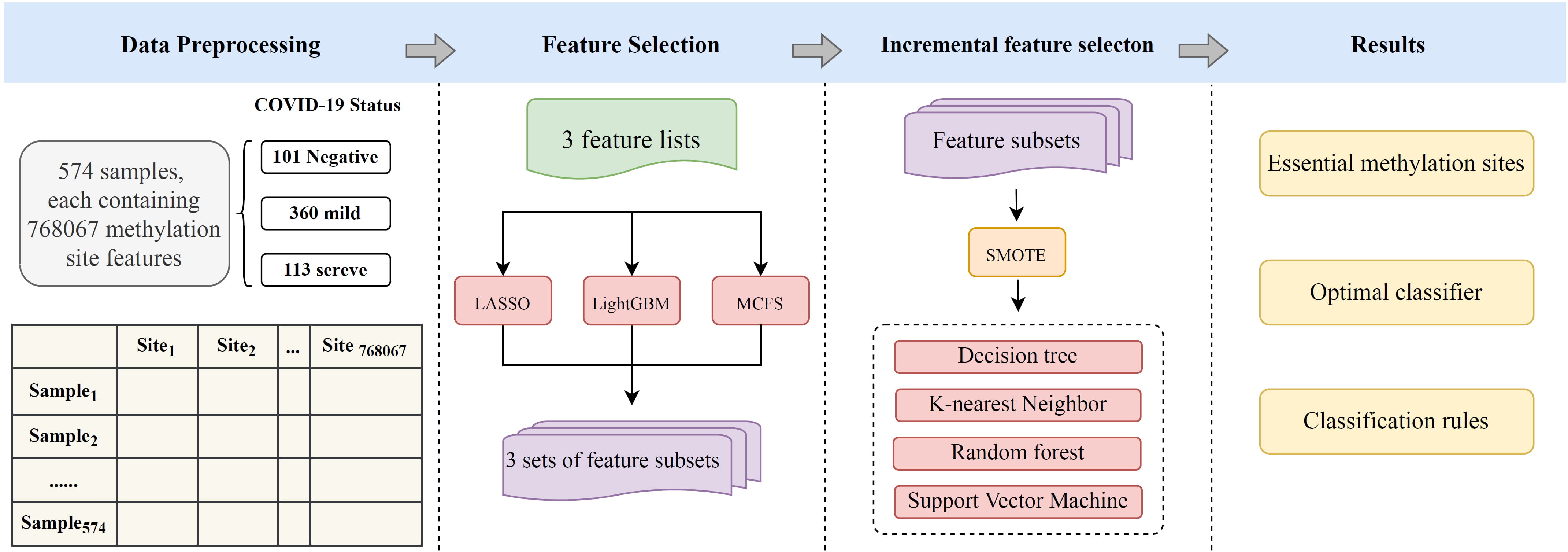
Fig. 1 illustrates the flow of the machine-learning method used in this study. The samples were grouped according to COVID-19 severity. The methylation sites were ranked using three feature-ranking methods. Then, the obtained feature-ranking list was fed into the incremental feature selection (IFS) framework, which contained four classification algorithms. In summary, we obtained key methylation sites that were strongly correlated with COVID-19 severity and a model that could predict the COVID-19 status of the samples. Quantitative classification rules were also summarized. This section describes the methods used in each segment.
 Fig. 1.
Fig. 1.Flow chart of the entire analytical process. Whole-blood DNA methylation data from patients with different COVID-19 severities were analyzed by machine-learning approach. The dataset contained 101 negative controls, 360 mildly infected individuals, and 113 severely infected individuals. The methylation-site features were analyzed by three feature-selection methods, namely, least absolute shrinkage and selection operator (LASSO), light gradient-boosting machine (LightGBM), and Monte Carlo feature selection (MCFS). The obtained feature lists were fed into the incremental feature-selection method, which combined decision tree (DT), k-nearest neighbor (kNN), random forest (RF), and support vector machine (SVM) to extract key site features and construct effective classifiers and classification rules.
The whole-blood DNA methylation data for the 574 used samples were obtained from the GEO database, with the accession number: 179325 [26]. The samples were divided into three groups according to the COVID-19 severity and included 101 negative controls, 360 mildly infected individuals, and 113 severely infected individuals. The DNA methylation sites in the samples were considered as features, and each sample contained 768,067 methylation sites.
The number of DNA methylation sites involved in the dataset was large, although only a very small number of sites were significantly methylated during COVID-19 development. All sites were screened and ranked using the least absolute shrinkage and selection operator (LASSO) [27], light gradient-boosting machine (LightGBM) [28], and Monte Carlo feature selection (MCFS) [29]. A higher site was ranked corresponding to its higher degree of association with the target variable. These methods are extensively accepted in the life sciences [30, 31, 32, 33].
LASSO is a regression analysis method that ranks features and performs preliminary feature screening. The method is internally designed with a penalty function that is bounded by an L1-type regular formula. The methylation-site features are assigned to independent variables, and the absolute values of the coefficients of the independent variables describe the degree of association with the target variable. By optimizing the function, the coefficients of some features become zero, indicating that they should be treated as irrelevant features. Afterward, the remaining features are ranked according to the absolute value of the coefficients. Finally, the dimensionality reduction and ranking of the data are completed.
LightGBM is based on gradient-boosting decision trees (DTs) for optimization, which consumes less memory and is suitable for high-dimensional datasets. It uses the gradient-based one-side sampling method to discard some samples with small gradients, and it bundles selected samples using an exclusive feature-bundling method to merge mutually exclusive features. It uses a leaf-wise strategy to construct the tree, extending only the more efficient branches. The importance of a feature is proportional to its involvement in the DT.
The MCFS method constructs a number of independent DTs. Each DT uses a different
subset of features and sample data sets. The features and samples used by each
tree are determined by independent random selection. For the selected sample
subsets, a division was performed
where
The use of IFS is well-established in machine learning research [33, 34]. When building a classification model, computational efficiency needs to be considered. For this purpose, we performed the IFS method to determine the optimum number of features required to build the classification model. It converted the list of methylation sites generated by the ranking method into a number of feature subsets. Depending on the set step size, the number of features contained in these subsets grew equally according to the ranking order. These subsets were used to train the downstream classification algorithm, where in reference to the performance of the obtained models, the finalized feature subset was the best feature subset, and the model at this point was the best classification model.
Looking at the sample dataset, a difference existed in the number of samples within the three classes, whereby there were 3.6 times more samples from the mildly infected individuals than from the negative controls. Moreover, directly training the model using an uneven dataset biased the results toward the majority classes. The SMOTE method generated new samples based on known samples. All samples were projected into the high-dimensional space, and one sample was randomly selected for the minority classes. Using Euclidean distance as a metric, SMOTE determined the k-nearest neighbors to that sample in the same class. Moreover, any point on the concatenation of this sample and any of its nearest neighbors can be selected as a new sample. The above process was repeated and the new samples were added into minority classes until the number of samples in each class was balanced.
Based on several feature subsets generated by IFS, this study used DT [35], kNN [36], RF [37], and SVM [38] algorithms to construct classification models. These algorithms have been approved by previous publications [34, 39, 40, 41, 42, 43, 44, 45].
As its name suggests, the DT algorithm constructs a tree structure that contains a root, branch, and leaf structure. The instances were inputted from the root, and the attributes of the instances were judged in the internal node. Based on the result, they were transported along the branches of the tree after several judgments until they reached the leaf structure. The leaves of the tree contained the final judgment of the algorithm on the class of the instance. As a white-box algorithm, the judgment process was transparent, thereby enabling the highly interpretable classification rules to be summarized.
The principle of the kNN algorithm was to determine the category of an unknown sample by comparing the distribution of known samples around a new sample in the feature space. The samples were mapped into a high-dimensional space based on the feature vectors; the k-nearest known samples for each new sample were selected, and the category labels for these nearest neighbors were referred to finally determine the category of the new sample.
RF was based on the DT algorithm. A number of independent DT models were constructed at once, and randomness was introduced into the selection of features and training samples used for judgment. Each DT constructed in this way had a different judgment process for the same sample, meaning the results often differed. For a sample, the classification results of each tree were combined, and the final classification results were obtained using the principle of majority rule.
The SVM algorithm utilized a kernel function that also mapped the samples into a high-dimensional space based on feature vectors. Through an optimization function, a hyperplane was determined in the space. This hyperplane partitioned the samples of different categories in space and ensured that the margin between each category of samples was maximized at this hyperplane. The model presented the best generalization ability at this time.
Using the IFS method and above four classification algorithms, we obtained several classification models and evaluated the performance of these models using 10-fold cross-validation [46]. The F1 measure is often used as a key metric to evaluate the performance of classification models, although it does not perform fairly enough in multiclassification problems. The current study required weighting the F1 measure.
where
Whole-blood DNA methylation data from 574 samples, each containing 768,067 methylation-site features, were analyzed using a machine-learning workflow. LASSO, LightGBM, and MCFS were used to rank all methylation sites and three ranked lists were obtained (Supplementary Table 1). LightGBM discarded the redundant features, meaning only 33,537 features were output in LightGBM for ranking. The lists involved numerous site features, although only a very small number were associated with COVID-19 levels. Thus, for subsequent analyses, only the top 10,000 site features in the list were used.
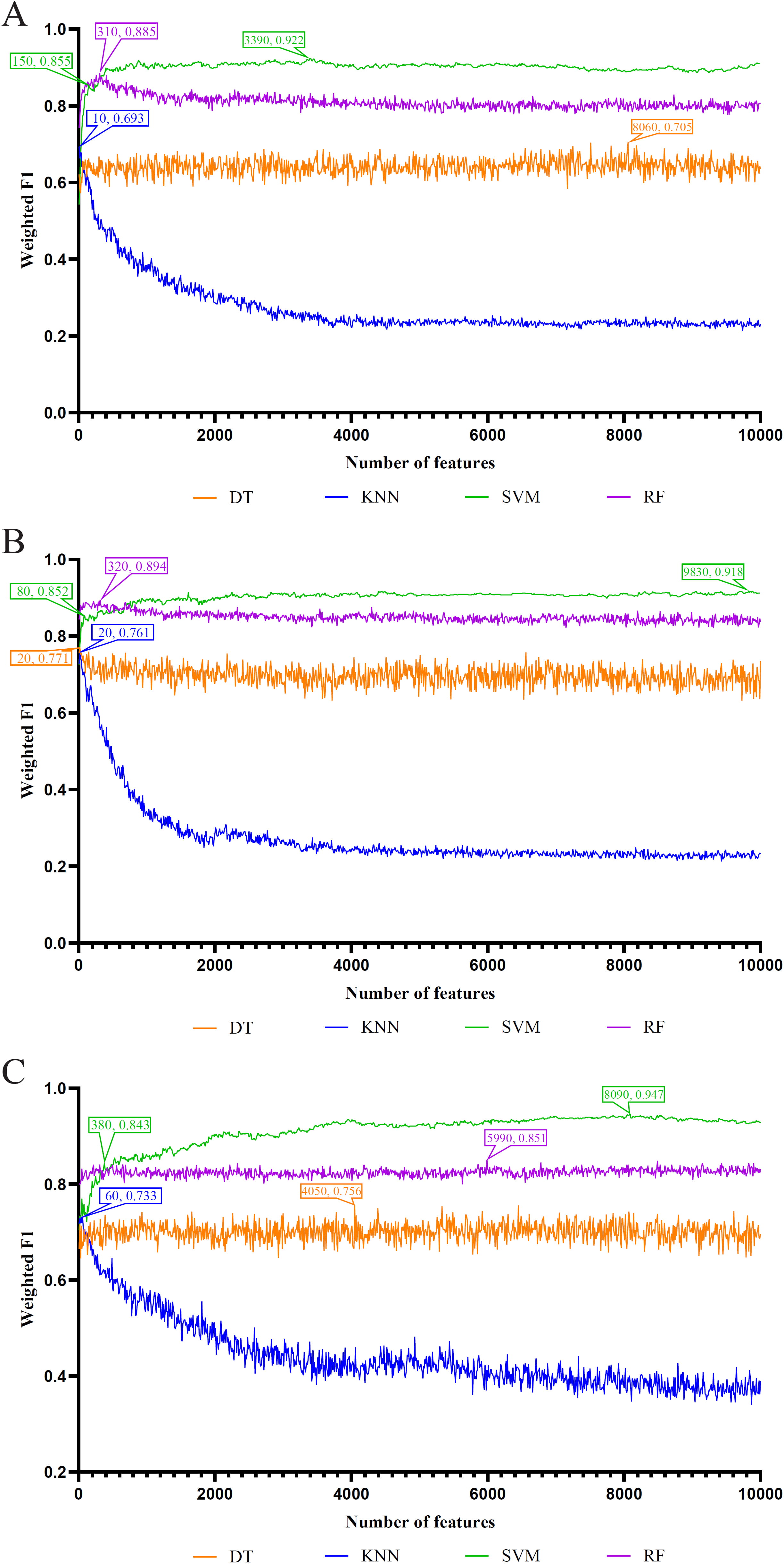
The first 10,000 methylation sites in the three lists were input into the IFS framework, and the step parameter was set to 10. Each list was transformed into a subset of 1000 sites, and these subsets of methylated sites were used to construct 4 classification models, namely, DT, kNN, RF, and SVM. Supplementary Table 2 describes the classification performance of these models. Based on the performance results, IFS curves were plotted using the number of features as the independent variable to depict the weighted F1 trend (Fig. 2). The IFS curve observation revealed that the SVM model always had the highest performance regardless of the list being used. The best SVM model used the top 3390, 9830, and 8090 methylation sites from the Lasso, LightGBM, and MCFS lists, respectively, to provide weighted F1 values of 0.922, 0.918, and 0.947, respectively. Their ACC and MCC results were also excellent, with ACC of 0.922, 0.916, and 0.946, respectively, and MCC of 0.862, 0.857, and 0.904, respectively (Table 1).
 Fig. 2.
Fig. 2.IFS curves for evaluating the performance of the three classification algorithms based on the weighted F1. Four classifier models were constructed under each algorithm. (A) IFS curves based on LASSO results. (B) IFS curves based on LightGBM results. (C) IFS curves based on MCFS results. IFS, incremental feature selection.
| Feature ranking algorithms | Number of features | Weighted F1 | MCC | ACC |
| LASSO | 150* | 0.855 | 0.761 | 0.852 |
| 3390** | 0.922 | 0.862 | 0.922 | |
| LightGBM | 80* | 0.852 | 0.753 | 0.848 |
| 9830** | 0.918 | 0.857 | 0.916 | |
| MCFS | 380* | 0.843 | 0.755 | 0.840 |
| 8090** | 0.947 | 0.904 | 0.946 |
* indicates the number of features at the inflection points of the IFS curve; ** indicates the number of features in the best feature subset. MCC, Matthew’s correlation coefficient; ACC, accuracy.
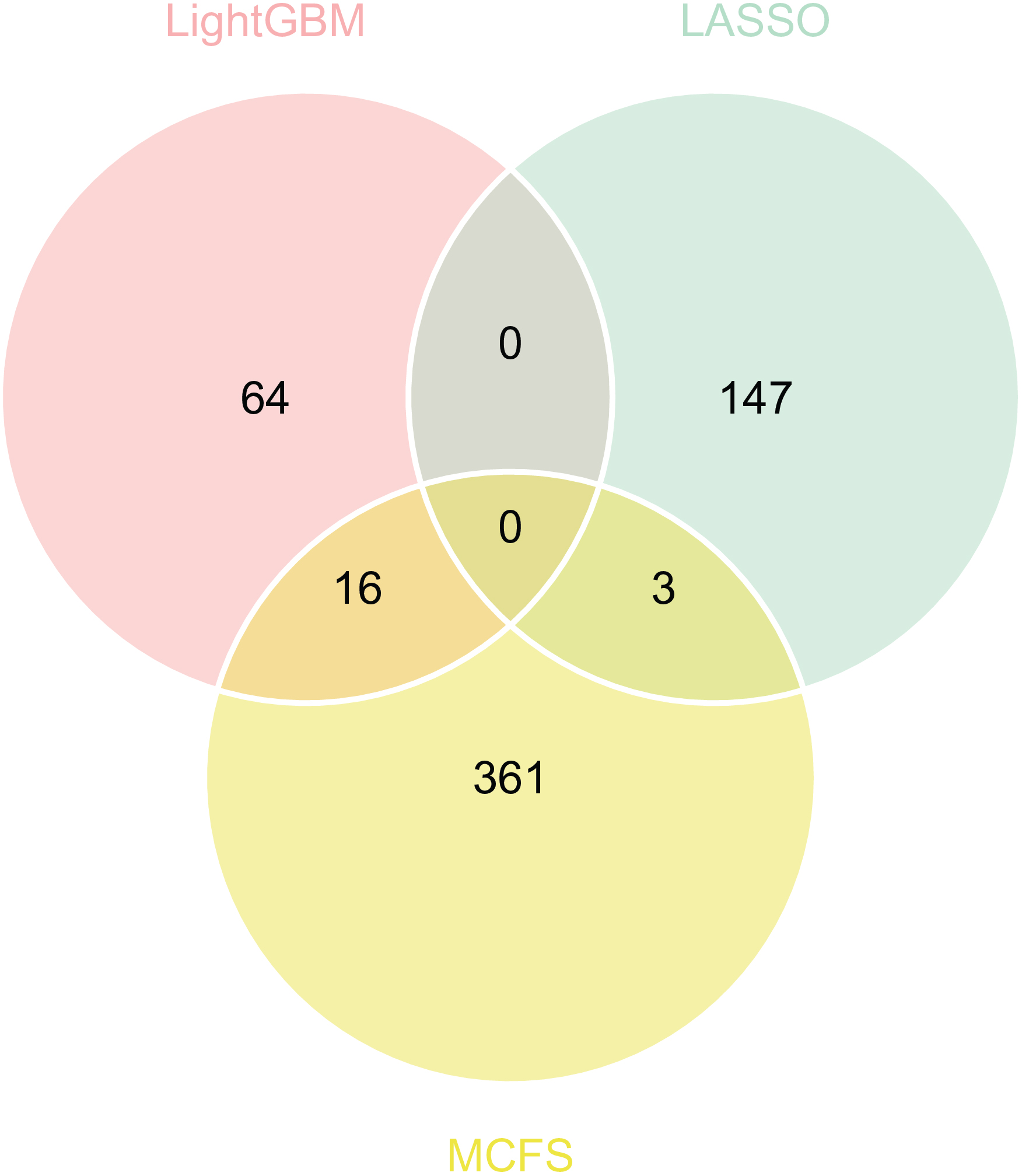
To extract the most critical sites among the methylation sites used by the best classifier, we observed the IFS curves of the SVM models and set the inflection points of the curves. The inflection points were 150, 80, and 380 for the LASSO, LightGBM, and MCFS lists, respectively. The performance evaluation of the model at the inflection points is displayed in Table 1. Recurring methylation sites tended to play a more important role. We calculated the intersection from the pre-inflection point sites and plotted a Wayne diagram (Fig. 3). We did not obtain methylation sites that appeared in all three lists but obtained 19 features that appeared in two lists. The specific intersection results are shown in Supplementary Table 3. A detailed analysis of these sites is presented later.
 Fig. 3.
Fig. 3.Venn diagram of the most critical subset of features obtained using LASSO, LightGBM, and MCFS. The overlapping circles indicate methylation sites that are identified as the most critical features by the different ranking algorithms.
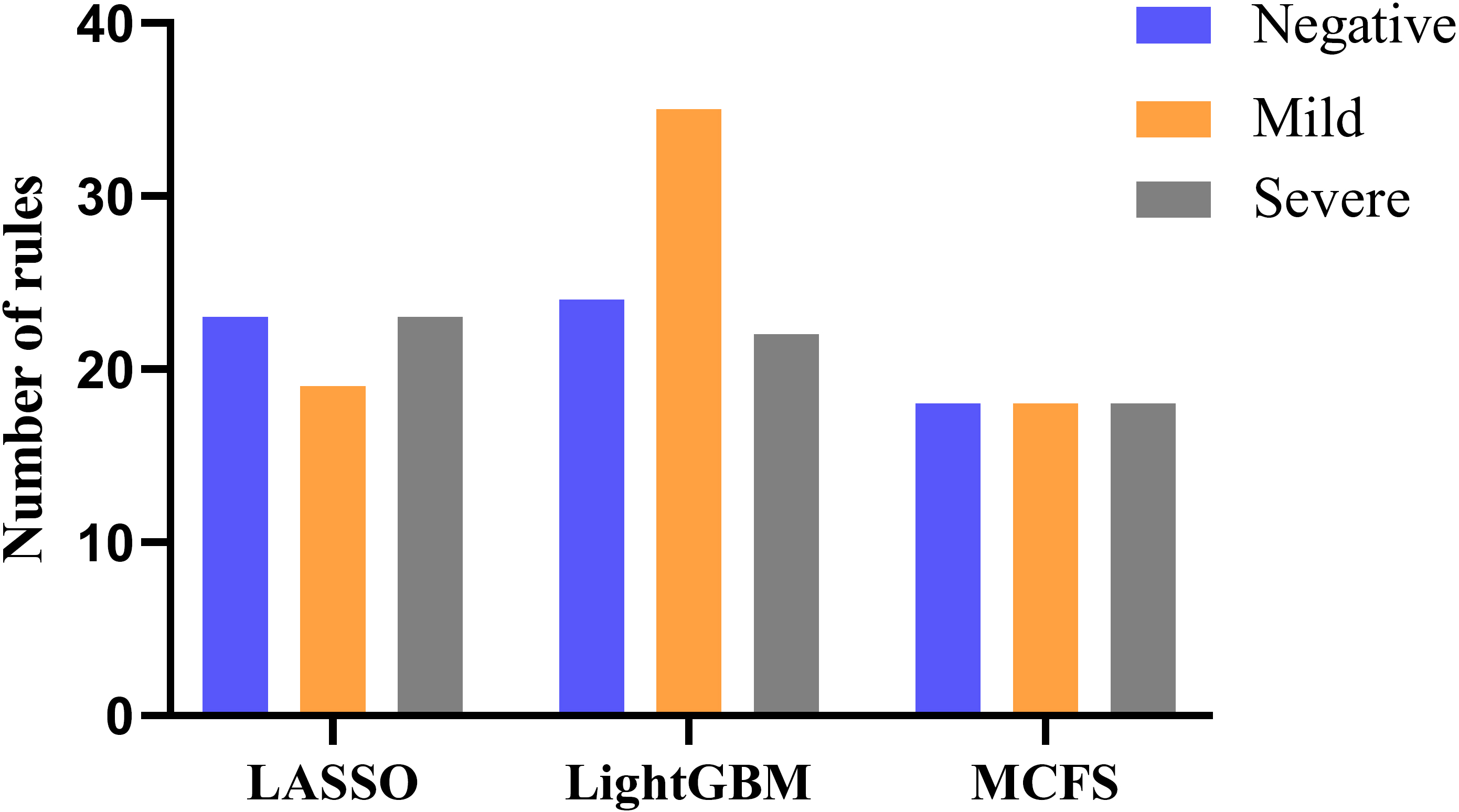
The DT model demonstrated a transparent decision process, which helped us obtain specific classification cues. Based on these cues, we summarized three sets of quantitative classification rules, which had implications for our understanding of the biological context of these key methylation sites. Supplementary Table 4 shows the specific classification rules. Each rule contained several parameters that represented the methylation levels of these sites. For the different lists, Fig. 4 shows the number of rules representing the different categories.
 Fig. 4.
Fig. 4.The number of rules for the three classes as summarized by the DT classification model, constructed using the three ranking methods.
This study had three groups: negative controls, mildly infected individuals, and severely infected individuals. Each algorithm presents its advantages, and taking the intersection of the results of multiple algorithms can increase the coverage and accuracy of the results better than one approach. Through the intersection of all analysis approaches, we identified some important methylation features. Combined with previous studies, summarizing the experimental evidence of the above-mentioned methylation features was necessary.
As shown in Fig. 3, using 2 algorithms identified 19 methylation sites, which were generally more essential than other methylation sites. Here, we selected four for detailed analysis and these are listed in Table 2 (Ref. [48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67]).
| Methylation locus | Gene symbol | Description | Algorithm | Reference |
| cg02481950 | IGSF6 | chr16:21650858-21663981 | LASSO, MCFS | [48, 49, 50, 51, 52, 53] |
| cg04332373 | CD38 | chr4:15778275-15853232, CpG island | LightGBM, MCFS | [54, 55, 56, 57, 58, 59, 60, 61] |
| cg03753191 | EPSTI1 | chr13:43566902, Shore region of CpG islands | LightGBM, MCFS | [62] |
| cg22930808 | DTX3L | chr4:15778275-15853232, CpG island | LightGBM, MCFS | [63, 64, 65, 66, 67] |
IGSF6 (cg02481950) is located at chr16:21650858-21663981 and belongs to
the immunoglobulin superfamily member 6, which is expressed in dendritic and
myeloid cells [48]. Studies have shown that IGSF6 is associated with
changes in the ratio of M0 macrophages and
CD38 (cg04332373) encodes a type II transmembrane glycoprotein that
synthesizes and hydrolyzes calcium-ion-mobilized messengers. The cg04332373
methylation site is located in the shore region of the CpG island. Studies have
shown that CD38 levels in white adipose tissue (WAT) and the liver increase with
age. Senescent cell signals promote the accumulation of CD38
cg03753191 is located at chr13:43566902 and in the Shore region of CpG islands. It corresponds to the EPSTI1 gene and encodes a protein that promotes tumor invasion and metastasis. Studies have reported that EPSTI1 expression levels were significantly upregulated in leukocytes and nasopharyngeal tissues of COVID-19 patients, compared to normal tissues. The downregulation of EPSTI1 also predicted poorer clinical outcomes in patients with COVID-19, such as intensive care unit hospitalization and increased viral load. Therefore, EPSTI1 may play an important role in antiviral immune regulation [62]. The expression profile of EPSTI1 can classify COVID-19 patients into different groups, whereby the younger patient population exhibited a stronger antiviral immune response, higher EPSTI1 expression level, and better clinical treatment effect. The expression level or methylation pattern of EPSTI1 may significantly affect the COVID-19 severity by modulating antiviral immune responses.
DTX3L (cg22930808) encodes Deltex E3 ubiquitin ligase 3L, which plays a role in DNA damage repair and interferon-mediated antiviral response [63, 64, 65, 66]. DTX3L plays a role in the antiviral response by mediating the “LS-48”-linked ubiquitination in the C3 protease of the cerebral myocarditis virus and human rhinovirus, thereby promoting their proteome-mediated degradation [65]. Dtx3l-related pathways include DNA damage and the innate immune system. The activation of the IFN response has been demonstrated to induce the adp nucleation of host proteins, which depend on PARP9 and its binding partner DTX3L. Expression of the large domain of SARS-CoV-2 nonstructural protein 3 (Nsp3) or deletion of PARP9 or DTX3L had no effect on IFN signaling. PARP9/DTX3L-dependent adp nucleation is a downstream effector of the host IFN response, and the SARS-CoV-2 Nsp3 macrodomain can hydrolyze the IFN signaling end product [67]. This study has added evidence to the relationship between DTX3L methylation patterns and disease severity in patients with SARS-CoV-2 infection, thereby providing more reference for the study of COVID-19 progression and pathogenesis.
In addition to identifying essential methylation sites by using various algorithms, we also obtained several classification rules, as listed in Supplementary Table 4. These rules can predict disease severity based on the methylation level of key DNA methylation sites. However, it is impossible to analyze them in detail because a huge number of rules were constructed. Herein, we focused on valuable features because they can identify important DNA methylation sites and demonstrate their important role as epigenetic-susceptibility sites in patients infected with SARS-CoV-2. We collected the scientific findings of other researchers and preliminarily summarized experimental evidence to conduct the discussion. The key methylation sites are listed in Table 3 (Ref. [68, 69, 70, 71]).
| Methylation locus | Gene symbol | Description | References |
| cg00197681 | TBC1D4 | chr13:76056419, CpG island | [68] |
| cg09623286 | TLR2 | chr4:154605468, CpG island | [69, 70, 71] |
TBC1D4 (cg00197681) belongs to TBC1 domain family member 4, and cg00197681 is located at chr13:76056419 and CpG island region. TBC1D4 encodes a rabbit-GTPase activating protein, which is involved in regulating glucose homeostasis through transporting glucose transporter 4 (GLUT4). Human genetic variants may influence SARS-CoV-2 infection and COVID-19 pathology. Researchers have applied machine-learning algorithms to predict phosphorylation-associated missense single nucleotide variants (pSNVs) and found that these variants are associated with the evolution of host defense systems [68]. Proteins with pSNVs include TBC1D4 and TRIM28, which are involved in the regulation of the viral life cycle and host antiviral response. TBC1D4 may also be associated with the dysregulation of glucose homeostasis in SARS-CoV-2 infection. In the present study, we found that TBC1D4 methylation levels were higher in patients with severe COVID-19 than in the normal population. Combined with previous studies, we hypothesized that SARS-CoV-2 infection induced an increase in the methylation levels of TBC1D4, thereby affecting the host antiviral immunity.
The methylation site cg09623286 is located at chr4:154605468 and the island region of CpG islands. Its related gene is Toll-like receptor (TLR) 2, which encodes products belonging to the TLR family. As a cell surface protein, TLR2 can form dimers with other TLRs to recognize pathogen-associated molecular patterns and activate host immune responses. Activation of different TLR pathways leads to the secretion of proinflammatory factors, including IL, tumor necrosis factor-alpha, and interferon, and is involved in SARS-CoV-2 invasion and infection. However, some TLRs, such as TLR2, may play a dual role in COVID-19 infection [69]. SARS-CoV-2 infection can cause severe disease features (e.g., cytokine storm) and organ failure (e.g., testicular and germ-cell damage). Studies have shown that exposure to SARS-CoV-2 envelope proteins causes TLR2 receptor-dependent testicular cytopaplasia [70]. TLR2 mRNA expression levels also significantly increase in patients with moderate to severe COVID-19. Moreover, the mRNA expression levels of CK-MB, ACE2, and neuroproteinase 1 receptors were positively correlated with TLR2 expression in all patients. The mRNA expression of TLR2 was positively correlated with renal biomarkers and cardiac enzymes in severe and moderate patients. These results suggested that it may be related to COVID-19 severity [71]. The present study showed that patients with moderate COVID-19 had higher methylation levels than normal controls; however, this was not observed in severe patients. Therefore, the TLR2 methylation level may serve as an indicator of COVID-19 severity, although the specific mechanism needs further verification.
This study aimed to determine the characteristics and patterns of methylation sites associated with COVID-19 severity through computational analysis. The results showed that the identified key features were validated in published academic studies. Important features may be involved in viral immune regulation through different methylation patterns or expression levels. Additionally, antiviral immunity was associated with age, which is consistent with our previous findings. The accuracy and credibility of the methylation characteristics of important groups were improved by mutual verification of various methods. On one hand, it laid the foundation for more accurate analysis methods in the future, while, alternatively, it provided a broad reference for research on the mechanism of serious diseases, such as COVID-19.
COVID-19, Coronavirus disease 2019; IFS, incremental feature selection; LASSO, least absolute shrinkage and selection operator; LightGBM, light gradient-boosting machine; MCFS, Monte Carlo feature selection; SMOTE, Synthetic Minority Oversampling Technique; DT, decision tree; kNN, k-nearest neighbor; RF, random forest; SVM, support vector machine; MCC, Matthew correlation coefficient.
The datasets analyzed during the current study are available in the [Gene Expression Omnibus] repository [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179325].
TH and YDC designed the research study. JXR, LC and KYF performed the research. FY, HPL, WG and FY analyzed the data. FY, JXR and HPL wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was funded by the National Key R&D Program of China [2022YFF1203202], Strate-gic Priority Research Program of Chinese Academy of Sciences [XDA26040304, XDB38050200], the Fund of the Key Laboratory of Tissue Microenvironment and Tumor of Chinese Academy of Sciences [202002], Shandong Provincial Natural Science Foundation [ZR2022MC072].
The authors declare no conflict of interest. YDC is serving as Editorial Board member of this journal and TH has served as the Guest Editor of the journal. We declare that YDC and TH had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Rosa Alduina.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.