
 , Xiangyu Zhong 1,*
, Xiangyu Zhong 1,*1 Department of Hepatopancreatobiliary Surgery, The Second Affiliated Hospital of Harbin Medical University, 150086 Harbin, Heilongjiang, China
2 Anhui Province Key Laboratory of Translational Cancer Research, Bengbu Medical College, 233030 Bengbu, Anhui, China
3 State Key Laboratory of Oncology in South China, Cancer Center of Sun Yat-Sen University, 510000 Guangzhou, Guangdong, China
4 Department of Pathology, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
†These authors contributed equally.
Abstract
Pancreatic cancer is a malignancy that affects the digestive tract and has a low 5-year survival rate of lower than 15%. Owing to its genetic mutation and metabolic complexity, pancreatic cancer is difficult to treat with surgical resection, radiotherapy, and chemotherapy. The predominant modality of pancreatic cancer is pancreatic ductal adenocarcinoma (PDAC), primarily attributed to mutations in KRAS gene. Ferroptosis, an iron-mediated reactive oxygen species (ROS)-elevated nonapoptotic cell death caused by lipid peroxidation, is distinct from any other known type of cell death. Ferroptosis is closely related to the occurrence and progression of different types of cancers, including PDAC. Previous research has demonstrated that ferroptosis not only triggers cell death in PDAC and hampers tumor growth but also enhances the effectiveness of antitumor medications. In our review, we mainly focus on the core mechanism of ferroptosis, reveal its interrelationship with PDAC, and illustrate the progress of ferroptosis in different treatment methods of PDAC.
Keywords
- ferroptosis
- pancreatic cancer
- pancreatic ductal adenocarcinoma
- PDAC
- treatment
- therapy
Pancreatic ductal adenocarcinoma (PDAC) is the most prevalent form of pancreatic cancer, characterized by its highly malignant nature in the digestive tract. With a low 5-year survival rate of lower than 15%, it ranks as the fourth leading cause of cancer-related death in both genders. The incidence and mortality rates of PDAC have been steadily increasing over the years [1, 2]. The primary mutant genes found in PDAC are KRAS, CDKN2A, TP53, and SMAD4, with KRAS gene mutations accounting for approximately 90% of PDAC cases [3]. Common risk factors associated with PDAC include age (being over 50 years old), smoking, drinking, obesity, and diabetes [4, 5, 6, 7, 8]. Despite significant advancements in cancer treatment methods such as surgical resection, chemotherapy, radiotherapy, and immunotherapy, PDAC remains relatively insensitive to these approaches [9]. Thus, it is pivotal to comprehend the molecular mechanisms involved in the formation and development of PDAC, and to identify more precise, safer, and effective therapeutic targets.
In recent years, programmed cell death (PCD) has seen significant advancements in cancer treatment, with breakthroughs in various forms including apoptosis, necroptosis, pyroptosis, and ferroptosis [10]. Among these, ferroptosis is a distinct nonapoptotic modality of programmed cell death which is characterized by iron accumulation, reactive oxygen species (ROS) production, and lipid peroxidation [11]. Extensive research has established a close link between ferroptosis and the occurrence and development in several diseases, including Alzheimer’s disease [12], chronic obstructive pulmonary disease [13], liver disease [14], and various tumors including PDAC [15, 16, 17]. Moreover, ferroptosis has been found to influence the effectiveness of chemotherapy, radiotherapy, and immunotherapy [18, 19, 20], and is being explored as a potential strategy to reverse drug resistance [21]. Despite the incomplete understanding of the regulatory mechanism of ferroptosis, it holds promise as a new target for PDAC therapy due to its interrelationship with tumors. Our review aims to thoroughly examine the fundamental mechanism of ferroptosis and its correlation with PDAC. Additionally, we explore the promising prospects of utilizing ferroptosis as a potential therapeutic approach for the treatment of PDAC.
Unlike other well-known modalities of cell death, such as apoptosis, necrosis,
and pyroptosis, ferroptosis is distinct and can be identified by the accumulation
of iron, the generation of ROS and lipid peroxidation. The induction of
ferroptosis can be achieved by the compound erastin, which inhibits the
cystine/glutamate antiporter (system X
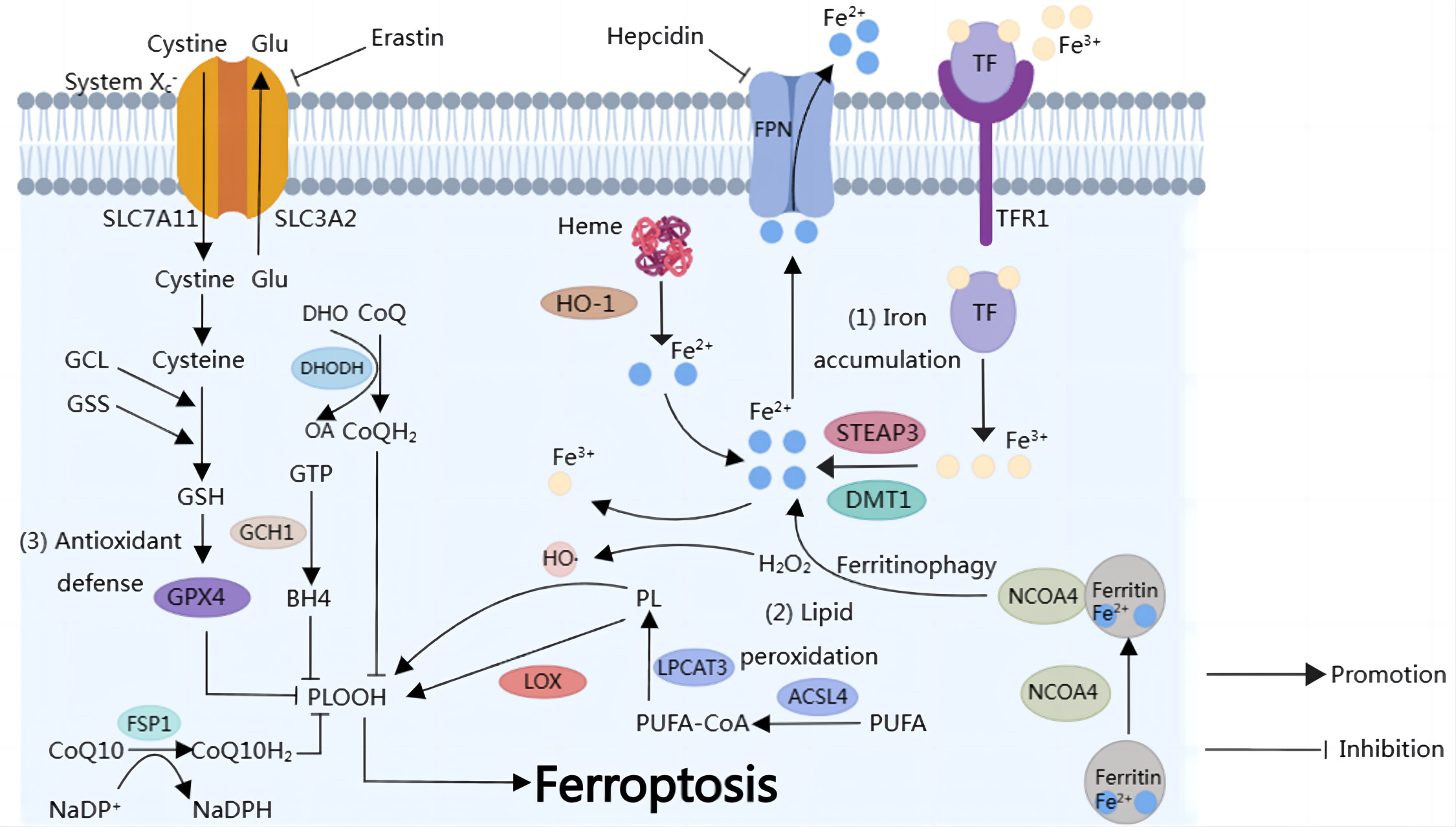
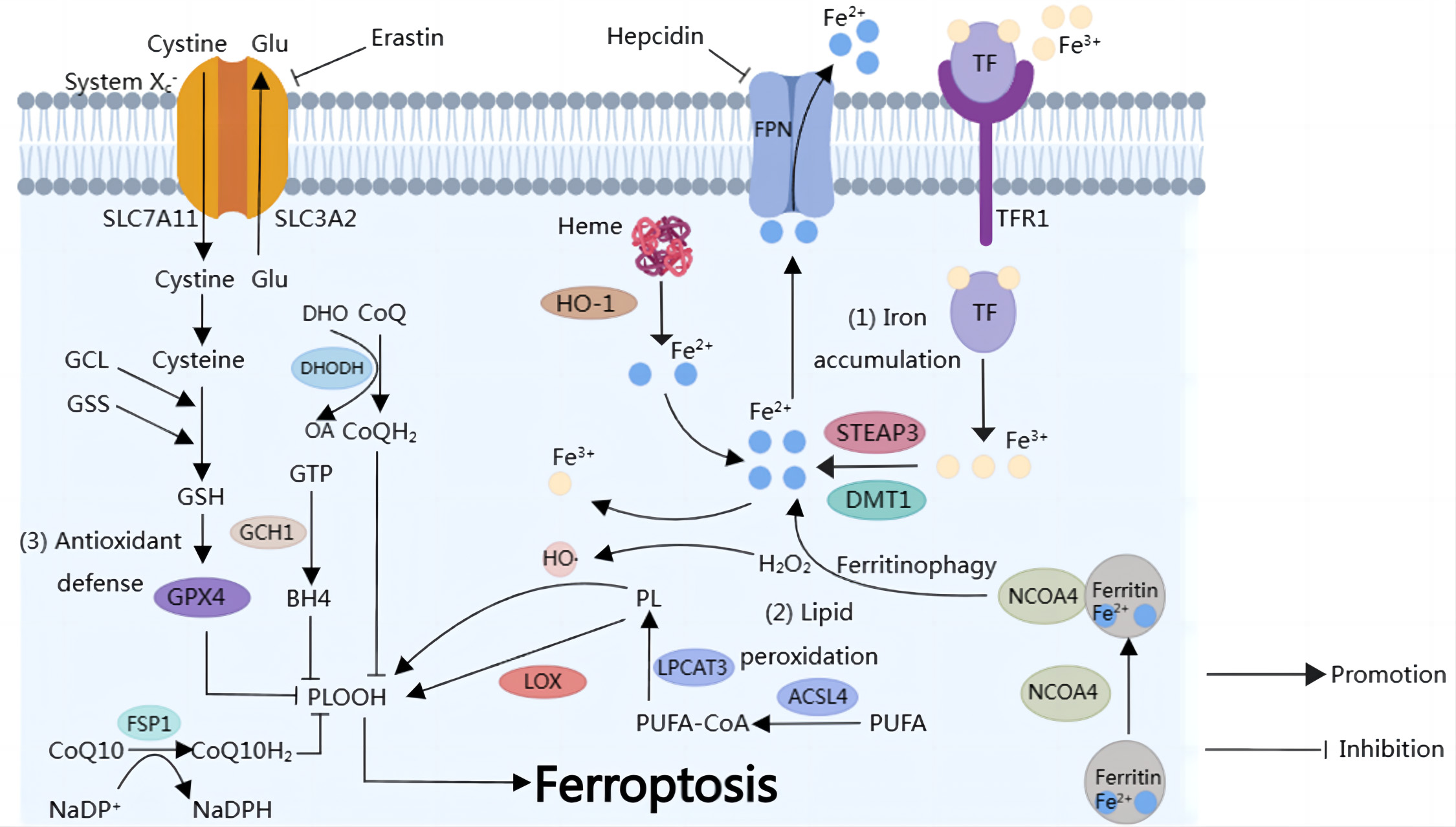 Fig. 1.
Fig. 1.Overview of ferroptosis. (1) Iron accumulation. Fe
Intracellular iron metabolism plays a direct role in modulating ferroptosis. The
biological contributions of iron rely on the electron transfer that occurs when
iron is transformed between its ferrous bivalent (Fe
ROS, which includes superoxide (O
Previous studies have indicated that PUFAs are highly susceptible to peroxidation during ferroptosis [33]. Phospholipids (PLs) play a crucial role in the composition of cell membranes, consisting primarily of glycerophospholipids and sphingomyelin. Glycerophospholipids contain PUFAs, particularly arachidonic acid (AA) and adrenaline acid (AdA), which are impressionable to peroxidation and can cause damage to the cell membrane [34, 35]. Before entering PLs, PUFAs require modification by acyl-CoA synthetases long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3).
Under the catalysis of ACSL4, AA and AdA react with coenzyme A (CoA) to produce
AA-CoA and AdA-CoA, respectively. These compounds then undergo esterification
reaction catalyzed by LPCAT3, resulting in the formation of AA-
phosphatidylethanolamine (PE) and AdA-PE. Finally, they enter PLs [36, 37, 38]. The
HO
Several antioxidant systems in cells can negatively regulate the progress of ferroptosis by suppressing lipid peroxidation. Among these systems, the antioxidant system mediated by glutathione peroxidase 4 (GPX4) is considered to be the most decisive.
System X
Coenzyme Q10 (CoQ10) is a crucial intracellular free radical scavenger that is
present in all cell membranes [45]. Ferroptosis suppressor protein 1 (FSP1) is an
independent suppressor of ferroptosis. FSP1 utilizes intracellular NADPH to
reduce CoQ10 to reduced CoQ10 (CoQ10H
Dehydrogenase (DHODH) is situated in the inner mitochondrial membrane and
catalyzes the conversion of dihydroorotate into orotate. It provides 2 electrons
to the electron transport chain (ETC). Coenzyme Q (CoQ) is reduced to reduced
Coenzyme Q (CoQH
GTP cyclohydrolase-1 (GCH1) serves as the enzyme that limits the rate of tetrahydrobiopterin (BH4) biosynthesis. Both BH4 and its oxidized form, dihydrobiopterin (BH2), can inhibit lipid peroxidation and ferroptosis. Additionally, BH4 indirectly promotes CoQ synthesis by converting phenylalanine to tyrosine. This process helps cells protect against ferroptotic death [49].
Therefore, the four parallel antioxidant pathways of GPX4, FSP1, DHODH, and GCH1 work together to resist lipid peroxidation and subsequently suppress ferroptosis.
Ferritinophagy is a vital pathway that contributes to the buildup of
intracellular iron. NCOA4, a cargo receptor responsible for ferritinophagy, is
overexpressed, resulting in the autophagic breakdown of ferritin and an increase
in Fe
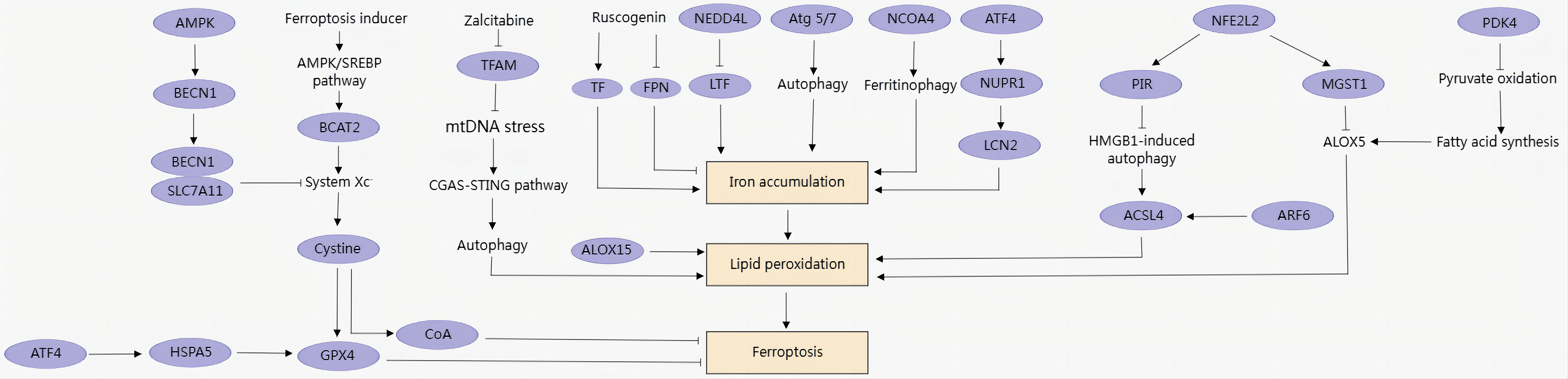
Lipid metabolism plays a vital role in ferroptosis in various malignancies, including pancreatic cancer. The combination of PUFAs and PLs, catalyzed by ACSL4, leads to the formation of PUFA-PL. ADP Ribosylation Factor 6 (ARF6) negatively regulates ACSL4 levels, thereby regulating the sensitivity of PDAC cells to RAS-selective lethal 3 (RSL-3)-mediated lipid peroxidation and influencing the occurrence of ferroptosis [55]. Pirin (PIR) is a nuclear redox-sensitive regulator and a target gene of nuclear factor, erythroid 2-like 2 (NFE2L2). It plays a role in inhibiting ferroptosis in cancer cells, specifically in PDAC, by restricting the autophagy induced by high-mobility group box 1 (HMGB1) and subsequently negatively regulating ACSL4 levels [56]. The oxidation of PUFA, catalyzed by LOX, is a critical step in ferroptosis. Knockdown of arachidonate 15-lipoxygenase (ALOX15) significantly reduces erastin- or RSL3-mediated ferroptosis, while overexpression or using an ALOX15 activator can enhance ferroptosis in PDAC cells [57]. Luciferase reporter gene assays demonstrated that microsomal glutathione S-transferase 1 (MGST1) is the target gene of NFE2L2, a crucial cellular antioxidant regulator. Activation of MGST1 by NFE2L2 contributes to its binding with arachidonate 5-lipoxygenase (ALOX5), resulting in reduced lipid peroxidation and suppression of ferroptosis in cancer cells, specifically in PDAC [58]. In the context of elevated glucose levels, downregulation of pyruvate dehydrogenase kinase 4 (PDK4) mediated by SLC2A1 promotes ferroptosis in pancreatic cancer cells. This promotion occurs via enhanced pyruvate oxidation-mediated fatty acid synthesis and ALOX5-dependent lipid peroxidation [59]. These studies emphasize the role of different modulators in regulating lipid metabolism in PDAC, offering a novel perspective for the prevention and treatment of PDAC.
The antioxidant system, including the GSH/GPX4 axis, is a protective mechanism
that counteracts lipid peroxidation and scavenges ROS. It acts a crucial function
in the regulation of ferroptosis. Ferroptosis inducers such as erastin,
sorafenib, and sulfasalazine can activate the AMP-activated protein kinase
(AMPK)/sterol response element binding protein 1 (SREBP1) pathway through
ferritinophagy. Branched-chain amino acid aminotransferase 2 (BCAT2), a pivotal
enzyme participated in sulfur amino acid metabolism, is overexpressed to enhance
glutamate synthesis, which in sequence increases the activity of system
X
| Compound | Positive or Negative regulators | Mechanism | Refs |
| ALOX15 | Positive | Promote ROS production | [57] |
| AMPK | Positive | Inhibit BCAT2 transcription | [60] |
| ARF6 | Negative | Negatively regulate ACSL4 level | [55] |
| Atg5/7 | Positive | Promote the formation of the autophagosome and ferroptosis | [50] |
| BCAT2 | Negative | Increase synthesis of glutamate | [60] |
| BECN1 | Positive | Bind to SLC7A11 and block system X |
[61] |
| CoA | Negative | cooperate with GSH to downregulate ferroptosis | [63] |
| DMT1 | Positive | Promote iron uptake | [24] |
| FPN1 | Negative | Promote iron export | [29] |
| GPX4 | Negative | Inhibit lipid peroxidation | [44] |
| Hepcidin | Positive | Induce FPN1 internalization | [29] |
| HSPA5 | Negative | Protect against degradation of GPX4 | [62] |
| LCN2 | Negative | Decrease iron accumulation | [54] |
| LTF | Positive | Increase iron accumulation | [53] |
| MGST1 | Negative | Bind to ALOX5 and limit lipid peroxidation | [58] |
| NCOA4 | Positive | Induce ferritinophagy | [26, 50] |
| NEDD4L | Negative | Promote LTF protein degradation | [53] |
| NFE2L2 | Negative | Mediate MGST1 upregulation during ferroptosis | [58] |
| NUPR1 | Negative | Promote LCN2 expression | [54] |
| PDK4 | Negative | Inhibit pyruvate oxidation and subsequent lipid peroxidation | [59] |
| PIR | Negative | Limit oxidative damage of DNA | [56] |
| SREBP1 | Positive | Inhibit BCAT2 transcription | [60] |
| TFAM | Negative | Inhibit autophagy-dependent ferroptosis | [51] |
| TFR1 | Positive | Increase iron level | [23] |
Abbreviations: PDAC, pancreatic ductal adenocarcinoma; ALOX15, 15-lipoxygenase; AMPK, AMP-activated protein kinase; ARF6, ADP Ribosylation Factor 6; Atg5/7, autophagy-related genes 5/7; BCAT2, branched-chain amino acid aminotransferase 2; BECN1, beclin 1; CoA, coenzyme A; DMT1, divalent metal transporter 1; FPN1, ferroportin 1; GPX4, glutathione peroxidase 4; HSPA5, heat shock 70 kDa protein 5; LCN2, lipocalin 2; LTF, lactotransferrin; MGST1, microsomal glutathione S-transferase 1; NCOA4, nuclear receptor coactivator 4; NEDD4L, neuronal precursor cell-expressed developmentally downregulated 4-like protein E3 ubiquitin ligase; NFE2L2, nuclear factor, erythroid 2-like 2; NUPR1, nuclear protein 1, transcriptional regulator; PDK4, pyruvate dehydrogenase kinase 4; PIR, pirin; SREBP1, sterol response element binding protein 1; TFAM, transcription factor A, mitochondrial; TFR1, transferrin receptor 1.
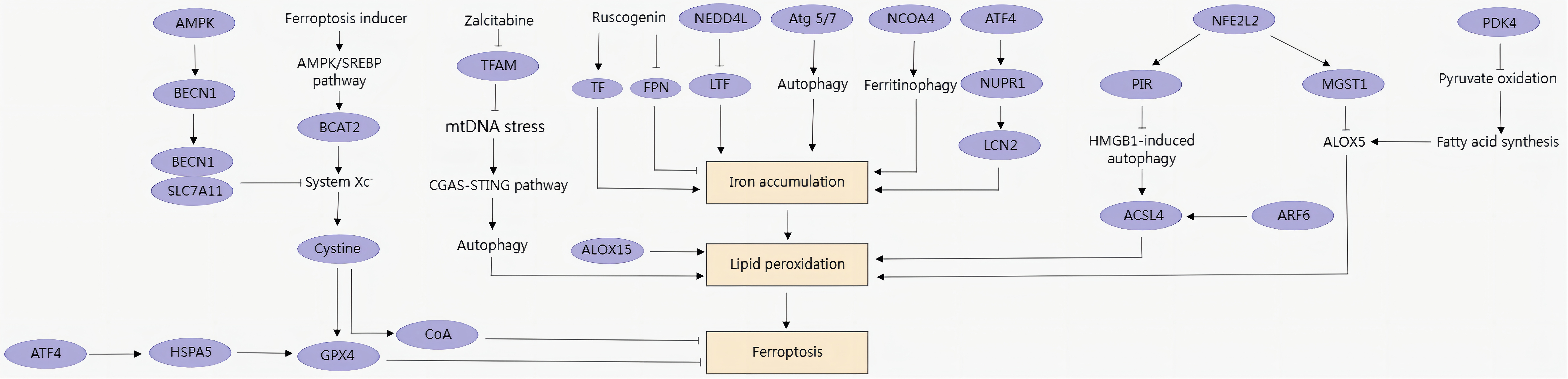 Fig. 2.
Fig. 2.Interrelationship between ferroptosis and PDAC.
Systemic therapy refers to the use of conventional cytotoxic and targeted agents to induce tumor cell death and exert anti-cancer effects. One important mechanism of tumor cell death is ferroptosis, which also inhibits tumor cell growth. Increasing evidence suggests that inducing ferroptosis in tumor cells could be a promising new therapeutic strategy. Furthermore, ferroptosis inducers have the potential to synergize with traditional antitumor drugs in order to inhibit tumor progression.
Despite the challenges of drug resistance and poor efficacy, gemcitabine remains the first-line chemotherapy drug for PDAC patients. Latest studies have verified that ferroptosis plays a crucial role in overcoming gemcitabine resistance [64]. Mechanistically, the overexpression of HSPA5 protects GPX4 from degradation, which in turn inhibits lipid peroxidation in ferroptosis. In pancreatic cancer, inhibiting the HSPA5-GPX4 pathway in a ferroptosis-mediated manner has been shown to reverse gemcitabine resistance [62]. Gene expression profiling revealed that F-box and WD repeat domain-containing 7 (FBW7) plays a crucial role in regulating ferroptosis in pancreatic cancer through its involvement in lipid peroxidation. Targeting FBW7 enhances the cytotoxicity of gemcitabine in PDAC by inducing both ferroptosis and apoptosis [65]. Therefore, inducing ferroptosis in pancreatic cancer cells could potentially reverse gemcitabine resistance and offer a new strategy for systemic therapy of pancreatic cancer.
Artemisinin, a Chinese-derived antimalarial drug, and its derivatives, such as artesunate (ART) and dihydroartemisinin (DHA), have shown multiple mechanisms of action, including ferroptosis, which exhibits potent anticancer activity [66, 67]. ART promotes the generation of ROS through a lysosomal iron-dependent manner, inducing ferroptosis in PDAC while not affecting non-neoplastic human pancreatic ductal epithelial cells [68]. In KRAS-mutant pancreatic cancer, the 78-kDa Glucose-regulated protein 78 (GRP78), an active molecular chaperone in the endoplasmic reticulum, has a negative regulatory effect on ART-induced ferroptosis [69]. DHA induces ferroptotic cell death by upregulating the expression of arachidonate 12-lipoxygenase (ALOX12), thereby inhibiting the proliferation of pancreatic cancer cells [70]. Therefore, artemisinin derivatives either alone or in conjunction with other medications may offer fresh treatment options for PDAC patients. However, further research is needed to explore whether the least toxic artemisinin derivatives have the ability to induce ferroptotic activity.
In addition to gemcitabine and artemisinin derivatives, there have been recent reports of more drugs affecting ferroptosis in pancreatic cancer cells. Baicalein, an efficient ferroptosis inhibitor in pancreatic cancer cells, not only inhibits the production of malondialdehyde (MDA), the end product of lipid peroxidation, but also prevents iron accumulation, protects against GSH depletion and GPX4 degradation, consequently preventing ferroptotic cell death [71]. Irisin, a multi-functional myokine, has been found to increase iron accumulation and the production of the lipid peroxidation end product MDA. It also downregulates GSH and inhibits the expression of SLC7A11, ultimately leading to ferroptotic cell death in PDAC [72]. QD394, a novel redox modulator, destabilizes the GPX4 protein and lessens the GSH/oxidized glutathione (GSSG) ratio, resulting in iron- and ROS-mediated ferroptosis in pancreatic cancer cells [73]. Cotylenin A (CN-A) and phenethyl isothiocyanate (PEITC), a plant growth regulator and a dietary antitumor phytochemical compound, have been found to synergistically trigger ferroptosis in human PDAC cells in a ROS-dependent pattern. However, CN-A or PEITC alone do not have the same effect [74]. Sorafenib, the first approved anticancer drug capable of inducing ferroptosis in different tissues, including liver and kidney cancers, is found to be less effective against PDAC [75, 76]. SLC1A5, a ferroptosis regulatory gene, has been positively associated with sorafenib sensitivity in pancreatic cancer cells [77]. Therefore, it appears that the induction of ferroptosis can help address the limitations of sorafenib in PDAC treatment. In conclusion, investigating novel ferroptosis inducers or combination therapies to enhance tumor sensitivity to ferroptosis could offer a fresh approach to PDAC treatment.
Immunotherapy, which utilizes immune checkpoint inhibitors (ICIs), has markedly
improved the prognosis for cancer patients. ICIs function by stimulating
cytotoxic T cells and enhancing antitumor immunity through the targeting of
specific proteins, including cytotoxic T lymphocyte-associated protein 4 (CTLA4),
programmed cell death protein 1 (PD-1), and its ligands programmed death-ligand 1
(PD-L1) [78]. Research studies have indicated that the release of interferon
gamma (IFN
The effect of radiation therapy in the treatment of locally advanced pancreatic
cancer (LAPC) has been a subject of controversy. However, studies have shown that
single-modality radiotherapy can provide potential benefits for LAPC patients
[85]. With a deeper understanding of the mechanism of ferroptosis, radiotherapy
may become more clinically significant in the therapy of PDAC. Research has
demonstrated that radiotherapy not only induces ferroptosis in various cancer
cells through promoting the ACSL4 expression and ROS production, but also
elevates the adaptability of SLC7A11 or GPX4, leading to radioresistance. It has
been observed that ferroptosis inducers can render tumor cells more sensitive to
radiotherapy. Furthermore, the combination of ferroptosis inducers and radiation
therapy synergistically promotes ferroptosis in various cancer cells such as lung
cancer, breast adenocarcinoma and fibrosarcoma [86]. Additionally, IFN
Nanotherapy involves the encapsulation of drugs in nanoparticles, enabling them
to effectively bypass biological barriers and deliver drugs directly to tumor
cells. Nanoparticles not only accumulate in tumors through the enhanced
permeability and retention effect (EPR), but also interact with tumor cells,
thereby increasing selectivity and reducing adverse drug reactions [89]. The
development of nanomedicine targeting ferroptosis is a promising approach to
enhance the effectiveness of existing ferroptosis inducers and improve the
prognosis of PDAC patients. In order to overcome the limitations of conventional
chemotherapy drugs, researchers prepared biocompatible MnFe
| Compound | Mechanism | Refs |
| ART | Promote ROS production in lysosomal iron-dependent manner | [68] |
| Baicalein | Limit iron accumulation and lipid peroxidation and suppresses GSH depletion and GPX4 degradation | [71] |
| CN-A and PEITC | Induce the generation of ROS | [74] |
| Cyst(e)inase | Synergistically promote lipid peroxidation with IFN |
[79] |
| DHA | Upregulate ALOX12 | [70] |
| Erastin | Inhibit system X |
[11] |
| GRP78 | Negatively regulate ART-induced ferroptosis | [69] |
| Irisin | Increase iron accumulation, ROS level and GSH depletion | [72] |
| MFC-Gem | Promote ROS generation and GSH depletion | [90] |
| PDBA@RSL-3 NPs | Induce GSH depletion and GPX4 inhibition and stimulate antitumor immunity | [92] |
| QD394 | Destabilize the GPX4 protein and decrease the GSH/GSSG ratio | [73] |
| Rapamycin | Induce autophagy-dependent ferroptosis | [93] |
| Ruscogenin | Increase intracellular iron | [52] |
| RSL-3@PVs | Induce ferroptosis and tumor vascular embolism | [91] |
| Zalcitabine | Trigger mitochondrial DNA stress and subsequent autophagy-dependent ferroptosis | [51] |
Abbreviations: ART, artesunate; CN-A, cotylenin A; DHA, dihydroartemisinin;
GRP78, glucose-regulated protein 78; MFC-Gem, MnFe
Ferroptosis, a lately discovered modality of non-apoptotic cell death, is distinguished by iron accumulation, ROS generation, and lipid peroxidation. Inducing ferroptosis in PDAC cells not only inhibits tumor growth but also enhances the sensitivity of PDAC to systemic therapy, immunotherapy, radiotherapy, and nanotherapy when combined with other antitumor drugs. This article focuses on the regulatory mechanisms of ferroptosis, its connection to PDAC, and the research progress in PDAC treatment. Despite the potential of ferroptosis in PDAC treatment, there are several noteworthy challenges that need to be addressed. Firstly, the comprehension of the underlying mechanisms of ferroptosis is still in its nascent stages. Although diverse major regulators of ferroptosis in PDAC have been identified, further research is required to explore potential signaling pathways and key regulators. Secondly, the development of drugs targeting the ferroptosis pathway and the exploration of ferroptosis’s role in immunotherapy and nanotherapy hold promise for advancing PDAC treatment strategies. However, although several drugs targeting the ferroptosis pathway have shown promising results in preclinical studies using cells and animal models, their effects on humans and the optimal dosages for inducing ferroptosis have not yet been determined. Furthermore, the efficacy and safety of combination therapy using ferroptosis inducers still require further investigation. Additionally, the development of resistance to ferroptosis inducers poses a challenge to their clinical application.
To address these issues, future research should focus on elucidating the underlying molecular regulation of ferroptosis and its specific targets for the treatment of PDAC. The induction of ferroptosis by anti-tumor drugs has shown potential in exerting anti-cancer activity. This discovery offers new possibilities and avenues for the clinical treatment of pancreatic cancer. Moreover, efforts should be made to minimize drug side effects and overcome drug resistance through the use of combination therapy and the development of nano-medicines. Overall, ferroptosis, as a recently discovered form of non-apoptotic cell death, demonstrates significant potential as an anticancer mechanism. We believe that targeting ferroptosis and its associated mechanisms will facilitate the discovery of new ferroptosis inducers and novel combination therapy strategies, ultimately enhancing the prognosis and quality of life for PDAC patients.
CY and QD collected the literatures and interpreted the data. CY and QD wrote the original manuscript. HB organized the tables. YG and ZX drew the figures. JL and XJ analyzed the contents from the reviewed literature. YX and XZ designed manuscript conception and critically revised manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We are grateful to the Department of Hepatopancreatobiliary Surgery, The Second Affiliated Hospital of Harbin Medical University for their help with this research.
This work was supported by the Opening Project of Shaanxi Provincial Key Laboratory of Infection and Immune Diseases (2023-KFMS-2); Opening Project of State Key Laboratory of Oncology in South China (HN2023-02); Key Laboratory of Human Development and Disease Research, Guangxi Medical University, Education Department of Guangxi Zhuang Autonomous Region (RTFY202301); Initiative Research Fund of Heilongjiang Postdoctoral Science Foundation; Beijing Xisike Clinical Oncology Research Foundation (Y-QL202201-0020); Opening Project of Key Laboratory of Tumor Immunology and Pathology, Army Medical University, Ministry of Education (2022jsz801); Opening Project of Anhui Province Key Laboratory of Translational Cancer Research, Bengbu Medical College (KFKT202301); Shenzhen Science and Technology Program (ZDSYS20210623092001003); Thematic Research Support Scheme of State Key Laboratory of Liver Research, The University of Hong Kong (SKLLR/TRSS/2022/08).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.