
- Academic Editors
Background: Atrial fibrillation is one of the most common cardiac
arrhythmias. Myocardial fibrosis is closely associated with
atrial remodeling, which leads to heightened risk of atrial fibrillation. This
study aimed to explore whether forkhead box protein O3 (FOXO3a) impacts
myocardial fibrosis incidence by regulating mitophagy. Methods:
Cell viability was assessed by cell counting kit-8 (CCK-8) assays. The expression
of vimentin and cytochrome C was detected by immunofluorescence assays.
Quantitative real-time polymerase chain reaction (PCR) was used to analyze the
relative mRNA level of FOXO3a. Expression of FOXO3a, phosphorylated FOXO3a,
Collagen I, Collagen III, alpha-smooth muscle actin (
Atrial fibrillation (AF) is one of the most common clinical arrhythmias and is associated with the risk of stroke and heart failure [1, 2]. Electrical and structural remodeling are two essential pathophysiologic hallmarks of AF [3], while myocardial fibrosis (MF) is the principal characteristic associated with structural remodeling [4, 5]. Cardiac fibroblasts (CFs) are the primary cell type in the myocardial interstitium, and can differentiate into myofibroblasts under conditions of myocardial ischemia or pressure stimuli [6]. Active myofibroblasts exhibit proliferative and invasive properties to initiate tissue repair, and they also secrete more collagen fibers to remodel the interstitium [7]. However, sustained activation of myofibroblasts can lead to MF and a salient characteristic feature of MF is excessive deposition of the extracellular matrix (ECM) [8]. The abnormal deposition of ECM contributes to the development and maintenance of AF by increasing the heterogeneity of atrial conduction [9, 10]. Hence, further insight into the molecular mechanism of MF may afford a new theoretical basis for novel AF therapies.
Recent research evidence suggests that alterations in mitochondrial function and metabolism play a crucial role in the differentiation of CFs into myofibroblasts [11]. Mitochondrial autophagy, also known as mitophagy, is a selective intracellular degradation process that helps eliminate dysfunctional mitochondria and maintain mitochondrial homeostasis [12]. Steps in this process include depolarization of damaged mitochondria, loss of mitochondrial membrane potential (MMP), formation of mitochondrial autophagosomes, fusion of autophagosomes with lysosomes, and degradation of the mitochondrial contents [13].
The phosphatase and tensin homolog (PTEN)-induced putative kinase 1
(PINK1)/Parkin signaling pathway has been widely reported to be involved in
mitophagy [14]. PINK1 is a serine/threonine kinase that plays a crucial role in
the regulation of mitochondrial function [15]. When the MMP is lost, PINK1
accumulates on the outer membrane of mitochondria and stimulates the recruitment
and activation of the E3 ubiquitin ligase Parkin. Activated Parkin is responsible
for binding ubiquitin molecules to adaptor proteins such as sequestosome-1 (p62), thereby
amplifying mitochondrial autophagic signaling [13]. Numerous groups have shown
that mitophagy plays a crucial role in regulating heart disease. For example,
Tong et al. [16] observed that Ulk1-Rab9-dependent replacement of
mitophagy protected the heart from obesity-related cardiomyopathy. Further, Cai
et al. [17] demonstrated that empagliflozin blocked cardiac
microvascular ischemia/reperfusion injury by activating FUN14 domain-containing 1
(FUNDC1)-dependent mitophagy. In addition, the work of Wang et al. [18]
suggested that the adenosine monophosphate-activated protein kinase catalytic
subunit alpha 2 (AMPK
Forkhead box protein O3 (FOXO3a) is a member of the forkhead transcription factor subfamily, and family members play important roles in cell cycle arrest, apoptosis, angiogenesis, DNA repair, and metabolism [19]. Several studies have highlighted the role of FOXO3a in controlling the occurrence and progression of certain diseases by regulating mitochondrial autophagy. For example, FOXO3a has been shown to be involved in diabetic retinopathy [20] and cardiac hypertrophy [21]. Additionally, FOXO3a has been implicated in insulin-like growth factor-1 receptor-mediated myocardial fibrosis and the development and maintenance of atrial fibrillation [22]. However, whether FOXO3a influences the process of MF by regulating mitophagy remains unknown. Therefore, the principal aim of this study was to determine whether FOXO3a could affect MF by regulating mitophagy to provide new insights for the treatment of MF and AF.
Neonatal (1–3 day old) rats were killed under anesthesia, disinfected with 75%
alcohol for twice, then the heart was dissected. Subsequently, the atrial tissue
was separated and washed with phosphate-buffered saline (PBS). Next, atrial
tissue was cut into fragments and digested with trypsin and type II collagenase.
The cells were collected and cultured in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum (FBS) for 2 h. Following this,
the CFs were obtained by differential adherence, and isolated CFs were identified
by immunofluorescence. Specifically, CFs were validated by Vimentin-positive
staining and also tested negative for mycoplasma. Cells are cultured in a
humidified incubator at 37 °C with 5% CO
To study whether FOXO3a affected the activation of CFs by promoting mitophagy and examine study the role of FOXO3a in this process, cells were transfected with negative control (si-NC) or FOXO3a-specific (si-FOXO3a) small interfering RNAs (siRNA) using Lipofectamine 2000 (11668019, Invitrogen, Carlsbad, CA, USA). Following this, rat CFs were divided into control (no intervention) and angiotensin II (AngII)-treated (10 µM AngII) groups and incubated for an additional 24 h. To verify whether FOXO3a affected the activation of CFs by promoting mitophagy, the si-FOXO3a cells were treated with 5 nM everolimus (RAD001), and this was termed the si-FOXO3a+RAD001 group.
Cells in each experimental group were treated as outlined. After removing the medium, cells were incubated with 100 µL of Cell Counting Kit-8 (CCK-8) (NU679, Dojindo, Tokyo, Japan) working solution at 37 °C for 4 h. The absorbance value at 450 nm was measured using a Bio-Tek microplate reader (MB-530, Heales, Shenzhen, Guangzhou, China).
CFs were applied to glass microscope slides and fixed with 4% paraformaldehyde.
Subsequently, the cells were permeated with 0.3% TritonX-100 and blocked with
5% bovine serum albumin (BSA). Next, the slides were immersed in primary
antibodies against vimentin (10366-1-AP, Proteintech, Chicago, IL, USA) or
cytochrome C (66264-1-Ig, Proteintech, USA) overnight at 4 °C. Subsequently, the
slides were incubated in secondary antibody (anti-Rabbit/Mouse IgG (H+L), 1:200, AWS0005c/AWS0004c, Abiowell, Changsha, Hunan, China) for 90 min.
4
Total RNA was extracted from cells using TRIzol reagent (15596026, Thermo
Scientific, Portsmouth, NH, USA). An mRNA Reverse Transcription kit (CW2569,
CWBIO, Taizhou, Zhejiang, China) was used to synthesize cDNA from total RNA. The
relative expression of the genes was examined by quantitative real-time
polymerase chain reaction (qRT-PCR) using UltraSYBR Mixture (CW2601, CWBIO, Taizhou, Jiangsu,
China) and a QuantStudio1 Real-Time PCR System (A40426, ABI, Foster city, CA,
USA). Relative mRNA abundance was calculated using 2
| Gene | Sequence | Length (bp) |
| R-FOXO3a | F TCTATAACTTTGTGCTGCTGCCG | 206 bp |
| R TACCTCGGCTCCTTCCCTTCA | ||
| R-GAPDH | F ACAGCAACAGGGTGGTGGAC | 252 bp |
| R TTTGAGGGTGCAGCGAACTT |
Total proteins were extracted from cells using radio immunoprecipitation assay (RIPA) lysis buffer (AWB0136, Abiowell, Changsha, Hunan, China) and quantified using a bicinchoninic acid assay (BCA) kit (AWB0104, Abiowell). The proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Invitrogen, USA). The primary antibodies were incubated with the membranes overnight at 4 °C. Subsequently, secondary antibodies were incubated with the membrane. Enhanced chemiluminescence (ECL) chromogenic solution (Chemiscope6100, CLiNX, Shanghai, China) was used to visualize protein bands. GAPDH was served as the internal control, and information regarding the antibodies used is displayed in Table 2.
| Name | Dilution rate | Cat number | Source | Company | Country |
| FOXO3a | 1:5000 | 10849-1-AP | Rabbit | Proteintech, Chicago, IL | USA |
| p-FOXO3a | 1:1000 | 28755-1-AP | Rabbit | Proteintech | USA |
| Collagen III | 1:5000 | ab7778 | Rabbit | Abcam, Cambridge | UK |
| Collagen I | 1:2000 | 14695-1-AP | Rabbit | Proteintech | USA |
| 1:2000 | 55135-1-AP | Rabbit | Proteintech | USA | |
| MMP9 | 1:1000 | ab228402 | Rabbit | Abcam | UK |
| LC3 | 1:2000 | 14600-1-AP | Rabbit | Proteintech | USA |
| PINK1 | 1:600 | 23274-1-AP | Rabbit | Proteintech | USA |
| Parkin | 1:2000 | 14060-1-AP | Rabbit | Proteintech | USA |
| p62 | 1:4000 | 18420-1-AP | Rabbit | Proteintech | USA |
| GAPDH | 1:5000 | 10494-1-AP | Rabbit | Proteintech | USA |
| PCNA | 1:5000 | 10205-2-AP | Rabbit | Proteintech | USA |
| HRP goat anti-Rabbit IgG | 1:6000 | SA00001-2 | / | Proteintech | USA |
Cell proliferation was assessed using a Cell-Light EdU Apollo567 Kit (C10310,
RiboBio, Guangzhou, Guangdong, China). After incubating cells with
5-Ethynyl-2
CFs were inoculated into 6-well plates and incubated until 100% confluent. The cell layer was then scratched in with the tip of a sterile pipette (T-200-Y, Axygen, Tewksbury, MA, USA). The cells were washed 3 times with PBS and serum free RPMI-1640 was added. Cells were subsequently examined at 0 h, 24 h and 48 h for wound healing. Cells were observed using an inverted microscope (DSZ2000X, Beijing Cnmicro Instrument Co., Ltd., Beijing, China).
Cells were treated with
5,5
Cells were incubated with 10 µM 2
Data was analyzed using GraphpadPrism9.0 (GraphPad, La Jolla, CA, USA) and
displayed as mean
As shown in Fig. 1A, the shape of isolated rat CFs was, as expected, fusiform in nature when examined in a light microscopy. IF was used to confirm the identity of isolated CFs, and, as expected, the expression of vimentin was positive (Fig. 1B). Subsequently, CFs were treated with AngII to establish the MF cell model. As expected, AngII treatment significantly promoted the proliferation of CFs (Fig. 1C). Furthermore, AngII treatment resulted in the upregulation of FOXO3a and downregulation of p-FOXO3a in cytoplasm of CFs (Fig. 1D). AngII treatment also led to increased levels of FOXO3a in nucleus (Fig. 1E). Collectively, these results suggested that FOXO3a expression was upregulated in AngII-induced MF cell model.
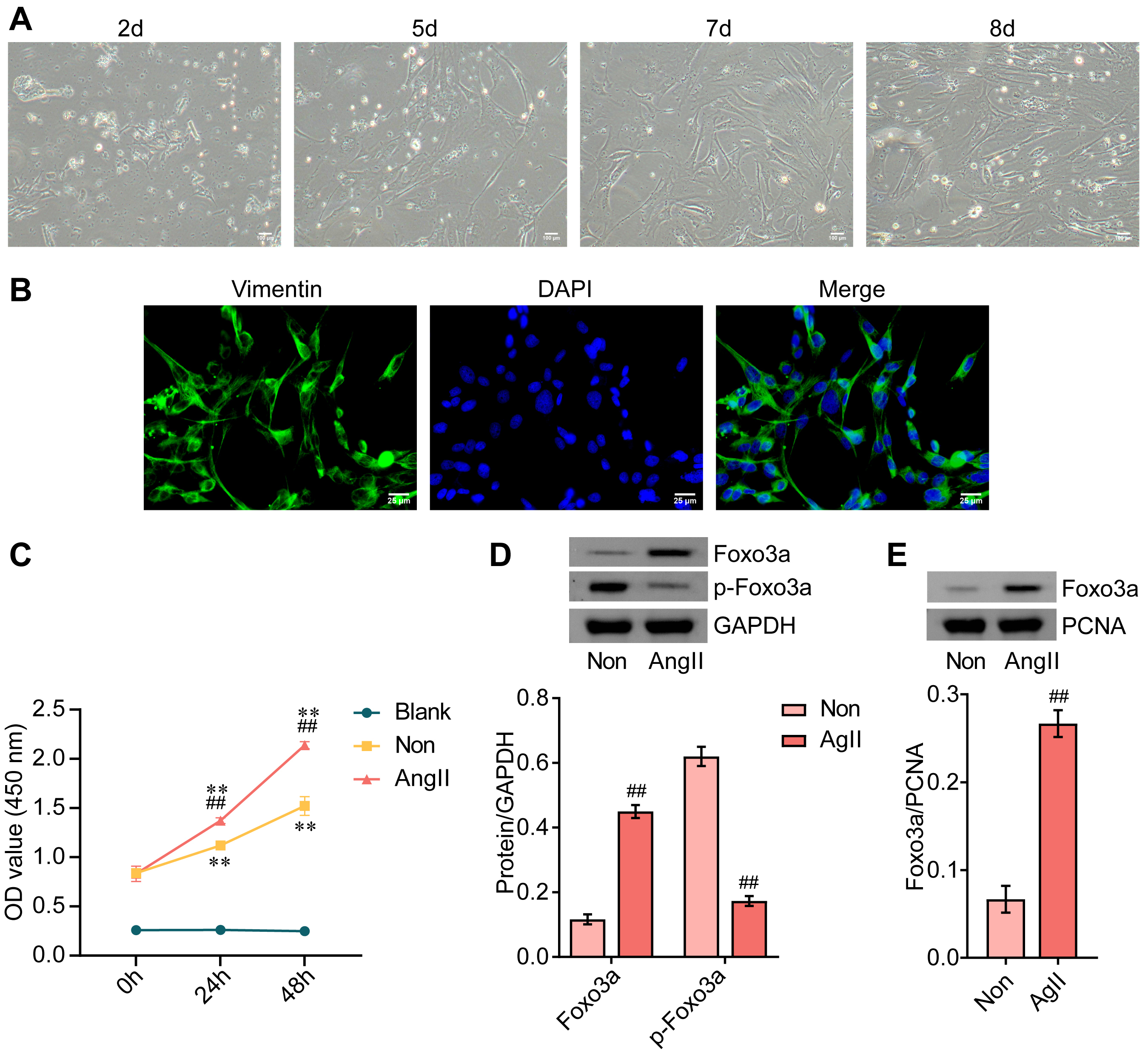 Fig. 1.
Fig. 1.Expression of FOXO3a is upregulated in AngII-induced myocardial
fibrosis cell model. (A) Photomicrograph of cultured cardiac fibroblasts. Scale bar = 100 µm. (B) IF
staining of vimentin, DAPI counter-staining, and a merged image is shown. Scale bar = 25 µm. (C)
Cell viability was measured by CCK-8 assays. (D) The expression of FOXO3a and
phosphorylated FOXO3a in the cytoplasm was examined by western blotting. (E) The
expression of FOXO3a in nucleus was assessed by western blotting. **p
Compared with control cells, the mRNA level of FOXO3a was elevated in response to AngII treatment. Further, relative to the si-NC group, the mRNA level of FOXO3a was decreased in cells transfected with si-FOXO3a. Changes in the protein level, as measured by WB, were consistent with mRNA measurements (Fig. 2A).
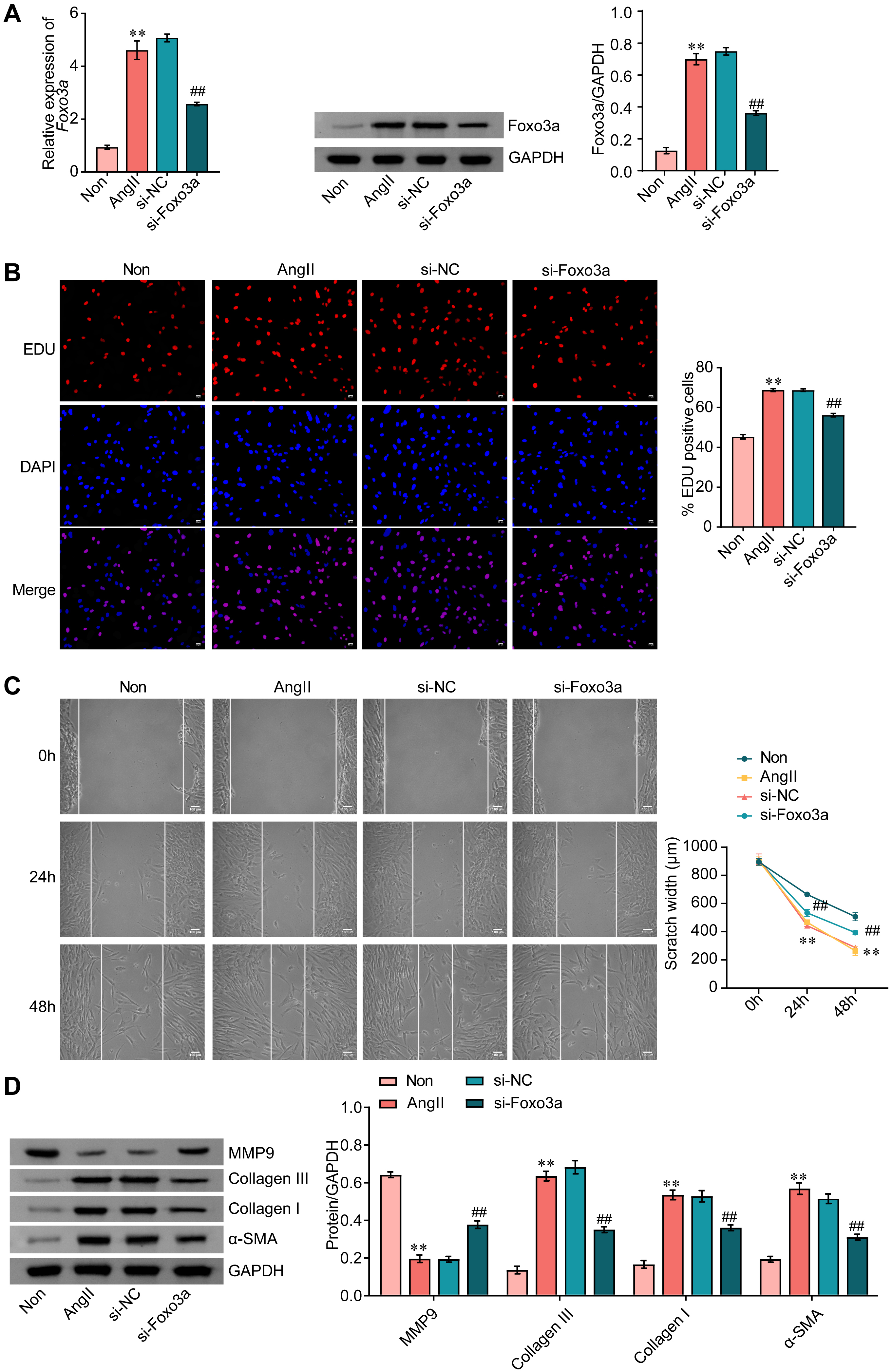 Fig. 2.
Fig. 2.FOXO3a knockdown inhibits AngII-induced activation of CFs. (A)
FOXO3a expression was measured by qRT-PCR and WB. (B) The proliferation
of CFs was assessed by EDU staining. Scale bar = 50 µm. (C) Wound healing assay was used to measure
cell migration. Scale bar = 100 µm. (D) The expression of MMP9, collagen Ⅲ, collagen Ⅰ, and
Subsequently, the effect of FOXO3a knockdown on CF proliferation was examined.
As shown in Fig. 2B, the AngII-induced proliferation of CFs was significantly
blunted in response to FOXO3a knockdown (Fig. 2B) and, similarly, FOXO3a
knockdown inhibited AngII-induced cell migration (Fig. 2C). FOXO3a knockdown also
reversed AngII-induced downregulation of MMP9 expression. In contrast,
upregulation of collagen III, collagen I and alpha-smooth muscle actin (
After treatment with AngII, the red fluorescence of cells decreased, while the green fluorescence was increased, indicating that the mitochondrial membrane potential of CFs decreased in response to AngII. Furthermore, FOXO3a knockdown effectively suppressed AngII-induced mitochondrial membrane potential reduction in CFs (Fig. 3A). We also noted that FOXO3a knockdown alleviated AngII-induced elevations in reactive oxygen species (ROS) (Fig. 3B). FOXO3a knockdown significantly reduced the release of mitochondrial cytochrome C in CFs following treatment with AngII (Fig. 3C). Furthermore, the ratio of mitophagy markers light chain 3 (LC3) Ⅱ and Ⅰ, and the expression of PINK1 and Parkin rose while expression of p62 was reduced in CFs treated with AngII, and these effects were blunted by FOXO3a knockdown (Fig. 3D). Our findings indicated that FOXO3a knockdown inhibited AngII-induced mitophagy in CFs.
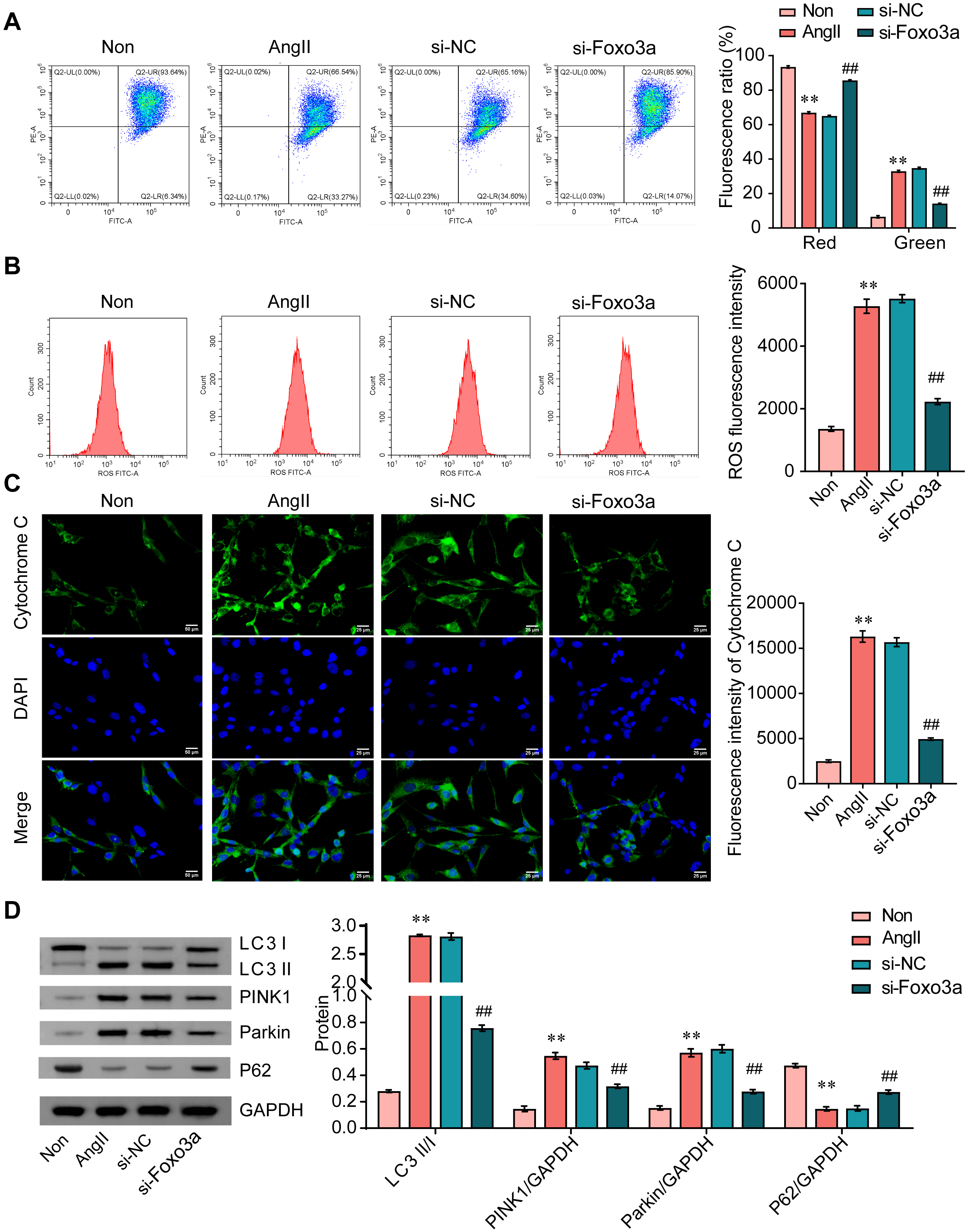 Fig. 3.
Fig. 3.FOXO3a knockdown inhibits AngII-induced mitophagy in CFs. (A)
JC-1 staining was conducted to measure mitochondrial membrane potential. (B) ROS
levels were measured by flow cytometry. (C) IF staining of cytochrome C. Scale bar = 25 µm. (D) The
ratio of LC3 II/I, and expression of PINK1, Parkin, and p62 was analyzed by WB.
**p
As shown in Fig. 4A, the mitophagy inducer RAD001 significantly attenuated the
proliferation inhibition caused by FOXO3a knockdown. RAD001 also rescued CF
migration inhibition in cells with FOXO3a knockdown (Fig. 4B). Finally, RAD001
reversed the upregulation of MMP9 and downregulation of collagen III, collagen Ⅰ
and
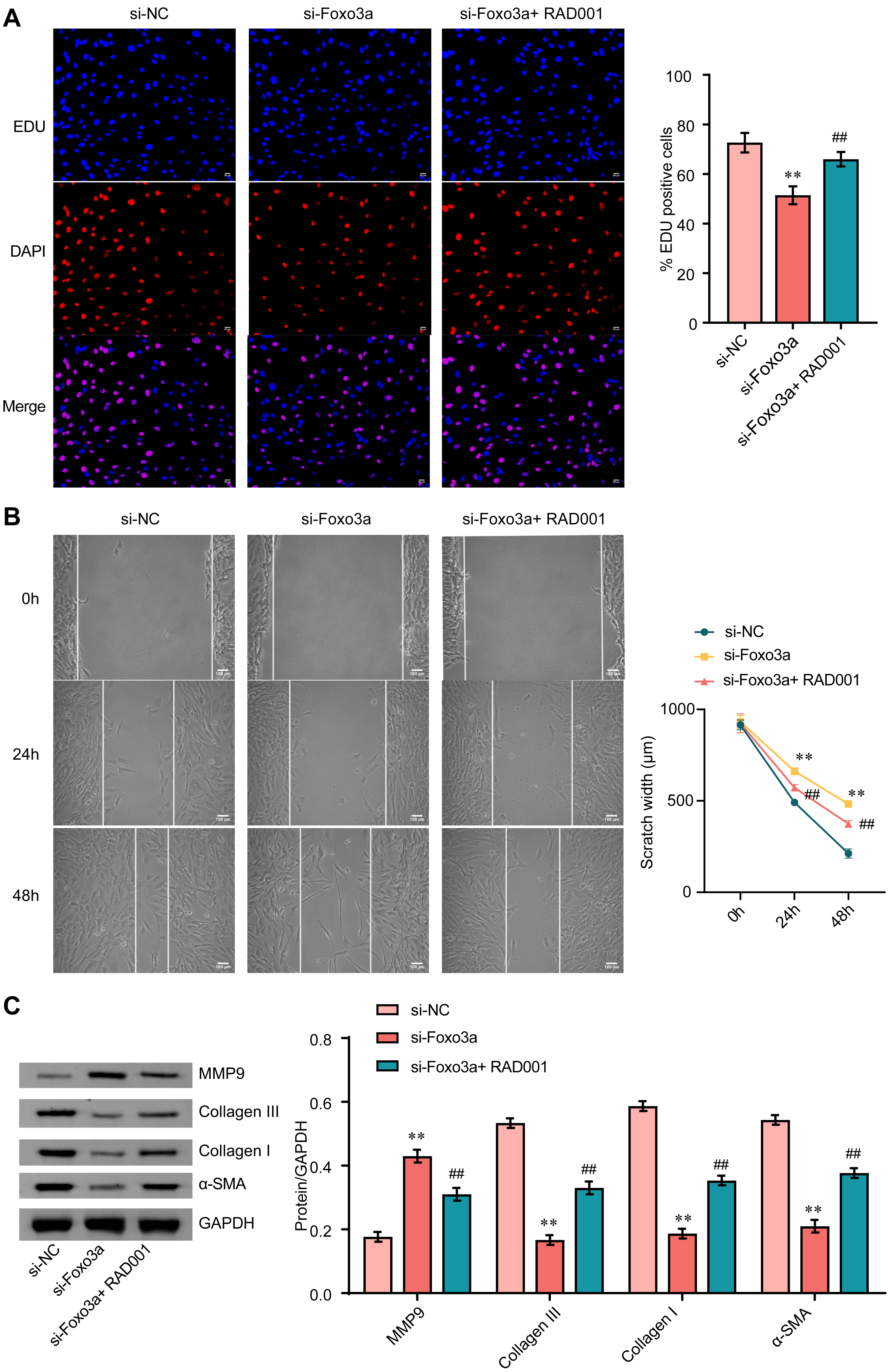 Fig. 4.
Fig. 4.FOXO3a blunts the activation of CFs by promoting mitophagy. (A)
The EDU proliferation assay was used to examine the proliferation of CFs. Scale bar = 50 µm. (B) A
wound healing assay was used to measure the cell migration. Scale bar = 100 µm. (C) The expression of
MMP9, collagen Ⅲ, collagen Ⅰ, and
MF is a critical pathological change that is associated with the development and maintenance of AF [23]. The content and distribution of fibrous tissue in patients with AF is also associated with the heightened risk of clinical complications and treatment failure [24]. Therefore, a better understanding of the molecular mechanisms that give rise to MF may provide new druggable targets and new therapeutic approaches. In this study, we demonstrated the role that FOXO3a plays in mediating MF. We provided in vitro evidence indicating that activated FOXO3a regulated mitophagy through the PINK1/Parkin pathway, thereby promoting ECM protein deposition and fibrosis (Fig. 5).
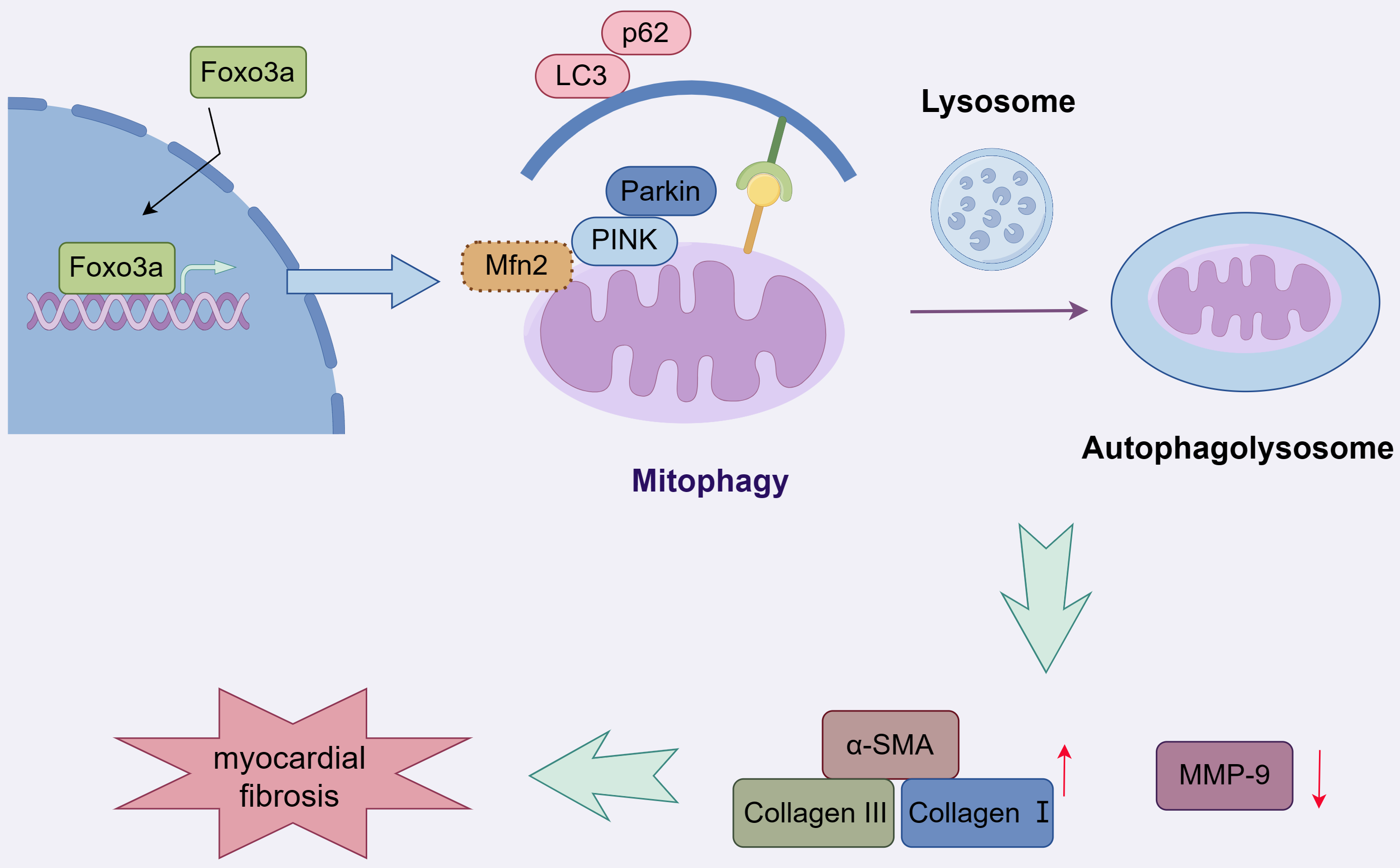 Fig. 5.
Fig. 5.A graphical abstract illustrates that Foxo3a promotes mitophagy and ultimately myocardial fibrosis via the PINK1/Parkin pathway. Mfn2, mitofusin-2. Fig. 5 was created by Figdraw (https://www.figdraw.com).
As a transcription factor, FOXO3a not only participates in the regulation of various cell functions such as apoptosis, proliferation, cell cycle, and DNA damage repair [25], but also contributes to organ fibrosis [26]. For example, the loss of IL-6 mitigated renal fibrosis by attenuating DNA methyltransferase 1 (DNMT1)-mediated FOXO3a inhibition [27]. 5-methoxytryptophan alleviated liver fibrosis by regulating autophagy mediated by the FOXO3a/microRNA-21 (miR-21)/autophagy-related gene 5 (ATG5) signaling pathway [28]. Additionally, resveratrol relieved renal interstitial fibrosis in rats with unilateral ureteral obstruction by inhibiting the protein kinase B (AKT)/FOXO3a pathway [29].
Recently, Zhang et al. [22] discovered that FOXO3a was upregulated in
activated CFs and was tightly associated with MF. Consistent with this work, we
also observed elevated expression of FOXO3a in the nucleus of AngII-induced CFs.
AngII is widely used to induce the activation of CFs into myofibroblasts [30],
and we noted that AngII also limited the phosphorylation of FOXO3a in cardiac
fibroblasts, an event linked to upregulating its transcriptional activity.
Further data showed that FOXO3a knockdown significantly inhibited AngII-induced
proliferation and migration of CFs. The pathological changes in the extracellular
matrix of cardiomyocytes are primarily caused by an imbalance between collagen
synthesis and degradation, and this phenomenon is characterized by excessive
collagen content and reduced MMP-mediated degradation [31]. In AngII-exposed CFs,
FOXO3a knockdown also decreased the expression of collagen III, collagen Ⅰ and
Recent studies have indicated that mitophagy may serve as a potential therapeutic target for mitigating MF [32]. Mitophagy is an important mitochondrial quality control mechanism that regulates the removal of damaged mitochondria [12]. At present, there are two principal mitophagic mechanisms: mitochondrial elimination mediated by B cell leukemia/lymphoma 2 (Bcl-2) interacting protein 3 (BNIP3)/BNIP3-like (NIX), and mitophagy mediated by PINK1/Parkin [33]. Surprisingly, both pathways are regulated by FOXO3a. Studies have validated that FOXO3a silencing dramatically prevents temozolomide-induced upregulation of BNIP3 and mitophagy [34]. Meanwhile, FOXO3a promotes mitophagy by upregulating the expression of Parkin [21]. In this study, we focused on the PINK1/Parkin mitophagic pathway. We observed that AngII induced mitophagy in CFs and that FOXO3a knockdown significantly suppressed AngII-induced increases in MMP, elevation of ROS levels, and release of cytochrome C. FOXO3a knockdown also significantly downregulated the ratio of LC3 II/I, and expression of PINK1 and Parkin, while upregulating the expression of p62. It has been found that activated Parkin drives the ubiquitination of mitochondrial fusion proteins, mitofusins (Mfn1 and Mfn2), thereby triggering mitochondrial degradation [35, 36]. Studies have shown that the loss of mitofusin-2 (Mfn2) prevents the recruitment of PINK1-associated Parkin and mitophagy in depolarization-induced cardiomyocytes [37, 38]. Previous research has reported that the PINK1/Parkin axis mediates mitophagy to promote cardiomyocyte ferroptosis and affect MF [39]. These findings suggested that FOXO3a promoted MF by accelerating mitophagy, and PINK1/Mfn2/Parkin signaling may be intimately involved in this process.
Interestingly, the role of mitophagy in MF has met with controversy in the literature. Zheng et al. [40] validated that fucoxanthin improved MF in streptozotocin (STZ)-induced diabetic rats by restoring mitophagy. Guan et al. [41] also concluded that ADAM17 knockout prevented MF by triggering mitophagy. While Wang et al. [42] indicated that microRNA-24-3p improved MF by inhibiting mitophagy in CFs. These findings may be attributed to the varying pathogenic mechanisms of different diseases, and/or the differential expression profiles of targets in these various pathological models. Given this, we explored the role of FOXO3a-mediated mitophagy in MF. We found that the mitophagy inducer RAD001 reversed the effects of FOXO3a knockdown. Specifically, in AngII-exposed CFs, supplementation with RAD001 increased cell proliferation, migration, and accelerated ECM protein deposition compared to cells transfected with FOXO3a siRNA alone. These results further indicated that silencing FOXO3a alleviated excessive activation of AngII-induced myofibroblasts by inhibiting mitophagy. Moreover, our results support the findings outlined in the study conducted by Zhang and co-workers [42].
However, there are some limitations in our study. Our in vitro evidence has not been validated in animal models and clinical samples. More evidence is needed to support the regulatory role of FOXO3a in mitophagy, such as mitochondrial superoxide production and changes in mitochondrial morphology. The role of Mfn2 in FOXO3a-mediated mitophagy requires further investigation as does the determination of whether the BNIP3/NIX pathway is involved in FOXO3a-mediated activation of myofibroblasts. Additionally, the mechanism of mitophagy and its crosstalk with other signaling pathways, such as the apoptotic signaling pathway in MF, are also worthy of further exploration.
In conclusion, our study provides in vitro evidence that limiting FOXO3a activity inhibited mitophagy in AngII-treated CFs and attenuated the excessive transformation of CFs into myofibroblasts. We also discovered that FOXO3a activated the PINK1/Parkin pathway to mediate mitophagy. Our findings suggest that targeting FOXO3a may have potential therapeutic implications for MF and AF.
All data found in this study is available upon request by contact with the first author or corresponding author.
DL and QS designed the experiments, participated in data analysis and interpretation of results, and drafted the manuscript. DL, HL, BD and ZH contributed to the experimental design and procedures, participated in data analysis and interpretation. LM, ZW and XW participated in data analysis and interpretation of results. QS designed the experiments, provided guidance on experimental procedures and data analysis, and primarily responsible for writing and revising the manuscript as the corresponding author. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the Medical Ethics Committee of the First Hospital of Changsha (2022-103).
Fig. 5 was created by Figdraw (https://www.figdraw.com). Thank this site for providing support.
This study was supported by funds from Hunan Provincial Health Commission (Project No. B202303016809 & 202103101790).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.