






 , Najeeb Ur Rehman 1, Abdul Samad 2, Wasim Ahmad 3
, Najeeb Ur Rehman 1, Abdul Samad 2, Wasim Ahmad 31 Department of Pharmacology & Toxicology, College of Pharmacy, Prince Sattam Bin Abdulaziz University, Al-Kharj 11942, Saudi Arabia
2 Pharmacy Department, Tishk International University, Erbil 44001, Iraq
3 Department of Pharmacy, Mohammed Al-Mana College for Medical Sciences, Dammam 34222, Saudi Arabia
Abstract
Background: Medicinal herbs are frequently used for the management of
gastrointestinal disorders because they contain various compounds that can
potentially amplify the intended therapeutic effects. Cuminaldehyde is a
plant-based constituent found in oils derived from botanicals such as cumin,
eucalyptus, myrrh, and cassia and is responsible for its health benefits. Despite
the utilization of cuminaldehyde for several medicinal properties, there is
currently insufficient scientific evidence to support its effectiveness in
treating diarrhea. Hence, the present investigation was carried out to evaluate
the antidiarrheal and antispasmodic efficacy of cuminaldehyde, with detailed
pharmacodynamics explored. Methods: An in vivo antidiarrheal
test was conducted in mice following the castor oil-induced diarrhea model, while
an isolated small intestine obtained from rats was used to evaluate the detailed
mechanism(s) of antispasmodic effects. Results: Cuminaldehyde, at 10 and
20 mg/kg, exhibited 60 and 80% protection in mice from episodic diarrhea
compared to the saline control group, whereas this inhibitory effect was
significantly reversed in the pretreated mice with glibenclamide, similar to
cromakalim, an ATP-dependent K
Keywords
- cuminaldehyde
- antispasmodic
- K+ channel opener
- Ca++ channel blocker
- glibenclamide
Gastrointestinal disorder is characterized by dysfunctions in intestinal motility, which can manifest as abdominal pain, constipation, or diarrhea [1, 2]. Gastrointestinal diseases, particularly constipation and diarrhea, affect 70% of the population worldwide [3]. Diarrhea is a significant cause of death in underdeveloped nations and is responsible for 1.5–2 million deaths in children under the age of five [4].
Medicinal herbs are commonly chosen for the treatment of gastrointestinal diseases due to the presence of several constituents that might enhance the desired effects or neutralize any potential negative effects [5, 6]. As a result, they are generally considered safe for long-term usage. Herbal medicines show potential uses in the future because, for most plants, their chemical composition and pharmacological activities have yet to be thoroughly explored [7]. Plant derivative extract has also been used as a remedy, and many clinically valuable drugs have been derived from plant materials [8, 9]. The seed husk of Plantago ovata, often known as psyllium husk, is a widely used plant-based medicine that both traditional healers and modern physicians favor as a complementary and alternative therapy [10]. Global interest in traditional medicine has surged, prompting initiatives to oversee and standardize herbal medications and traditional medicine [11]. Due to their perceived safety, natural products are increasingly favored over synthetic items, leading to a decline in the use of synthetic drugs [12].
Cumin seeds are sourced from the herb Cuminum cyminum Linn, which is a member of the Apiaceae family [13]. The seeds of cumin (C. cyminum L.) are popularly utilized as a spice due to their characteristic aroma. Additionally, they are frequently employed in traditional medicine to address various ailments such as chronic diarrhea, dyspepsia, acute gastritis, diabetes, and cancer [13]. The literature provides extensive evidence of the biological and medicinal effects of cumin, which are mostly attributed to its bioactive components, including terpenes, phenols, and flavonoids [14, 15]. An investigation was conducted to evaluate the effect of cumin seed extract on diarrhea in albino rats induced with castor oil [16]. A natural monoterpenoid, cuminaldehyde, is a component of cumin oil and oils such as eucalyptus, myrrh, and cassia [17]. The health-promoting effects of cumin are attributed to cuminaldehyde [18], which has other functionalities such as anticancer [19], antidiabetic [20], neuroprotection [21], and anti-inflammation [22].
Although cuminaldehyde has been employed for several therapeutic purposes, there still needs to be more scientific evidence to substantiate its efficacy in the treatment of diarrhea and the detailed mechanism(s) involved. Therefore, the present study was designed to investigate the effectiveness with an exploration of the precise mechanism(s) involved in the potential gastrointestinal inhibitory effects of cuminaldehyde using in vivo, ex vivo, and in silico experiments.
Cuminaldehyde, carbamylcholine (CCh), loperamide, acetylcholine perchlorate (ACh), isoprenaline, verapamil, and papaverine were obtained from Sigma (St. Louis, MO, USA). Reagents (salts) to prepare a physiological buffer solution (Tyrode) were procured from Merck (Darmstadt, Germany). All chemicals used were of analytical grade, with the exception of castor oil, which was procured from a local pharmacy.
The Wistar albino rats (180–200 g) and Swiss albino mice (26–30 g) were
obtained from the Animal Care Unit at the College of Pharmacy, Prince Sattam bin
Abdulaziz University (PSAU), Saudi Arabia. They were kept at an optimal
temperature of 22
A previously reported method was followed to test the possible antidiarrheal effect of cuminaldehyde [24]. A total of thirty-five mice were divided randomly into groups, ensuring that each group consisted of five mice. After fasting for twenty-four hours, the mice in the first group were given a saline solution (10 mL/kg) through oral gavage, and they were labeled as the negative control group. Following the pilot screening to determine the effective dose, the second and third groups (test groups) were subjected to two doses of cuminaldehyde (10 and 20 mg/kg). The mice in the fourth group were exposed to cromakalim (10 mg/kg). The mice in the fifth, sixth, and seventh groups were given a pretreatment of glibenclamide (3 mg/kg) one hour before receiving cuminaldehyde or cromakalim. One hour after administration, all mice were orally administered castor oil (10 mL/kg) using a 1 mL syringe. Each mouse was then placed in an individual cage with a blotting sheet on the floor. After 4 hours, each cage blotting sheet was examined for the presence or absence of characteristic diarrheal droppings by a blind observer. The presence of protection was observed when no instances of diarrhea were detected, as previously documented by Jebunnessa et al. [25].
Prior to conducting ex vivo studies, rats underwent a 24-hour fasting
period. Subsequently, their cervical dislocation was carried out under mild
anesthesia. The ileum tissue was isolated using a previously described procedure
[26]. After being isolated, specific sections of ileum tissues measuring 2–3 cm
in length were thoroughly cleansed to remove nearby tissues and fecal matter.
These cleaned tissues were then placed in an emkaBath apparatus (Paris, France)
and connected to a transducer and IOX software (version 2.9.10.6, emka
technologies, SAS, Paris, France). The tissue baths were filled with 20 mL of
fresh tyrode solution (pH 7.4) and gassed with carbogen. The temperature was
adjusted to 37 °C. A force of 1 gram was exerted by turning the
transducer knob in a clockwise direction, and the tissues were allowed to
stabilize for 30 minutes while being exposed to numerous doses of acetylcholine
(0.3 µM). Following stabilization, sustained contractions were
induced using low (25 mM) and high (80 mM) K
After examining the relaxation effect of cuminaldehyde on high levels of
K
In order to get a molecular understanding of the predominant involvement of potassium channel activation by cuminaldehyde observed in the ex vivo assays, in silico molecular modeling studies were performed on three distinct isoforms of ATP-dependent potassium channels with Protein Data Bank (PDB) IDs 7MJP, 7S61, and 6C3O. The protein structures of the receptors under consideration for study were obtained from the RCSB Protein Data Bank (RCSB PDB) website (https://www.rcsb.org/). The co-crystalline ligands of each receptor were isolated and redocked to validate the docking process. As indicated in our prior study work [32], Autodock Vina [33] was utilized for docking. The interaction between the docking confirmations and potassium channel receptors was observed using the Biovia Solutions Discovery Studio 4 programs [34]. The downloaded proteins were treated to remove water molecules, insert polar hydrogens, add Coleman charges, and store them in PDBQT format for docking. The structures of cuminaldehyde and cromakalim were retrieved in Stromal cell-derived factor (SDF) format from PubChem. In the next step, they were converted from SDF to PDB format and then written in the appropriate PDPQT format.
A chemical’s ADMET (absorption, distribution, metabolism, excretion, and toxicity) characteristics are critical for developing it as a lead medicine in the drug discovery cascade. The ADMET characteristics of cuminaldehyde were determined using the pkCSM software (https://biosig.lab.uq.edu.au/pkcsm/prediction), a web-based application [35]. Because this software allows smiles, the smiles were copied from PubChem and then sent to pkCSM for comprehensive profile prediction of the cuminaldehyde molecule.
The results were provided as the mean
In mice, cuminaldehyde exhibited dose-dependent protection from diarrhea evoked by castor oil, while the saline group did not produce any protection. Preincubated mice with cuminaldehyde (10 and 20 mg/kg) exhibited 60% and 80% protection, whereas cromakalim (positive control group) produced 100% protection from diarrhea at 10 mg/kg. Glibenclamide (3 mg/kg) preincubation suppressed the antidiarrheal actions of cuminaldehyde and cromakalim, as detailed in Table 1.
| Treatment (p.o.) | Dose (mg/kg) | No. of mice with diarrhea (out of 5) | % protection |
| Saline + Castor oil (mL/kg) | 10 | 5 | 0 |
| Cuminaldehyde + Castor oil | 10 + 10 | 2 |
60 |
| Cuminaldehyde + Castor oil | 20 + 10 | 1 |
80 |
| Cromakalim + Castor oil | 10 + 10 | 0 |
100 |
| Cuminaldehyde + Glibenclamide + Castor oil | 10 + 3 + 10 | 4 |
20 |
| Cuminaldehyde + Glibenclamide + Castor oil | 20 + 3 + 10 | 2 | 60 |
| Cromakalim + Glibenclamide + Castor oil | 10 + 3 + 10 | 4 |
20 |
When checked against low and high K
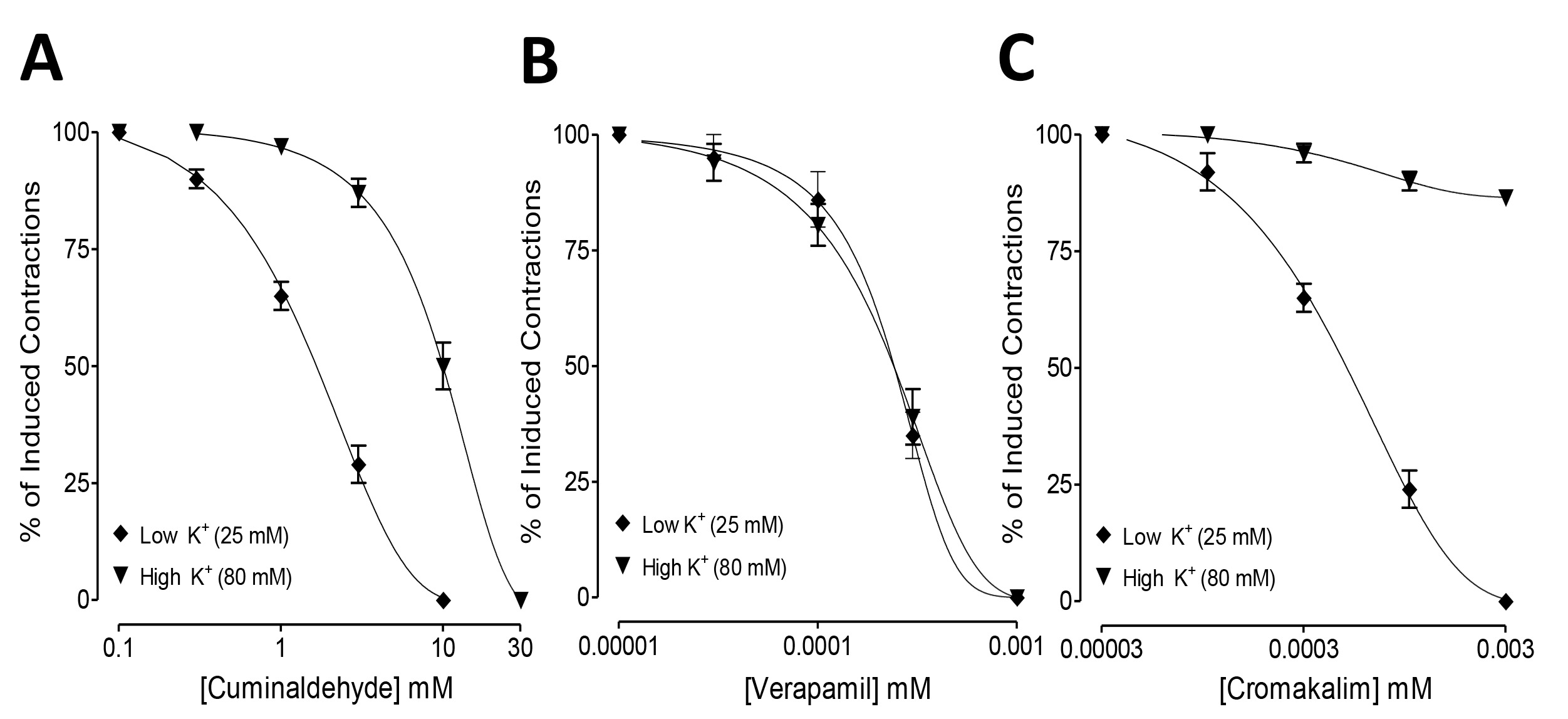 Fig. 1.
Fig. 1.Inhibitory concentration-response curves showing the comparison
of (A) cuminaldehyde, (B) cromakalim, and (C) verapamil for the inhibitory effect
against low K
The inhibitory actions of cuminaldehyde and cromakalim against low K
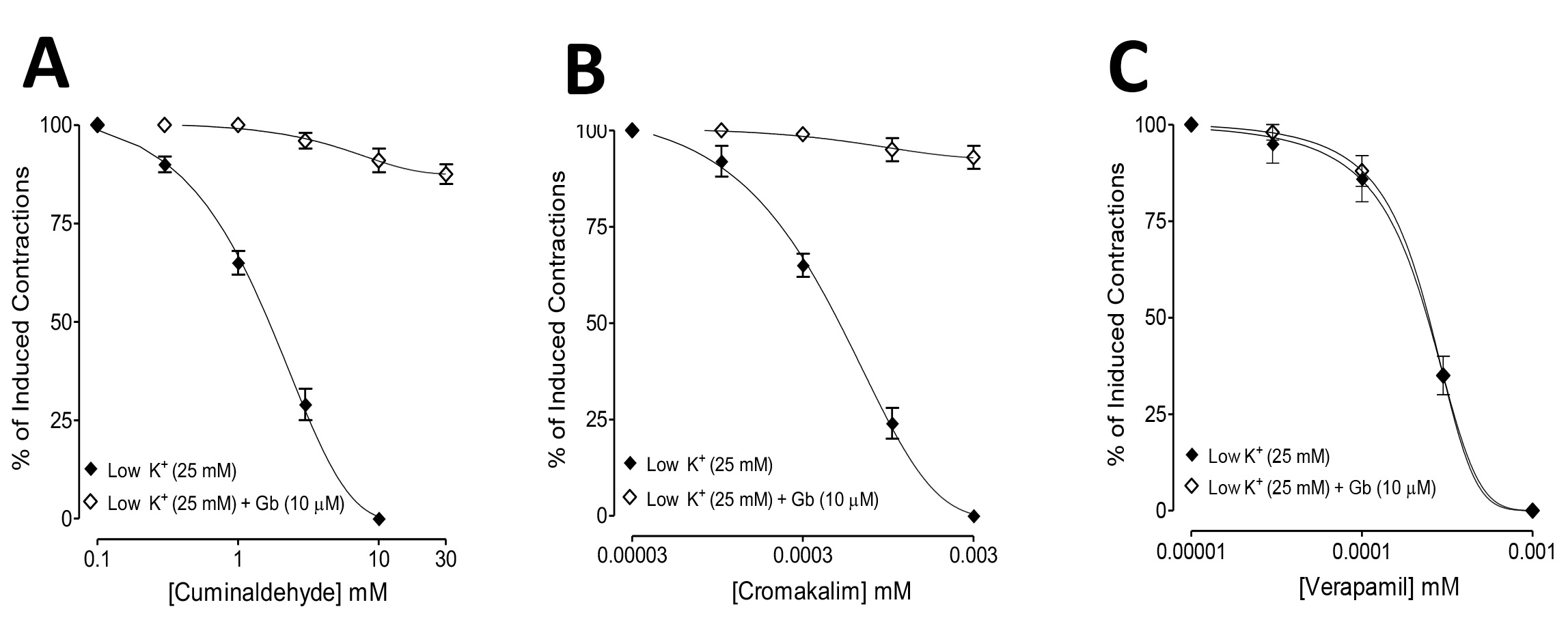 Fig. 2.
Fig. 2.Comparisons of the inhibitory concentration-response curves of
(A) cuminaldehyde, (B) cromakalim, and (C) verapamil against low K
To further confirm the Ca
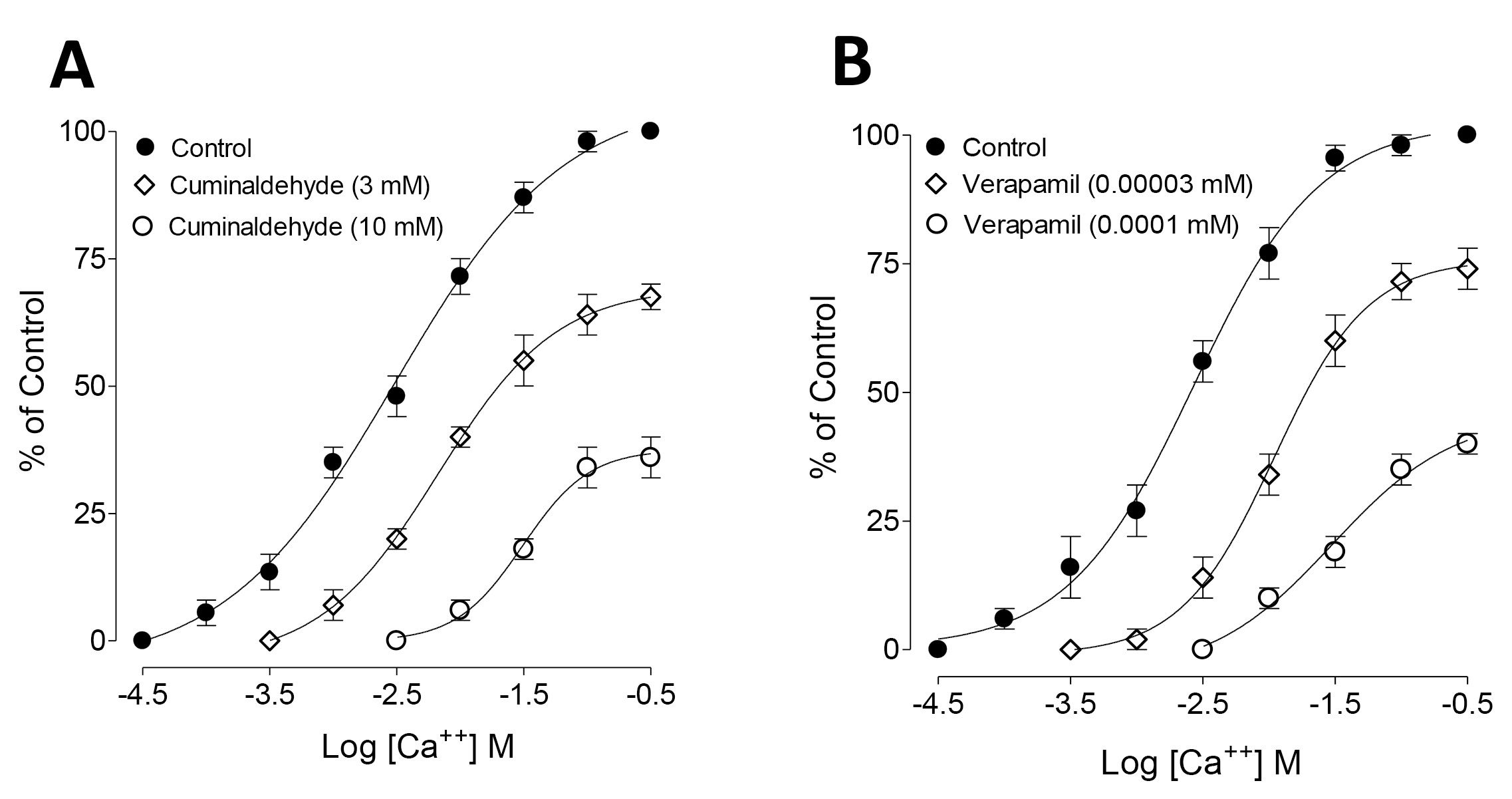 Fig. 3.
Fig. 3.Concentration-response curves of Ca
ATP-dependent potassium channel receptors are complex macromolecules consisting of sulfonylurea receptor (SUR) and inward rectifier Kir subunits, and they have been reported in RCSb in propeller and quatrefoil forms. The pore opening in Kir is linked to coordinated structural changes in the ATP-binding site and the channel gate. The first chosen protein, 7S61, when subjected to molecular docking with cuminaldehyde and cromakalim, exhibited binding energies of –5.8 and –6.8 kcal/mol, respectively. For further confirmation, another human KATP (ATP-sensitive potassium channel) bound to ATP and ADP in quatrefoil form was downloaded and docked with the same two compounds considered in the study. The binding energies exhibited by cuminaldehyde and cromakalim this time were –6.8 and –7.8 kcal/mol, respectively. For further legitimization of this study, another ATP-dependent potassium channel, 7MJP, was considered for the docking study of the molecules in question, and they exhibited binding energies of –5.0 and –6.9 kcal per mole. The pattern of binding energy among all three receptors was found to be more or less similar. Cuminaldehyde and cromakalim have exhibited various forms of molecular interactions with the KATP proteins, as shown in Table 2 (Conventional H-Bond, Vander Waal, pi-pi stacking, Amide-pi stacking, Sigma-Pi stacking). Cuminaldehyde exhibited three conventional hydrogen bond interactions with the protein 7S61, while cromakalim exhibited only one. The hydrogen bond distance is also considered a crucial parameter for strong binding between ligand and receptor, the lesser the stronger. Cuminaldehyde showed shorter H-bond distances with 7S61 {a-1.91 (Ser721), b-2.32 (Lys720), c-2.48 (Ser722)}, on the other hand, cromakalim had longer {2.67 (Ser409)}. As shown in Fig. 4A–D, cuminaldehyde showed interactions with 6 residues (Trp-E689, Ser-E1483, Phe-E1482, Ser-E722, Ser-E721, Lys-E720), and cromakalim showed interactions with only 5 residues (Trp-E689, Ser-E1483, Phe-E1482, Ser-E409, Ser-E721). The KATP protein 6C3O, when overviewed for such interaction, cuminaldehyde exhibited interactions (Vander Waal, Sigma-Pi stacking) with 6 residues (Val679, Ile703, Ala726, Arg766, Phe851, Val784), while cromakalim showed interactions (Conventional H-Bond, carbon H-Bond, pi-pi stacking, Anion-Pi stacking) with 4 residues (Trp688, Glu1479, Ser1482, Gln1483), as shown in Fig. 5A–D. Interestingly, cuminaldehyde made two Vander Waal interactions with bond distances of 2.07 and 2.93 with Arg766. At the same time, cromakalim exhibited a conventional H-bond but with a larger distance of 2.99 with Gly1483. Likewise, when the KATP protein 7MJP was overviewed cuminaldehyde exhibited interaction (Conventional H-Bond, Vander Waal, Sigma-Pi stacking) with 7 residues (Ile199, Leu206, Ile271, Leu277, Ile280, Phe208, Ala355) while cromakalim shown interaction (Conventional H-Bond and Sigma-Pi stacking) with 4 residues (Thr190, Leu191, Arg216, Ile220) as shown in Fig. 6.
| Receptor | Ligands | Binding Energy | Type of Interactions | H-Bond Distance | Residues |
| 7S61 | Cuminaldehyde | –5.8 | Conventional H-Bond, Vander Waal, pi-pi stacking, Amide-pi stacking, Sigma-Pi stacking | a-1.91 (Ser721), b-2.32 (Lys720), c-2.48 (Ser722) | Trp-E689, Ser-E1483, Phe-E1482, Ser-E722, Ser-E721, Lys-E720 |
| Cromakalim | –6.8 | Conventional H-Bond, Vander Waal, carbon H-Bond, pi-pi stacking, Sigma-Pi stacking | a-2.67 (Ser 409) | Trp-E689, Ser-E1483, Phe-E1482, Ser-E409, Ser-E721 | |
| 6C3O | Cuminaldehyde | –6.8 | Vander Waal, Sigma-Pi stacking | a-2.07 & 2.93 (Arg: 766) | Val679, Ile703, Ala726, Arg766, Phe851, Val784 |
| Cromakalim | –7.5 | Conventional H-Bond, carbon H-Bond, pi-pi stacking, Anion-Pi stacking | a-2.99 (Gly1483) | Trp688, Glu1479, Ser1482, Gln1483 | |
| 7MJP | Cuminaldehyde | –5 | Conventional H-Bond, Vander Waal, Sigma-Pi stacking | a-2.69 (Phe208) | Ile199, Leu206, Ile271, Leu277, Ile280, Phe208, Ala355 |
| Cromakalim | –6.9 | Conventional H-Bond, Sigma-Pi stacking | a-2.23 (Ile220), b-2.86 (Thr190), c-2.29 (Leu191) | Thr190, Leu191, Arg216, Ile220 |
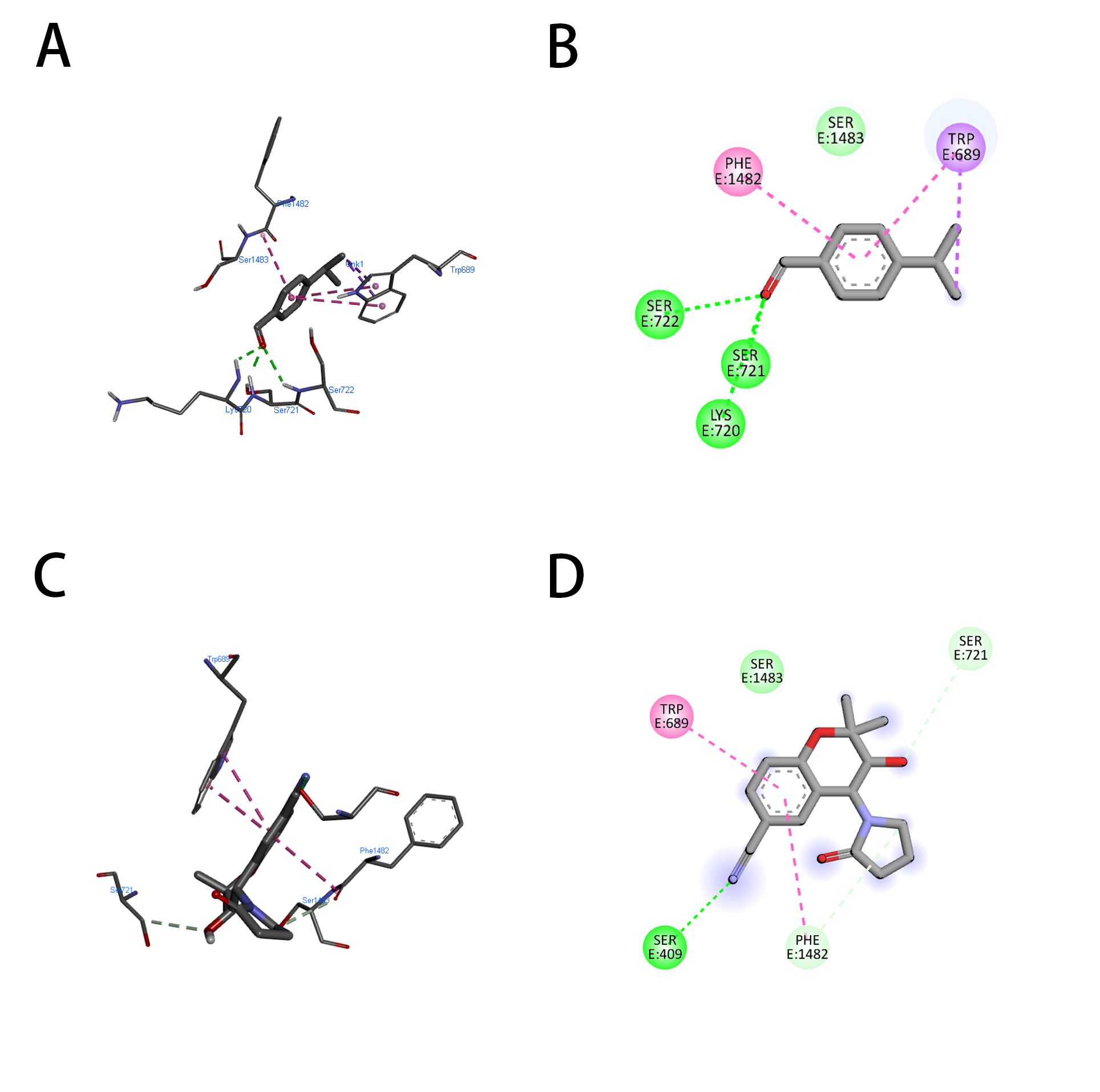 Fig. 4.
Fig. 4.Representation of the molecular interaction of cuminaldehyde (A,B) and cromakalim (C,D) with KATP (7S61). (A,C) 2-Dimensional and (B,D) 3-Dimensional.
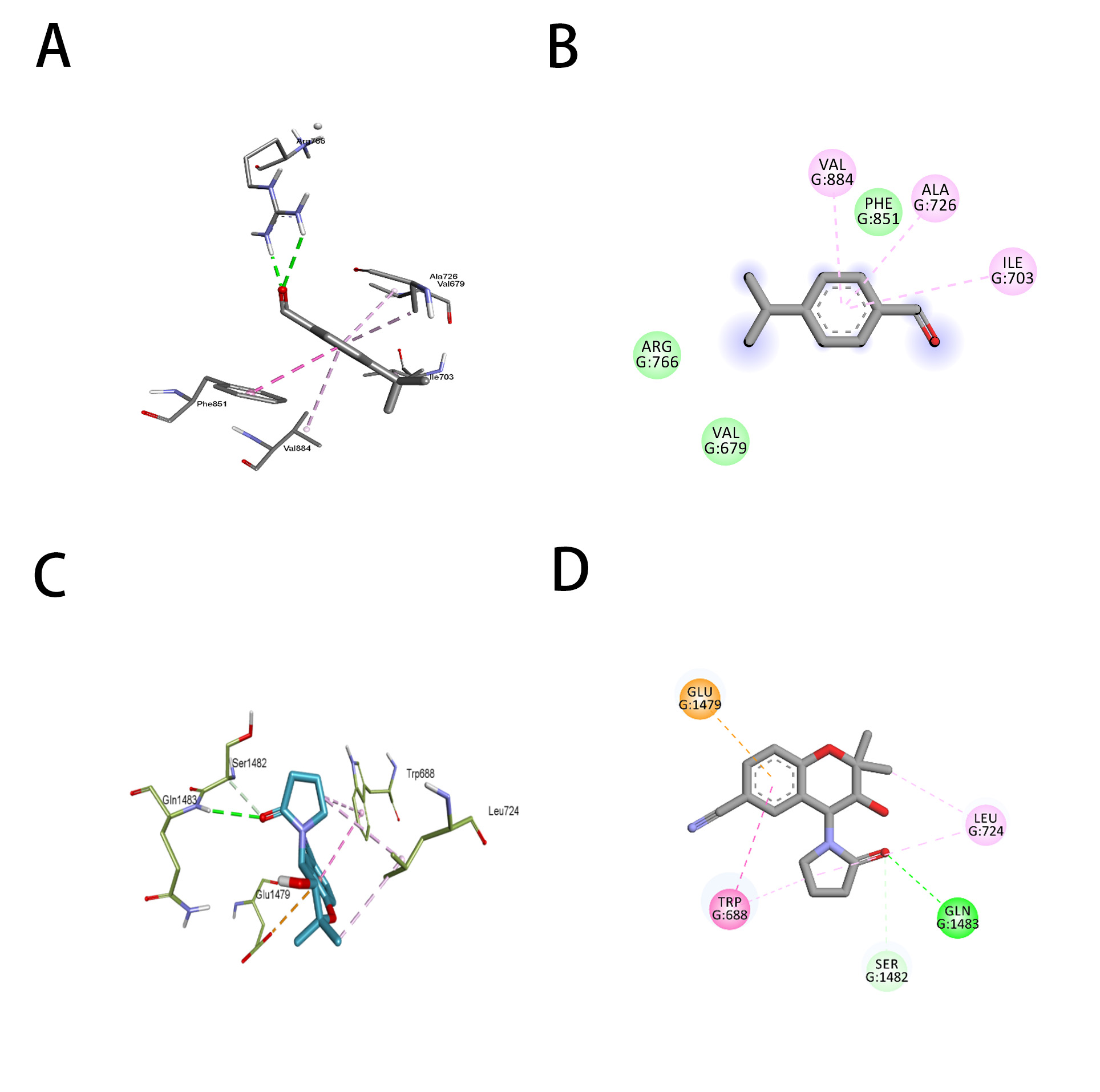 Fig. 5.
Fig. 5.Representation of the molecular interaction of cuminaldehyde (A,B) and cromakalim (C,D) with KATP (6C3O). (A,C) 2-Dimensional and (B,D) 3-Dimensional.
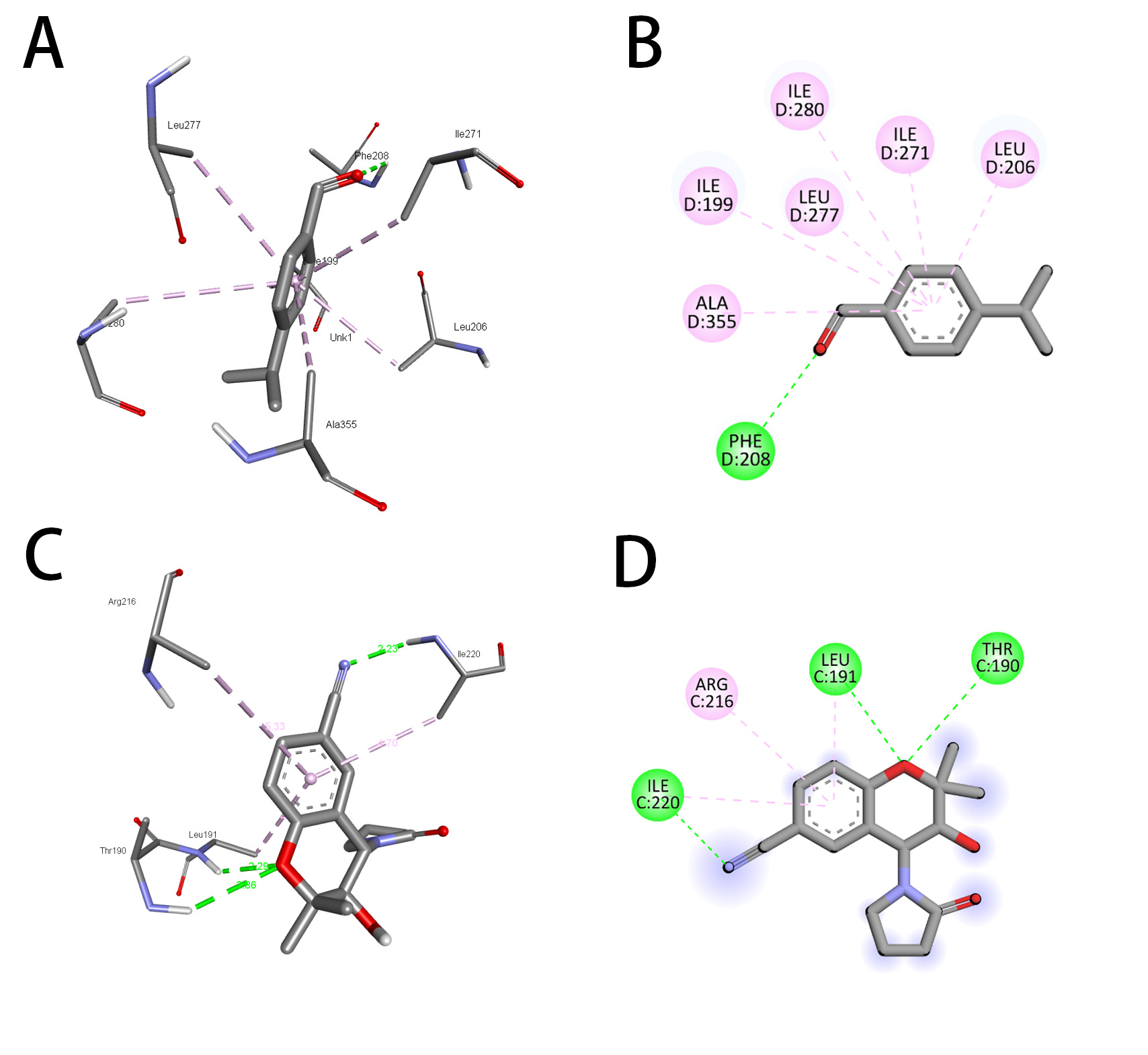 Fig. 6.
Fig. 6.Representation of the molecular interaction of cuminaldehyde (A,B) and cromakalim (C,D) with KATP (7MJP). (A,C) 2-Dimensional and (B,D) 3-Dimensional.
As shown in Table 3, cuminaldehyde has exhibited acceptable ADMET profiling. ADMET profiling is a must for establishing the drug-like properties of any molecule. The ADMET properties of cuminaldehyde were calculated by PKCSM software and exhibited an intestinal absorption close to 95%, and volume distribution (steady state) was found to be 0.324 (log L/kg). The log BB quotient for blood-brain barrier permeability was found to be 0.438, showing good BBB permeability. Cuminaldehyde does not inhibit any family of CYP450 enzymes except CYP1A2 and has a safe toxicity profile, except for being sensitive to the skin.
| Property | Model Name | Predicted Value | Unit |
| A | Water solubility | –2.966 | Numeric (log mol/L) |
| Caco2 permeability | 1.609 | Numeric (log Papp in 10 | |
| Intestinal absorption (human) | 95.844 | Numeric (% Absorbed) | |
| Skin Permeability | –1.196 | Numeric (log Kp) | |
| P-glycoprotein substrate | No | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | No | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | No | Categorical (Yes/No) | |
| D | VDss (human) | 0.324 | Numeric (log L/kg) |
| Fraction unbound (human) | 0.263 | Numeric (Fu) | |
| BBB permeability | 0.438 | Numeric (log BB) | |
| CNS permeability | –1.485 | Numeric (log PS) | |
| M | CYP2D6 substrate | No | Categorical (Yes/No) |
| CYP3A4 substrate | No | Categorical (Yes/No) | |
| CYP1A2 inhibitor | Yes | Categorical (Yes/No) | |
| CYP2C19 inhibitor | No | Categorical (Yes/No) | |
| CYP2C9 inhibitor | No | Categorical (Yes/No) | |
| CYP2D6 inhibitor | No | Categorical (Yes/No) | |
| CYP3A4 inhibitor | No | Categorical (Yes/No) | |
| E | Total Clearance | 0.227 | Numeric (log mL/min/kg) |
| Renal OCT2 substrate | No | Categorical (Yes/No) | |
| T | AMES toxicity | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.839 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | Categorical (Yes/No) | |
| hERG II inhibitor | No | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 1.7 | Numeric (mol/kg) | |
| Oral Rat Chronic Toxicity (LOAEL) | 2.194 | Numeric (log mg/kg_bw/day) | |
| Hepatotoxicity | No | Categorical (Yes/No) | |
| Skin Sensitisation | Yes | Categorical (Yes/No) | |
| T.Pyriformis toxicity | 0.766 | Numeric (log μg/L) | |
| Minnow toxicity | 0.819 | Numeric (log mM) |
ADMET, absorption, distribution, metabolism, excretion, and toxicity.
The current study tested cuminaldehyde, a plant-based derived compound, for its
antidiarrheal and antispasmodic actions in the in vivo, ex vivo
and in silico models. Diarrhea is one of the leading diseases of the
gastrointestinal system worldwide [36]. The castor oil-evoked diarrhea model,
considered one of the well-accepted models for antidiarrheal screening drugs
[37, 38], was followed. Cuminaldehyde exhibited a dose-dependent protection,
similar to cromakalim, a K
As cuminaldehyde was also found active against high K
Furthermore, to have a greater insight into cuminaldehyde and potassium channel activation binding, the molecular docking studies with the three receptors, namely 7S61, 6C3O, and 7MJP, proved to be of great importance. Cuminaldehyde has exhibited binding affinity to these receptors comparable to the cromakalim. In prima facie, the binding energy of cuminaldehyde was lesser than cromakalim, imparting the impression that the former has a lesser affinity. However, when the residues, binding interactions, and bond distances were considered, cuminaldehyde outweighed the molecular interaction. For instance, in docking with the receptor 7S61, the cromakalim showed only one conventional hydrogen bonding with a distance of 2.67 with the residue Ser-409. At the same time, the cuminaldehyde exhibited three hydrogen bonds {1.91 (Ser721), 2.32 (Lys720), 2.48 (Ser722)} with much lesser distance. Likewise, in docking with 6C3O, cromakalim showed conventional hydrogen bonding but with a distance of 2.99. At the same time, the cumin aldehyde exhibited Wonderwall interactions with the distances 2.07 and 2.93 (Arg766), which again proved cumin aldehyde to be a better alternative. Moreover, let us compare the size of the two molecules. Cuminaldehyde is a much smaller molecule, having only one hydrogen acceptor and without a donor. At the same time, the cromakalim consists of five hydrogen bond acceptors and one donor, imparting higher chances of interaction with the residues of an active pocket of KATP. This also explains why cuminaldehyde, being a smaller molecule, exhibited significant potassium channel activation.
These findings show that cuminaldehyde possesses antidiarrheal and antispasmodic
effects predominantly mediated by activation of ATP-sensitive K
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
MNA, NUR, and WA designed the research study. MNA, NUR, AS, and WA performed the research. NUR and AS analyzed the data. MNA, NUR, AS, and WA wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study has obtained approval from the Bio-Ethical Research Committee (BERC) at PSAU, with the reference number BERC-004-12-19.
The authors are thankful to the Department of Pharmacology and Toxicology, College of Pharmacy, PSAU, for providing the facility to perform the study.
The authors extend their appreciation to Prince Sattam Bin Abdulaziz University for funding this research work through the project number (PSAU/2023/03/25802).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.