1. Introduction: Brain Protein Aggregates - A Traditional View
Protein aggregation is a common feature of many neurodegenerative diseases
(NDs), including Parkinson’s disease (PD) and Alzheimer’s disease (AD). According
to traditional view, each ND is characterized by aggregation of one or two
specific proteins, which are therefore considered disease-specific biomarkers and
their detection and characteristic distribution pattern in the brain allow for a
definitive disease diagnosis. Prior to aggregating, these proteins undergo
conformational rearrangements which give them a propensity to clump and form
toxic species that can impair brain functions. For instance, aggregates of
aberrant -synuclein (-syn) are found in a group of
pathologies referred to as -synucleinopathies, which include, in
addition to PD, also dementia with Lewy bodies (DLB) and multiple system atrophy
(MSA). The classification of these conditions depends on the clinical
presentation and the spatiotemporal accumulation of pathological -syn
[1]. In PD, aggregates of -syn are found within neurons of the
substantia nigra and the basal forebrain but also in the cortex, olfactory bulb
and limbic system [2, 3]. These species trigger the activation of glial cells and
the release of pro-inflammatory cytokines which can result in cell death [4]. In
DLB, -syn often deposits within neurons of the neocortex, limbic
system, brainstem and amygdala [5] while in MSA -syn preferentially
aggregates in oligodendrocytes, forming the typical glial cytoplasmic inclusions
(GCIs) [6]. The involvement of -syn in multiple disorders (PD, DLB and
MSA) has allowed for the demonstration that this protein might exists as
“strains”, which are characterized by different aberrant structures and toxic
properties, similarly to what has been widely described for prion diseases
[7, 8, 9, 10, 11, 12, 13]. Different strains of -syn are also believed to be responsible
for the phenotypic heterogeneity of the same pathology, as in the case of PD
[14, 15] and MSA (that can present with parkinsonism (MSA-P) or cerebellar ataxia
(MSA-C)) [16, 17], although further studies are needed. Prion diseases are caused
by the misfolding of the cellular prion protein (PrP) into a toxic species
able to self-replicate named prion (PrP) which accumulates in the brain.
PrP can adopt variable aberrant and toxic conformations that give rise to
different diseases or disease-phenotypes, including the sporadic
Creutzfeldt-Jakob disease (sCJD) and the recently discovered variably
protease-sensitive prionopathy (VPSPr) [13, 18, 19].
Aggregates of amyloid-beta (A) and tau are the main hallmarks of AD:
A forms extracellular plaques whose deposition follows a spatiotemporal
pathway beginning in the neocortex and then spreading to the allocortex, basal
ganglia, midbrain, pons and cerebellum [20]; while tau forms intraneuronal
neurofibrillary tangles (NFTs) composed of hyperphosphorylated protein which
spread from the transentorhinal region to the entorhinal region, hippocampus,
temporal neocortex and superior temporal neocortex [20, 21, 22]. A
deposition precedes tau aggregation by several years or decades and preclinical
studies have shown that A is able to trigger the formation of NFTs,
which more closely correlate with cognitive decline [23, 24, 25]. Interestingly,
recent studies have suggested that the phenotypic heterogeneity of AD may be
linked to the existence of various A strains capable to affect distinct
brain regions, as observed in the case of -synucleinopathies [26, 27, 28, 29, 30].
Studies with F-florbetapir positron emission tomography (PET) imaging
performed on healthy subjects showed A deposition in the cortex which
increased with age and these findings were in accord with postmortem analyses,
which showed an age-dependent increase of A plaques in the brain of
healthy individuals [31, 32, 33].
Aggregates of tau, TDP-43 or FUS (FUsed in Sarcoma protein) characterize a group
of diseases known as frontotemporal lobar degeneration (FTLD) that can be
classified in tauopathies, TDP-43 proteinopathies or FUS proteinopathies,
respectively. Tauopathies (40% of FTLD cases) can be further divided in
primary tauopathies which include corticobasal degeneration (CBD),
progressive supranuclear palsy (PSP), primary age-related tauopathy (PART),
argyrophilic grain disease (AGD), tangle only dementia (TOD) and Pick’s disease
(PiD) [34]; or secondary tauopathies, where tau is not the unique
pathological feature, and include AD and chronic traumatic encephalopathy (CTE)
[35]. Primary and secondary tauopathies are characterized by the accumulation of
abnormal tau protein in the form of neuropil threads, neurofibrillary tangles
(NFTs) or tau-positive astrocytes [36, 37, 38, 39, 40, 41, 42]. The tau protein is usually associated
to microtubules in neuronal cells and regulates their maintenance and shape as
well as the axonal transport. Tau can be found also in astrocytes and
oligodendrocytes. Once this protein undergoes aberrant conformational
rearrangements, it aggregates most of the time under hyper-phosphorylated forms.
Interestingly, the biochemical composition of tau which aggregates differ between
diseases. For instance, PiD is characterized by the presence of tau isoform
containing three microtubule-binding repeats (3R); while CBD, PSP and AGD show
the presence of tau with four microtubule-binding repeats (4R). In the case of
AD, the aggregates of tau are composed by both 3R and 4R isoforms [43, 44, 45].
As previously mentioned for -syn and A, the pathogenesis of
tauopathies may involve the formation of distinct tau strains which can create
unique patterns of neuropathology for each disease [46]. TDP-43 proteinopathies
(45% of FTLD cases), include FTLD-TDP as well as amyotrophic lateral sclerosis
(ALS) and limbic-predominant age-related TDP-43 encephalopathy (LATE) which are
instead characterized by the presence of neuronal intranuclear and cytoplasmic
inclusions of TDP-43, which is often hyper-phosphorylated, N-terminally truncated
and ubiquitinated [47, 48, 49, 50, 51, 52, 53, 54]. Interestingly, mutations in progranulin protein
(PGRN) cause FTD with TDP-43 pathology [55]. Aggregates of toxic C-terminal
TDP-43 species are found in the cytoplasm of neurons (in brain and spinal cord)
of more than 97% of amyotrophic lateral sclerosis (ALS) cases and seems to
impair neuronal proteostasis while promoting progressive loss of motor function
[56, 57, 58, 59, 60]. The 90% of ALS are sporadic while approximately the 10% are caused by
mutations in superoxide dismutase 1 (SOD1), C9orf72, fused in sarcoma (FUS)
genes, or TAR DNA binding protein (TARDBP) [61]. In particular, there are several
mutations affecting the TARDBP protein in different regions: N-terminal domain
(e.g., D89E), RNA recognition motifs (e.g., K145Q, D219E) and C-terminal region
(e.g., Q331K, M337V, Q343R, N345K, R361S, and N390D) [62].
Finally, FUS proteinopathies are the least common subtype of FTLD (5% of FTLD
cases) and are characterized by the accumulation of FUS in neuronal cells
[61, 63, 64]. The exact mechanisms through which A, tau, -syn,
TDP-43 undergo misfolding and acquire disease-specific structures are now being
elucidated. In addition to genetic proclivity to accumulate misfolded proteins,
exposure to environmental toxins is now known to result in amino acid
misincorporation and subsequent protein misfolding [65, 66, 67]. Certainly, these
proteins are key players for the onset and progression of NDs and their detection
as aggregated species in the brain is crucial for an accurate disease diagnosis.
Through a prion-like mechanism, A, tau, -syn, TDP-43 spread
from cell to cell in the brain thus allowing the propagation of pathology [68].
However, numerous studies have shown that some individuals show co-occurrence of
more than one ND. In particular, aggregates composed of proteins which are not
primarly involved in the pathological process of each ND can be found in
postmortem brains (Fig. 1). These are considered secondary protein aggregates and
may influence the clinical and neuropathological features of the NDs, although
their role remain unknown. This work provides an up-to-date review of the
literature on the presence of secondary aggregates in NDs.
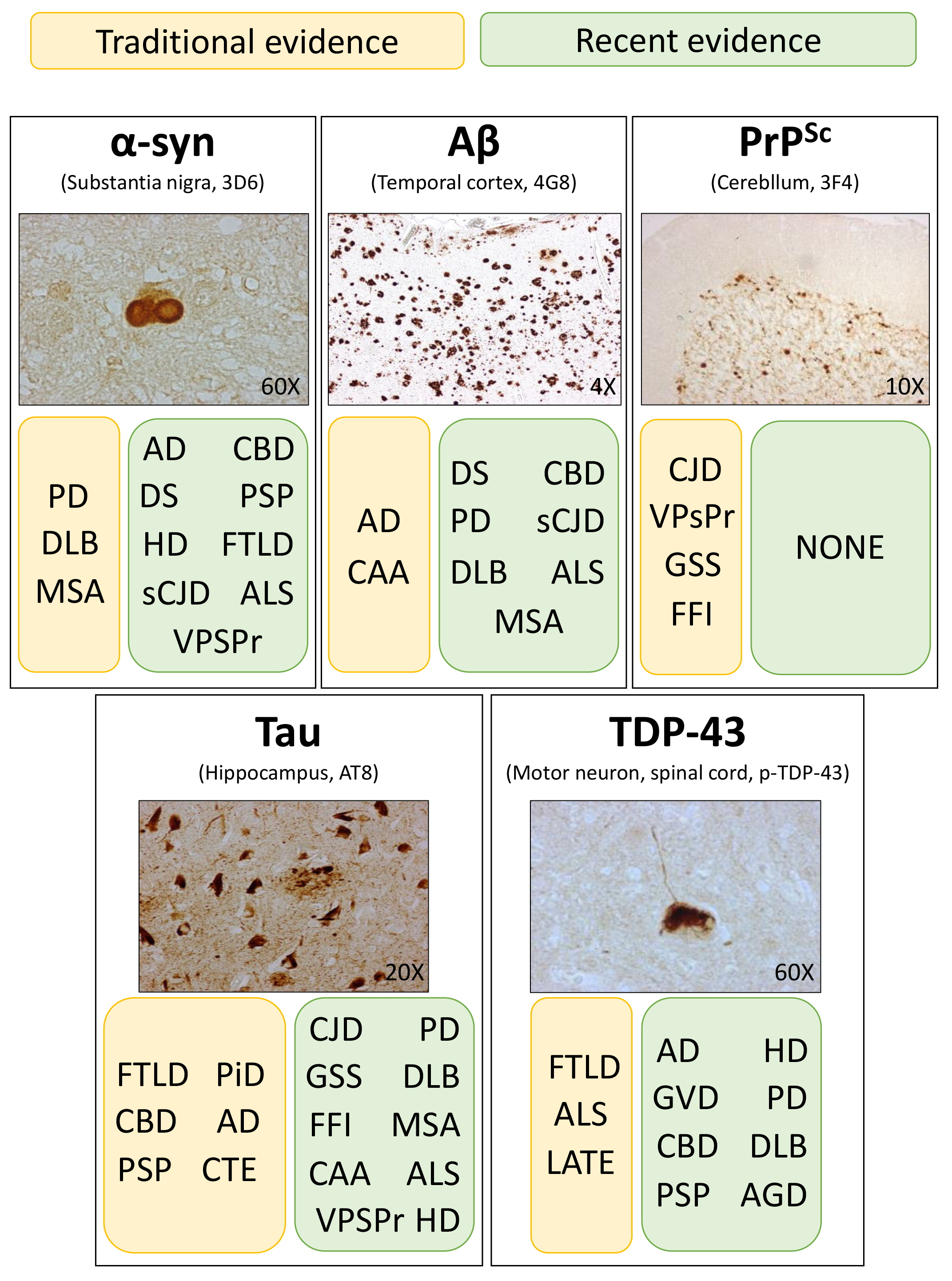 Fig. 1.
Fig. 1.
Schematic representation of the traditional (yellow boxes) and
recent (green boxes) evidence regarding protein aggregates in the central nervous
system of patients with various neurodegenerative diseases. Histological stains
show the typical morphological features of -synuclein (-syn),
amyloid- (A), prion (PrP), tau and TDP-43 aggregates
found in the brain of patients with PD, AD, CJD, AD and ALS, respectively. PD,
Parkinson’s disease; AD, Alzheimer’s disease; CJD, Creutzfeldt-Jakob disease;
ALS, amyotrophic lateral sclerosis.
2. Alpha-Synuclein Aggregates in Non--Synucleinopathies
Aggregates of misfolded -syn are commonly found in the brains of
patients with -synucleinopathies, such as PD, DLB and MSA [69].
However, several works confirmed their presence, as secondary aggregates, in the
brains of patients with other neurodegenerative diseases, including AD, Down
syndrome (DS), Huntington’s disease (HD), prion diseases, primary tauopathies
(CBD and PSP), ALS, FTLD, parkinsonism-dementia complex of Guam [70, 71, 72] and
neurodegeneration with brain iron accumulation type 1 [73, 74]. The frequency of
-syn in most of these NDs is high and is considered the rule rather
than the exception. Regarding AD, -syn deposits were found in almost
50% of postmortem brains [75, 76, 77]. These aggregates were distributed in the
neocortex, limbic system (being the amydgala highly vulnerable to -syn
pathology [78]) and substantia nigra of patients with sporadic and familial
(PSEN1, PSEN2 or APP gene mutations) forms of the
disease [75, 76, 79, 80, 81, 82, 83, 84]; often colocalizing with tau [75, 77, 80, 82] and less
frequently with A aggregates [81, 83, 85]. Therefore, -syn
pathology often accompanies AD pathology and, most importantly, it occurs in the
brain of patients with specific clinical symptoms, including hallucinations,
unexplained falls and extrapyramidal signs, constituting the nosologic entity of
dementia with Lewy bodies (DLB) [5, 21, 75, 76, 77, 79, 80, 82, 86, 87, 88, 89, 90]. Remarkably, the
presence or absence of -syn aggregates in AD may further contribute to
the phenotypic heterogeneity of the disease, making its clinical diagnosis even
more challenging. In the case of DS, -syn aggregates were identified
mainly in the amygdala of more than 50% patients with behavioral decline and
concomitant AD pathology and were absent in DS patients who lacked AD pathology
or with preserved cognitive functions [91, 92, 93]. Huntington’s disease (HD) is an
autosomal dominant inherited neurodegenerative condition characterized by the
presence of an abnormal form of huntingtin (mHTT) protein which forms toxic
intraneuronal inclusions, leading to choreatic movements, behavioral disturbances
and dementia [94]. Aggregates of -syn colocalized with mHTT inclusion
of HD patients, although both proteins formed independent homomeric filaments.
Thus, mHTT and -syn and can cross-talk, with the latter being recruited
as a mediator of toxicity in HD. In particular, -syn may promote
oxidative stress, mitochondrial disfunctions and brain inflammation [95, 96, 97, 98, 99, 100, 101],
which are all implicated in disease pathogenesis [95, 96, 102, 103, 104]. In the case of
prion diseases, -syn aggregates were found with low prevalence
(9–23%) in the brains of patients with sCJD and VPSPr [105]. The aggregates
were observed in different brain areas, including the substantia nigra, locus
coeruleus, dorsal motor nucleus of the vagus and cortical neurons [106],
brainstem and amygdala [107] or even in glial cells of the striato-nigral-system
[108]. They often co-deposited with other amyloidogenic proteins, including
prion, A and tau [105, 109, 110, 111, 112, 113]. Notably, the presence of copathology
was associated with a prolonged survival in patients with CJD [114]. Recent
studies, performed under extreme and artificial conditions, showed that
aggregates of -syn can even cross-seed the aggregation of the prion
protein, leading to the onset of prion diseases [115]. In the case of
tauopathies, few studies reported the presence of -syn aggregates in
(1) CBD brains (sometimes in patients who had longer survival) [116, 117, 118, 119], (2)
PSP brains (10–12% and mainly distributed in substantia nigra, basal
forebrain, amygdala and frontal cortex) [120, 121, 122, 123, 124] and (3) a subgroup of sporadic
or genetic FTD brains, especially in cases with concomitant motor neuron disease
(FTD/ALS) [125, 126, 127]. -syn aggregates were found to colocalize with
SOD1 in familial ALS and it was shown that -syn could accelerate SOD1
aggregation [128]. Moreover, -syn aggregates deposited in astrocytes
and Schwann cells in the spinal cords and bulbar regions, with a morphology
resembling that of Lewy bodies found in PD [129]. The presence of -syn
aggregates in ALS supports their contribution to motor neuron disease pathology,
as also observed in animal models. This also rules out PD as incidental pathology
[129, 130, 131, 132, 133, 134, 135, 136, 137]. Remarkably, -syn aggregates were described in 11–15% of
ALS cases with TDP-43 pathology [138, 139] and several works have shown that
-syn can directly interact with tau [140, 141, 142, 143] and TDP-43 [144, 145]. In
particular, emerging evidence from preclinical models showed that -syn
and TDP-43 can form hybrid fibrils whose toxic properties are more severe
compared to those of homotypic fibrils of -syn or TDP-43 [145].
Finally, aggregates of -syn were detected in the brain of cognitively
healthy elderly subjects (9–10%) [120, 146] and these findings have
allowed for the formulation of some hypotheses about their meaning: (1) they are
inert species, (2) they represent common findings in normal ageing or (3) they
represent an asymptomatic pathology in subjects at early disease stage
[146, 147, 148]. The presence of -syn aggregates in the brains of
non--synucleinopathies has the potential to provide new insights into
the complex neuropathology underlying NDs.
3. Amyloid-Beta Aggregates in Non AD Conditions
A primarily occur in the brain of AD patients, but they can also occur
in some non-AD conditions. For instance, A deposition is present in the
brains of nearly 30% of cognitively normal older individuals, as well as in
patients with mild cognitive impairment (MCI) and dementia. In particular,
neuropathological studies involving a small cohort of elderly non-demented
subjects showed the presence of numerous A deposits in the temporal
cortex and large and round deposits in most cortical areas, similar to what is
observed in AD brains [149, 150]. Even more interestingly, the neuropathological
analysis of the brain of centenarians, revealed that the highest stages of
A pathology were found in the brains of high performing subjects
[151, 152]. Therefore, A deposition may be a common feature of brain
aging and may contribute to predict the cognitive decline in some non-AD
individuals [153]. The most conservative interpretation is, therefore, that these
represent early stages of AD pathology not (yet) inducing cognitive impairment.
A deposits were also found in the brain of individuals with DS who have
an increased expression of the amyloid precursor protein (APP) gene located on
chromosome 21. Trisomy 21 leads to a dose-dependent increase in APP and
A production [154, 155]. As a result, individuals with DS develop
A deposits in their brains at young age, and these deposits invariably
lead to the development of AD later in life. In fact, the majority of DS
individuals between the ages of 50 and 60 have an increased risk of developing
dementia due to AD [156, 157]. PET data analyses suggested that, similar to AD,
A deposits first occur in the striatum and gradually affect various
cortical regions (affecting the rostral prefrontal and cingulate-parietal
cortices) and finally the parahippocampal cortex, thalamus, and amygdala
[158, 159]. Aggregates of A were found in the brains of some patients
with DLB, suggesting that A could contribute to the cognitive decline
and alterations in attentive/executive and language functioning [160, 161].
However, there is often an overlap between AD and DLB pathologies, thus other
proteins (e.g., tau) may contribute to influence disease progression and dementia
onset. Recent findings have suggested that there may also be some overlap between
AD and PD. Specifically, some research has found evidence of A plaques
in the brains of individuals with Parkinson’s disease, particularly in those who
have cognitive impairment or dementia (PDD) in addition to their motor symptoms.
In 2008, Kalaitzakis et al. [162], in a cohort of 30 patients (16 PDD,
14 PD), found that A deposition was significantly more prevalent in the
striatum (which includes the caudate nucleus, putamen, and nucleus accumbens) in
PDD cases than in PD cases. One year later, the same research group described
that A deposition in the claustrum was associated with the occurrence of
dementia in PD and DLB patients. The physiological role of the claustrum is
mainly unknown, but it has been linked to the presence of visual hallucinations
in DLB and cognitive impairment in AD [163]. Current evidence as post-mortem,
biochemical, and imaging analyses, increasingly indicate the involvement of
A in PD, and recent studies indicate a possible role of A in
gait disturbances and cognitive impairment in PD patients [164]. However, the
role of A in cognitive decline in PD requires further investigations
[165, 166]. Similarly, deposits of A were described in the brain of a
patient with MSA-C and in some cases of CBD and sCJD [167, 168, 169, 170, 171, 172, 173]. Even more
intriguingly, altered level of APP, the A precursor, was found in the
brain, cerebrospinal fluid (CSF) and skin of patients with ALS and is considered
an early indicator of neuromuscular junction instability and denervation [174].
In vitro studies have shown that A interacts with SOD1 (an
enzyme linked to ALS) and this lead to an impaired SOD1 enzymatic activity. Thus,
A may modulate ALS progression [175]. Another condition characterized by
the presence of A is the cerebral amyloid angiopathy (CAA), a
cerebrovascular disease commonly observed among the elderly [176]. In this case,
the deposition of A occurs mainly within cortical and leptomeningeal
arteries, capillaries and arterioles and may cause spontaneous intracerebral
hemorrhages (ICHs), other neurologic symptoms or may remain asymptomatic [177].
While CAA is characterized by amyloid deposition in vasculature with posterior
microbleeds and vascular cognitive impairment, AD involves amyloid deposition in
brain parenchyma and also leads to cognitive impairment. CAA is recognized as a
leading cause of subarachnoid hemorrhage [178] and the co-occurrence of CAA and
AD pathology is often observed in the same brain, with a higher incidence in
symptomatic AD patients. However, no significant correlation was found between
the severity of AD and CAA pathology [179, 180]. Interestingly, CAA can be
observed in cognitively normal subjects who test positive for A on PET
imaging [181]. This suggests that CAA may occur independently of AD [180, 182].
4. Tau Aggregates in the Brain of Non-Primary Tauopathies
Aggregates of misfolded tau are commonly found in the brains of patients with
primary and secondary tauopathies, including AD, CTE, CBD, PSP AGD, TOD, PART and
PiD. However, tau aggregates can be found also in other neurodegenerative
diseases. For instance, tau aggregates could be observed in the brain of patients
with prion diseases, including cases of PrP cerebral amyloid angiopathy,
Gerstmann-Sträussler-Scheinker (GSS), fatal familial insomnia (FFI), variant
Creutzfeldt-Jakob disease (vCJD) and VPSPr [183, 184, 185]. In sCJD the frequency of
tau deposition is not unusually high but it does not often relate to prion
deposition [186]. However, in GSS tau deposition was found to parallel PrP
aggregation, with a cortical distribution that can reach the deepest neuronal
layers. This specific pattern of tau deposition is mainly observed in GSS cases
with a stop-mutation in the PRNP gene at codons 145, 198 and 217 which are
therefore characterized by a higher degree of neuronal degeneration [187]. Tau
deposits were found in the brain of patients with -synucleinopathies.
In particular, tau aggregates were found in dopaminergic neurons of the
nigrostriatal region in patients with PD, and PD with dementia; while in the case
of DLB, tau and -synuclein have been found to codeposit in the same
neuronal populations [188, 189]. Interestingly, in DLB, there is a frequent
overlap among -synuclein, tau, and -amyloid pathologies which
supports the combined contribution of each protein in disease onset and
progression [190]. A few reports have shown the presence of tau aggregates in the
brain of patients with MSA with longer disease duration. In these cases, tau and
-synuclein were found to co-occur in neuronal and glial cytoplasmic
inclusions (NCIs and GCIs, respectively) [191]. In vitro studies have
shown that, in some cases, tau enhances -synuclein toxicity [192, 193].
Tau aggregates were also observed in the astrocytes of a subgroup of ALS cases
with cognitive impairment [194]. However, tau aggregation often occurs in Western
Pacific variant of ALS in which ALS, parkinsonism, and dementia co-occur. In this
variant, tau-immunoreactive neuronal inclusions with the morphology of
neurofibrillary tangles (NFTs) are found in the II and III layers of the frontal
cortex [195]. Remarkably, tau pathology is present in many cases of CAA [196].
These findings provide evidence for a cohesive pathological mechanism in which
the accumulation of amyloidogenic peptides within the vasculature initiates a
complex sequence of pathological events, ultimately resulting in the aggregation
of tau and subsequent neurodegeneration [197]. Tau aggregates are also found in
the brain of patients with HD and mouse models of the disease [198, 199].
Remarkably, unique expression patterns of tau isoforms were described in the
cortex and putamen of individuals affected by HD [200]. Interestingly, the
presence of an attenuated motor phenotype of HTT transgenic mice with genetic tau
reduction indicates a role of tau in HD pathogenesis [201].
Even with the physiological aging, there is a normal phosphorylated-tau
deposition in the brain [202]. The PART, describes a group of conditions commonly
observed in the brains of aged subjects that are characterized by the presence of
neurofibrillary tangles (NFT) that are indistinguishable from those of AD, in the
absence of A plaques [39]. PART represents a pathologic continuum which
spans from a condition of focal distribution of neurofibrillary tangles (NFT) in
cognitively normal aged individuals, to pathological situations that include the
tangle-predominant senile dementia (TPSD), the TOD, the preferential development
of NFT without senile plaques, and the senile dementia of the neurofibrillary
tangle type (SD-NFT) [39].
5. TDP-43 Aggregates in Non TDP-43 Proteinopathies
TDP-43 is mainly localized in the nucleus and is involved in RNA regulation,
including transcription, splicing and stabilization [203, 204]. The protein can
undergo several post-translational modifications (e.g., hyperphosphorylation,
cleavage, ubiquitination) which lead to its cytoplasmic aggregation [62, 205].
TDP-43 inclusion bodies are commonly observed in neurons and glial cells of
patients with ALS, FTLD-TDP and LATE [62, 206]. However, several studies showed
that aggregates of TDP-43 occurs in the brain of patients with other
neurodegenerative diseases as well as neurologically normal subjects. For
instance, TDP-43 pathology is present in up to 57% of AD cases [207, 208].
Interestingly, TDP-43 species were found to colocalize with A and tau
aggregates and were responsible for a more severe AD pathology, including greater
brain atrophy and memory loss [209]. TDP-43 deposition was common in limbic
predominant and typical AD subtypes (67% and 59%, respectively), but less
prevalent in the hippocampal sparing subtype (21%) [210]. This suggests that
TDP-43 can either influence AD progression and clinical features (representing a
risk factor for developing dementia) or be the results of neuropathological
changes occurring in advanced AD. Regardless of the AD subtypes, the presence of
TDP-43 aggregates always correlate with worse clinical progression. The
deposition of TDP-43 follows a specific staging scheme which involves: amygdala
(stage 1); enthorhinal cortex and subiculum of the hippocampus (stage 2); dentate
gyrus of the hippocampus and occipitotemporal cortex (stage 3); insular cortex,
basal forebrain, inferior temporal cortex and ventral striatum (stage 4);
brainstem nuclei (stage 5) and basal ganglia and middle frontal cortex (stage 6)
[211]. In vitro and in vivo studies have shown that oligomers
of A or tau are able to cross-seed the polymerization of TDP-43 into
pathological aggregates [212] and TDP-43 may regulate A clearance
[208, 213]. Granulovacuolar degeneration (GVD) may occur as AD co-pathology and is
characterized by the accumulation of TDP-43 along with other proteins associated
with AD [214].
In the case of PSP, a study published in 2016 showed that out of 945 cases of
pathologically confirmed cases, 56 (7%) of them were found to have
TDP-43 aggregates mainly affecting the amygdala or hippocampus, or both.
Interestingly, the progression pattern of TDP-43 aggregates was very similar to
that observed in AD, more than that typically observed in ALS or FTLD-TDP [215].
A more recent study showed that 10 out of 26 spinal cord samples of PSP patients
contained aggregates of TDP-43 (38%), mainly in motor neurons [216]. The
aggregates were composed of the insoluble C-terminal part of TDP-43.
Compared to PSP, CBD patients are more vulnerable to TDP-43 pathology. In
particular, in a study published in 2018, of the 187 CBD cases the 45% showed
TDP-43 aggregates, often involving brainstem, pons, subthalamic nucleus,
posterior hypothalamus, superior frontal gyrus and cingulate gyrus [217]. A more
recent study confirmed the presence of TDP-43 aggregates in spinal cord samples
of CBD patients [216]. In contrast, previous studies reported lower coincidental
deposition of TDP-43 in CBD cases, spanning from 9% to 24%, but this
discrepancy was associated with differences in the screening methods used
[138, 218, 219]. Interestingly, the higher prevalence of TDP-43 pathology in CBD
than PSP patients can help to distinguish these diseases, especially in patients
presenting with PSP syndrome. Indeed, TDP-43 pathology significantly influences
CBD clinical features. For instance, the presence of TDP-43 aggregates in the
midbrain tectum of CBD patients may lead to a clinical PSP presentation. Unlike
AD, the amygdala of CBD cases was less affected by TDP-43 aggregation, while the
midbrain, subthalamic nucleus and pons were found to be the most vulnerable
regions [217].
TDP-43 aggregates with oval, round and ellipsoid shapes were found to colocalize
with hungtingtin (HTT) inclusions in cortex and basal ganglia of HD cases
[220, 221]. Both TDP-43 and HTT proteins are involved in transcriptional
regulation and similar to TDP-43 [222], HTT inclusions occur in cytoplasm and
nucleus [220].
TDP-43 copathology has been reported also in the brain of patients with MSA, PD
and DLB. In particular, TDP-43 aggregates occur infrequently in MSA (7%
of the cases analyzed) and mainly localize in the medial temporal lobe of aged
patients. TDP-43 aggregates were found also to colocalize with
-synuclein in GCIs indicating a possible interation between the two
proteins [223]. Similarly, TDP-43 inclusions were reported in the 7% of PD cases
and in the 19% of PD cases with dementia [224]. Conversely, in the context of
DLB, TDP-43 inclusions are observed with divergent prevalence spanning from 0%
to 56% [224, 225, 226, 227] and their distribution mainly affect the amygdala and
hippocampal structures, as observed in AD. Interestingly, cingulate and insular
cortices were not involved, unlike AD [228]. In vitro studies have shown
that the co-occurrence of TDP-43 and -synuclein lead to a more severe
-synuclein pathology [229, 230], likely because TDP-43 is able to
enhance the toxicity of -synuclein [231].
Aggregates of TDP-43 were also observed in the brain of patients with AGD,
especially in cases with severe grain pathology. This study suggests that
abnormal accumulation of TDP-43 may be involved in AGD pathological process and
disease progression. However these findings are still controversial since other
studies postulated that TDP-43 pathology does not significantly impact the
clinical presentation of AGD [232]. Interestingly, in most AGD cases, TDP-43
pathology was consistent with LATE [233].
Finally, TDP-43 aggregates were found in the brain of aged cognitively normal
individuals with an incidence ranging between 11% and 36% and increasing with
age [225, 234, 235], suggesting that this phenomenon may be age-dependent but its
interpretation is still controversial.
6. Discussion
The concept of identifying a specific neurodegenerative disease based on the
accumulation of a particular protein in the brain (such as tau, TDP43,
-syn) is appealing and has been crucial for molecular classification.
However, this idea is becoming increasingly challenged by the fact that, more
often than not, multiple proteins aggregate in a single brain, leading to
copathologies that stratify upon the original disease [236]. In some cases, it
may even be unclear which condition was the original one.
We have presented a detailed list of protein aggregates that have been reported
to associate with the main proteinopathy in different neurodegenerative diseases.
It is therefore clear that the occurrence of copathologies represents almost the
rule rather than the exceptions in this field.
This may represent the molecular basis of the extreme phenotypic variability
that has been extensively described for example in Alzheimer disease. AD may
coexist with -synuclein pathology in DLB, with TDP-43 pathology in
LATE. The coexistence of AD pathology and PART may be postulated and would be
impossible to resolve as tau pathology of AD and PART are not distinguishable.
Our knowledge on this field is limited by the difficulties of performing
large-scale post mortem neuropathological studies that are the only way to obtain
detailed description of protein-aggregates topography and burden.
7. Conclusions
This review highlights the complexity of protein aggregation in
neurodegeneration and suggests potential common mechanisms and interactions
underlying different diseases. The traditional view of each neurodegenerative
disease (ND) being exclusively associated with a single protein aggregate is
being challenged, and the coexistence of multiple proteinopathies may contribute
to the phenotypic heterogeneity observed in these conditions. In this scenario,
achieving a precise understanding of the pathological processes occurring during
a ND in a living patient can be accomplished through specific biomarkers tailored
to each proteinopathy. Over the past few years, numerous methods have been
developed to analyze promising peripheral biomarkers, such as seed amplification
assays, which have the potential to enhance the clinical diagnosis of
neurodegenerative diseases (NDs). However, considering the complexity of NDs, it
is likely that multiple approaches would be necessary to identify novel and
reliable biomarkers for a paradigm shift towards a more precise biological-based
diagnosis which overcomes the limits of clinical interpretation. Further research
in this field will likely lead to more comprehensive and accurate disease
classifications and therapeutic approaches aimed at targeting the shared
pathological mechanisms across multiple neurodegenerative disorders. Ultimately,
the hope is that these efforts will pave the way for improved treatments and
better outcomes for patients affected by these devastating diseases.
Author Contributions
FM and GG wrote the manuscript and designed the figure. AC, ILD, AL, NC, GB and FAC contributed in manuscript preparation, conceptualization, data analysis and interpretation. All the authors critically reviewed the manuscript and approved its final version.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of Interest
The authors declare no conflict of interest. Given their role as Guest Editor member of FBL, Fabio Moda and Giorgio Giaccone had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Antoni Camins.


 , Arianna Ciullini 1
, Arianna Ciullini 1