





 , Maria Paula Kwesiga 2, Jiachen Lou 3,4, Ai Lyn Tan 4,5, Michael F McDermott 4,*
, Maria Paula Kwesiga 2, Jiachen Lou 3,4, Ai Lyn Tan 4,5, Michael F McDermott 4,*1 Cancer Biology Research Center, Cancer Institute, Tehran University of Medical Sciences, 14166-14178 Tehran, Iran
2 Department of Biomedical Sciences, Grand Valley State University, Allendale, MI 49401, USA
3 Faculty of Biology, Medicine and Health, The University of Manchester, M13 9PL Manchester, UK
4 Leeds Institute of Rheumatic and Musculoskeletal Medicine, University of Leeds, LS9 7TF Leeds, UK
5 NIHR Leeds Biomedical Research Centre, Chapel Allerton Hospital, Leeds Teaching Hospitals NHS Trust, LS7 4SA Leeds, UK
Abstract
Rheumatoid arthritis (RA) is a relatively common systemic autoimmune disease with an estimated prevalence of approximately 1% worldwide. Patients present predominantly with symmetrical small joint inflammatory arthritis, which involves dysregulated immune responses, leading to bone and cartilage deformities due to extensive erosive damage. The introduction of biological based therapies for the management of this life-altering condition, over the past three decades, has led to marked improvements in patients’ quality of life. A wide range of both innate and adaptive immune cells are involved in the pathogenesis of RA, with a complex interplay of cytokines, T-cells, B-cells, and dendritic cells. Some of these cells have been successfully targeted in the treatment of RA by the use of biologics-based therapies. For example, rituximab therapy blocks B cell activation and abatacept effectively blocks T cell activation in patients with RA. Despite these advances, there remain some patients who are resistant to all current therapeutic options, which has encouraged further research into understanding the primary signal transduction pathways that mediate the disease. In this review we discuss the roles of the main signalling pathways, including metabolic reprogramming that have been implicated in RA disease progression, in order to develop a conceptual framework for more precise deployment of existing therapies, and to provide a rationale for producing molecular inhibitors of these pathways. Improved knowledge of the many intracellular signalling pathways in RA will complement current precision medicine strategies, particularly for the patients with difficult-to-treat RA, and especially in those with multidrug resistance disease.
Keywords
- rheumatoid arthritis
- intracellular signalling pathways
- ER stress
- treatment response
- multidrug resistance
- autoimmune/autoimmunity
- inflammation
- refractory RA
- metabolic reprogramming/metabolites
Rheumatoid arthritis (RA) is a systemic autoimmune and inflammatory disease, characterised by recurrent episodes of inflammatory arthritis, which are often symmetrical, involving peripheral joints with the hands, feet and wrists being the most commonly affected. Active disease in RA implicates both innate and adaptive elements of the immune system with persistent inflammation of synovial tissue and extra-articular organ involvement, more frequently seen in patients with severe disease. Regulatory mechanisms normally used by the body to contain self-directed immune responses may be insufficient to regulate the production of self-reactive autoantibodies like rheumatoid factor (RF) and anti–citrullinated protein/peptide antibodies (ACPA) also known as anti-cyclic citrullinated peptide antibodies (anti-CCP) [1, 2]. This inappropriate activation of B cells and dysregulated immune responses may eventually result in marked deformities of bone and cartilage, due to extensive erosive damage, mainly in the smaller joints of the hands and feet, with associated systematic symptoms, such as fatigue, general malaise and musculoskeletal pain. The opportunity to target these pathways has provided patients with therapeutic hope. However, despite the increasing knowledge in treating patients with RA, disappointingly, there are still patients who do not have favourable responses to existing therapies. In this review, we explore the pathogenesis of RA, and discuss the roles of the main signalling pathways, including metabolic reprogramming, that have been implicated in RA disease progression in order to develop a conceptual framework for more precise deployment of existing therapies, and also to provide a rationale for producing molecular inhibitors of these pathways. Improved knowledge of the many intracellular signalling pathways in RA will complement current precision medicine strategies, particularly for patients with difficult-to-treat RA, and especially for those with multidrug resistance disease.
Different databases, including Pubmed, Web of Science, SCOPUS and Google Scholar, were used for the keywords of ‘Rheumatoid arthritis, Intracellular signalling pathways, endoplasmic reticulum (ER) stress, Treatment response, Multidrug resistance, Autoimmune/autoimmunity, Inflammation, Refractory RA, Metabolic reprogramming/metabolites’. Inclusion criteria included all of the following keywords; RA pathogenesis, inflammation, immune tolerance, janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, stress-activated protein kinases/mitogen-activated protein kinases (SAPK/MAPK) pathway, PI-3K/AKT/mTOR pathway, Interferon signalling pathway, ER stress and the unfolded protein responses (UPR) pathway, vascular endothelial growth factor (VEGF) signalling pathway, spleen tyrosine kinase (SYK) signalling pathway, Wingless/Integrated (Wnt) signalling pathway, Notch signalling pathway, metabolites and pathogenesis, response to treatment and drug resistance. Exclusion criteria were the studies that included other rheumatological diseases apart from RA.
RA is a heterogenous condition and ACPA/anti-CCP autoantibodies, which are a hallmark of RA, are present in approximately 70% of these patients, in most reported series [3]; the specificity of ACPA/anti-CCP antibodies for RA ranges between 87% to 98% which makes their presence an excellent diagnostic indicator/marker of this disease. Shared epitope positive HLA-DR alleles are associated with anti-citrullinated peptides and with genetic susceptibility to developing RA. However, the molecular basis for these two associations is unknown.
The process of citrullination refers to post-translational conversion of arginine to citrulline residues by peptidylarginine deiminase enzymes (PADs) and is a key factor in the pathogenesis of RA [4]. The mechanisms controlling the citrullination process in RA are both dysregulated and upregulated. Under normal circumstances, the citrullinating enzymes are controlled by levels of calcium and redox conditions in order to limit their hyperactivation. Perforins and bacterial toxins induce prominent calcium influx which induces the hyperactivation of PADs and hypercitrullination in the RA joint. In addition, the extracellular environment of synovial joints in RA is in a state of oxidative activation [5], which is known to inactivate PADs: efficient extracellular citrullination in RA is likely to require the constant release of active enzymes from dying cells, which may be accelerated in the presence of PAD-activating autoantibodies.
PADs are inactivated extracellularly, and the presence of PAD activating co-factors, such as anti-PAD activating antibodies, and the continuous release of active PADs enzymes from dying and activated cells are necessary for efficient extracellular citrullination in RA. Therefore, targeting the signalling pathways that lead to PAD enzyme hyperactivation has the potential to prevent the development and progression of the RA citrullinome, with consequent clinical improvement in patients with RA [6].
An early study has suggested that one citrullinated peptide from vimentin, Vim R70Cit, could bind shared epitope positive with greater affinity than shared epitope negative HLA-DR molecules thus presenting citrullinated peptides to T cells that help production of IgG ACPA [4]. However, a thorough investigation of the binding of 180 peptides, either citrullinated or native, encompassing the entire alpha and beta chains of human fibrinogen did not find that citrullinated peptides bound shared epitope positive HLA-DR molecules better than their native counterparts. Besides, half of the patients with RA have IgG autoantibodies to the peptidyl arginyl deiminase 4 (PAD4) enzyme. This suggests the presence, in these patients, of helper T cells recognizing peptides from PAD4, which could provide help to B cells specific for citrullinated epitopes present on proteins being citrullinated by PAD4, by a hapten-carrier mechanism [7].
The main cause(s) of RA is unknown; however, some genetic and environmental factors are definitely involved in initiating disease onset [8]. Studies on patients with RA have revealed genetic factors that underlie the disease, such as the human leukocyte antigen (HLA)–DRB1 locus, where some alleles contain a five amino acid motif sequence (QKRAA), in residues 70–74 in the HLA-DRB1 region, named the shared epitope. Other factors include selection of certain predisposing T-cell repertoires, and antigen presentation; alteration in peptide affinity may also lead to autoreactive immune responses in RA [9]. The prognosis in patients with RA who are seropositive for ACPA and/or RF, is worse than those with ACPA-negative disease [10]. Neuro-immunological factors also influence disease development [8].
The environmental risk factors which are reported in patients with RA include smoking, exposure to silica, periodontitis, and gut microbiota. Another risk factor in RA is gender, as the disease is three times more common in women than in men [9].
The genetic and environmental risk factors as well as epigenetic elements lead
to post-transcriptional modifications and alteration in self-protein
citrullination. Anti-citrullinated peptides such as
Variations in specific microbiota are proven to be associated with both disease development and response to drugs. In this regard, specifically targeting the gut microbiota in each individual patient may alleviate RA disease activity, increase drug efficacy and improve the outcome [14]. The different gut microbiota composition in patients with RA, compared to healthy controls, shows the importance of gut dysbiosis in the onset of RA. One of the most important environmental factors that modulate the gut composition is nutritional content of the diet [15].
To prevent and manage the degree of inflammation in RA, modulation of the gut microbiota through diet is recommended. For instance, the Mediterranean diet has beneficial effects on the gut microbiota and may improve the outcome in RA [16]. The use of Lactobacillus acidophilus and Lactobacillus casei in probiotic products are also helpful in reducing disease activity while bacillus spp. and other lactobacillus spp. are increased in quantity in the gut of patients with RA. It seems that different lactobacilli have different roles in RA pathogenicity and diseases activity [14]. In addition, faecal microbiota transplantation (FMT) from healthy individuals to patients with RA may also reduce disease activity [17]. Furthermore, the content of gut microbiota could potentially be considered as biomarkers for RA diagnosis, especially in early onset disease. The abundance of beneficial intestinal bacteria, such as Bacteroidetes, is decreased in early RA while Prevotella copri are increased and take the place of other beneficial bacteria [18]. Not only gut microbiota but also the oral microbiota are important in RA development. Dysbiosis of oral microbiota may lead to periodontitis and the inflamed gingiva contains citrullinated proteins and ACPA. Porphyromonas gingivalis, a common microorganism in gingiva, can citrullinate the proteins for ACPA development, and is involved in the onset of RA disease [19].
A normal synovium contains mesenchymal-derived fibroblast-like synovial cells (FLS) and macrophages. In RA the FLS acquire specific characteristics, including increased proliferation, apoptosis resistance, as well as secretion of high-level disease-associated cytokines and chemokines, adhesion molecules, matrix metalloproteinases (MMPs), and tissue inhibitors. FLS are directly involved in cartilage destruction and chronic inflammation [20].
Considerable insights into the pathogenesis of RA may be gained from study of the mechanisms of action of drugs shown to be effective in RA. Methotrexate (MTX) is a conventional synthetic disease modifying antirheumatic drug (csDMARD) that is often used as a first line treatment for RA. MTX is a folic acid antagonist that inhibits the synthesis of both DNA and RNA and exerts anti-inflammatory effects on many pathways involved in RA. Furthermore, MTX works synergistically with different anti-TNF therapies, independently of its property of reducing the titres of anti-drug antibodies [21].
The balance between pro- and anti-inflammatory cytokine levels is disturbed in
rheumatoid joints, leading to the process of autoimmunity, chronic inflammation,
and associated joint damage. Inflammation of the synovium begins with
infiltration, accumulation, and migration of leukocytes into synovial
micro-vessels, which induce the expression of adhesion molecules such as
integrins, selectins, in addition to members of the immunoglobulin superfamily
and chemokines. These adhesion molecules and chemokines facilitate leukocyte
rolling, adhesion, and transmigration into the synovial tissue. Within the
synovium, leukocytes release proinflammatory cytokines, such as TNF,
IL-1
Cytokine production is essential to the development of RA. The early stages of
disease show cytokine expression patterns of IL-4, 13, and 15 TNF and IL-6 and
during inflammation the other cytokines, chemokines and adhesion molecules are
also activated. DCs also have an important role in the initiation and
perpetuation of RA through presentation the arthritogenic antigens to T-cells.
Two major DC subsets, myeloid DCs (mDCs) and pDCs, in RA synovial tissue are
reported to have different cytokine profiles [29]. In RA synovial tissues, both
pDCs and mDCs release IL-15 and IL-18, while pDCs express IFN
When loss of immune tolerance to self-antigens occurs in diseases such as RA,
protective immune mechanisms switch to auto-aggressive immunity in
dedifferentiated synoviocytes, with T cell-mediated joint damage and loss of
tissue-protective macrophage populations. The Treg subpopulation of helper
T-cells prevents the development of autoimmunity and maintains immune tolerance
by using a variety of mechanisms, including elimination of self-reactive T cells
and production of anti-inflammatory cytokines, such as IL-10 and TGF-
In effect, Treg cells are divided into three categories according to their
origin and differentiation: they are produced by immature T lymphocytes during
thymus development, and with a phenotype is CD4+CD25+Foxp3+ T cells, which are
called natural Treg (nTreg) cells that constitutively express CD25 and express
the specific nuclear transcription factor, Foxp3. Upon peripheral antigen
stimulation or immunosuppressive factor induction, mature CD4+CD25- T cells are
transformed into acquired Treg (iTreg) cells, including Tr1 and Th3 subsets; the
former mainly secretes IL-10 and TGF-
In the thymus, nTreg cells are derived from hematopoietic progenitor cells and migrate into the peripheral blood to maintain immune tolerance and to prevent autoimmunity [33].
During the development of peripheral Treg cells, naïve CD4+ T cells first
migrate into the peripheral blood without any TCR activation. Once stimulated by
antigens in the peripheral blood, naïve CD4+ T cells can differentiate into
Foxp3+ Treg cells in the presence of both TGF-beta (TGF-
There is an additional CD4+ T cell subset that mediates immunosuppression
in vitro and which is characterised by secretion of TGF-
The mechanisms of self-tolerance are complicated, however, and dysfunction of Treg cells and dendritic cells (DCs) are among the proposed mechanisms underlying the breakdown of self-tolerance in the pathogenesis of RA. Furthermore, the ratio of Treg/Th17 and Th1/Th2 cells is vitally important and the imbalance between these specialised subpopulations of T cells induces the initiation of RA pathology [35]. The anti-TNF biologic, etanercept, in combination with MTX has been shown to ameliorate disease in RA by normalising the distribution of Th17 and Treg [36] cells. In a number of chronic autoimmune diseases, enhancement of Treg function by the use of tolerogenic dendritic cells or stem cell transplantation are both potentially effective treatments to re-establish immune tolerance [37, 38]. Moreover, tolerogenic vaccine platforms to deliver the self-antigens to specific antigen-presenting cell subtypes are other therapeutic strategies that may be employed for treat RA [22].
In autoimmunity, it has been postulated that citrullination of peptides and self-antigens may expose cryptic peptides by modifying protein structure and proteolytic cleavage, with subsequent changes to antigen processing by the MHC-II, which may, in turn, lead to loss of immune tolerance to self-antigens because of exposure of these cryptic peptides previously hidden from immune system [6].
Janus kinases (JAKs) belong to the family of tyrosine kinases (TYKs); the basic structure of all JAKs consists of four structural domains consisting of seven homologous regions [JH1-7] [39]. The signal transducer and activator of transcription (STATs) are recruited by their corresponding JAKs to act as transcription factors to orchestrate their diverse biological functions (Fig. 1). In summary, the whole signalling module provides a rapid membrane-to-nucleus transmission of the cytokines’ signal from the cellular receptor (e.g., IL-2R) and JAK/STAT signalling is a key intracellular cascade in response to cytokines and growth hormones [40]. This pathway is initiated upon binding the type I/II cytokine to its receptor. Type I/II cytokine receptors are composed of different chains that oligomerize after binding the cytokines [41]. The oligomerization of the four chains separates the intracellular subunits of cytokine receptors, thereby cleaving receptor-associated JAKs from each other and reversing their constitutive inhibition and activation [39]. Autophosphorylation of JAKs converts the inactive form of cytosolic STAT monomers to their active forms, as either homodimers, heterodimers, or tetramers. Active STATs then translocate to the cell nucleus where they function as transcription factors and regulate gene expression [41]. In RA, proinflammatory cytokines, including IL-2, IL-3, and IL-19/IL-20, are the main effectors of the JAK/STAT pathway. However, IL-17A, IL-19, and IL-20 also play a significant role in the context of the JAK/STAT pathway activation. IL-17A has been shown to increase matrix metalloproteinases (MMPs)-1, -2, -3, -9, and -13 which are important mediators of the degradation of articular cartilage extracellular matrix (ECM) proteins in RA [42]. MMP-3 plays a central role in this regard as it promotes structural damage in the joints of patients with RA due to its ability to cleave ECM proteins [42]. The expression of the enzyme matrix metalloproteinase-3 (MMP-3) is observed in FLS which is involved in joint destruction in patients with RA. The MMP-3 enzyme degrades collagen types II, III, IV, IX and X, proteoglycans, fibronectin, laminin, and elastin. In addition, MMP-3 can also activate other MMPs such as MMP-1, MMP-7, and MMP-9, rendering MMP-3 crucial in connective tissue remodelling. MMP-3 is abundant in the serum of patients with RA and considered a biomarker of the condition [43, 44].
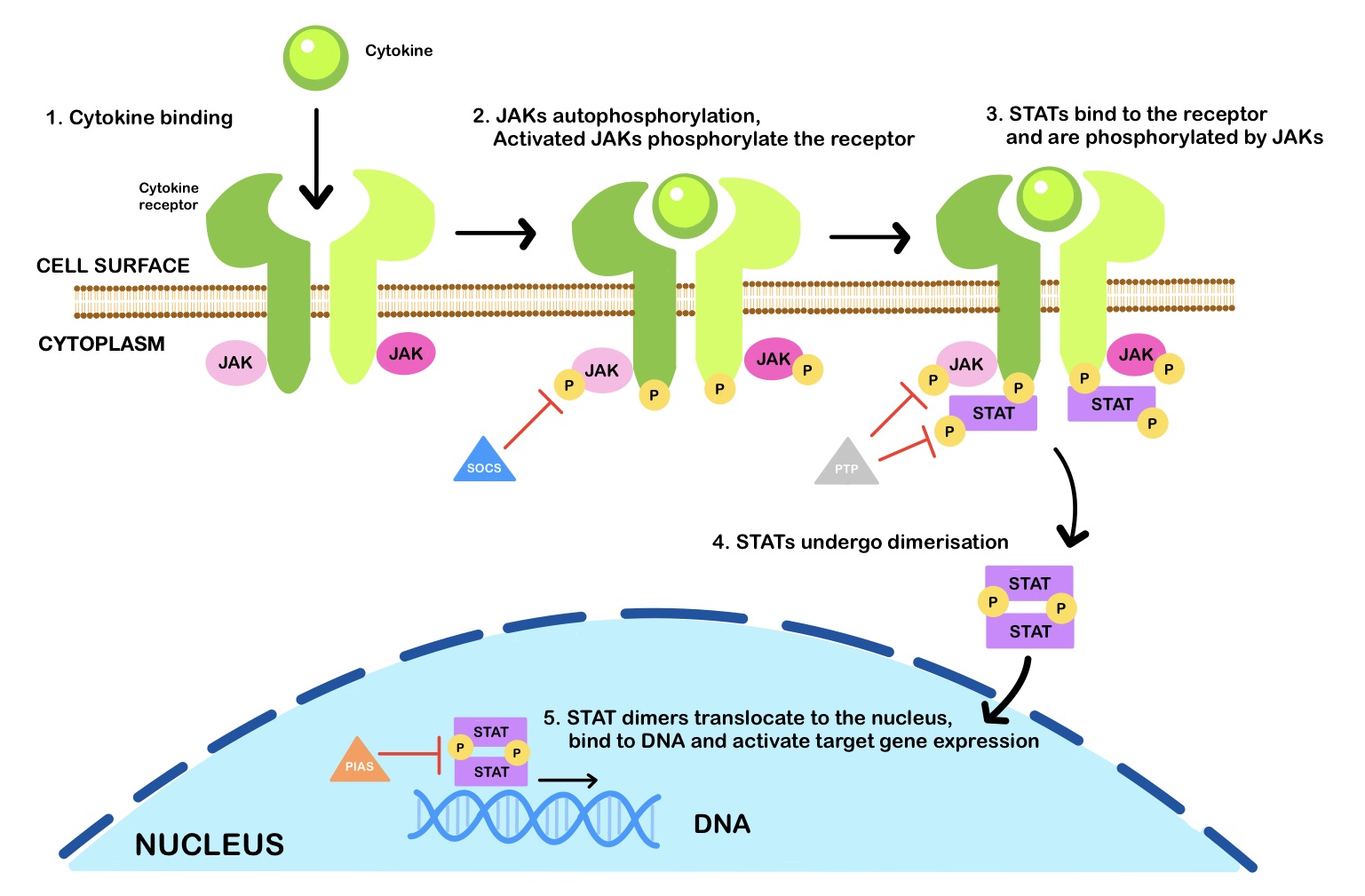 Fig. 1.
Fig. 1.The JAK-STAT signalling pathway. Janus kinases (JAKs) are activated upon cytokine binding and phosphorylate signal transducer and activator of transcription (STAT) proteins, which dimerise and translocate to the nucleus to regulate gene expression. Inhibitors of the JAK-STAT signalling pathway include suppressor of cytokine signalling (SOCS) proteins, protein tyrosine phosphatases (PTPs), and protein inhibitor of activated STAT (PIAS). [P] is for phosphate.
As with the use of the biologic-based (bDMARDs) drugs, the targeted synthetic (tsDMARDs) therapies have also transformed the management of RA. The orally bioavailable, synthetic small-molecule Janus kinase (JAK) inhibitors (JAKi) are examples of tsDMARDs used to treat RA. Clinical trials have shown that tofacitinib, a JAK inhibitor, used either as a monotherapy or in combination with csDMARDs, is effective in the treatment of patients with RA [45]. However, an increased incidence of major adverse cardiovascular events (MACEs) and malignancy has been reported with tofacitinib compared to TNF inhibitors [46]. A dose-dependent increase in venous thromboembolism (VTE) risk was also observed with tofacitinib; these findings have resulted in the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) restricting the use of all JAKi to patients with RA who had failed TNF inhibitors ((i) Janus Kinase inhibitors (JAKi) European Medicines Agency (europa.eu) and (ii) Janus Kinase (JAK) inhibitors: Drug Safety Communication - FDA Requires Warnings about Increased Risk of Serious Heart-related Events, Cancer, Blood Clots, and Death j FDA).
Mitogen-activated protein kinases (MAPKs) are important signal transduction pathways involved in several intracellular mechanisms such as differentiation, proliferation, and migration in response to extracellular stimuli. MAPK signalling pathways have a dual role in survival and apoptosis which depend on the cell type and the stimulus (Fig. 2). The MAPK family contains at least four subfamilies including, extracellular signalling kinases (ERK)-1/2, Jun amino-terminal kinases (JNK1/2/3), p38 proteins (p38a/b/g/d) and ERK5. MAPKs are regulated by a three-step cascade consisting of MAPKKK (MAP3K, MEK kinase), MAPKK (MAP2K, MKK, MEK), and MAPK1 [47]. JNKs and p38 signalling pathways contribute to both pro-apoptotic and anti-apoptotic mechanisms and can also regulate the autophagy pathway [47]. Autophagy has a dual role in cell survival and death during nutrient starvation [48]. In addition, JNK activation decreases ER stress activity and cell death by inducing the expression of anti-apoptotic proteins [49]. The ERK1/2 cascade is a signalling pathway that regulates a large number of cellular processes, mainly by anti-apoptotic functions, including cell proliferation and differentiation [50]. However, there are also several examples of ERK1/2 signalling exerting a role in pro-apoptotic function [51, 52].
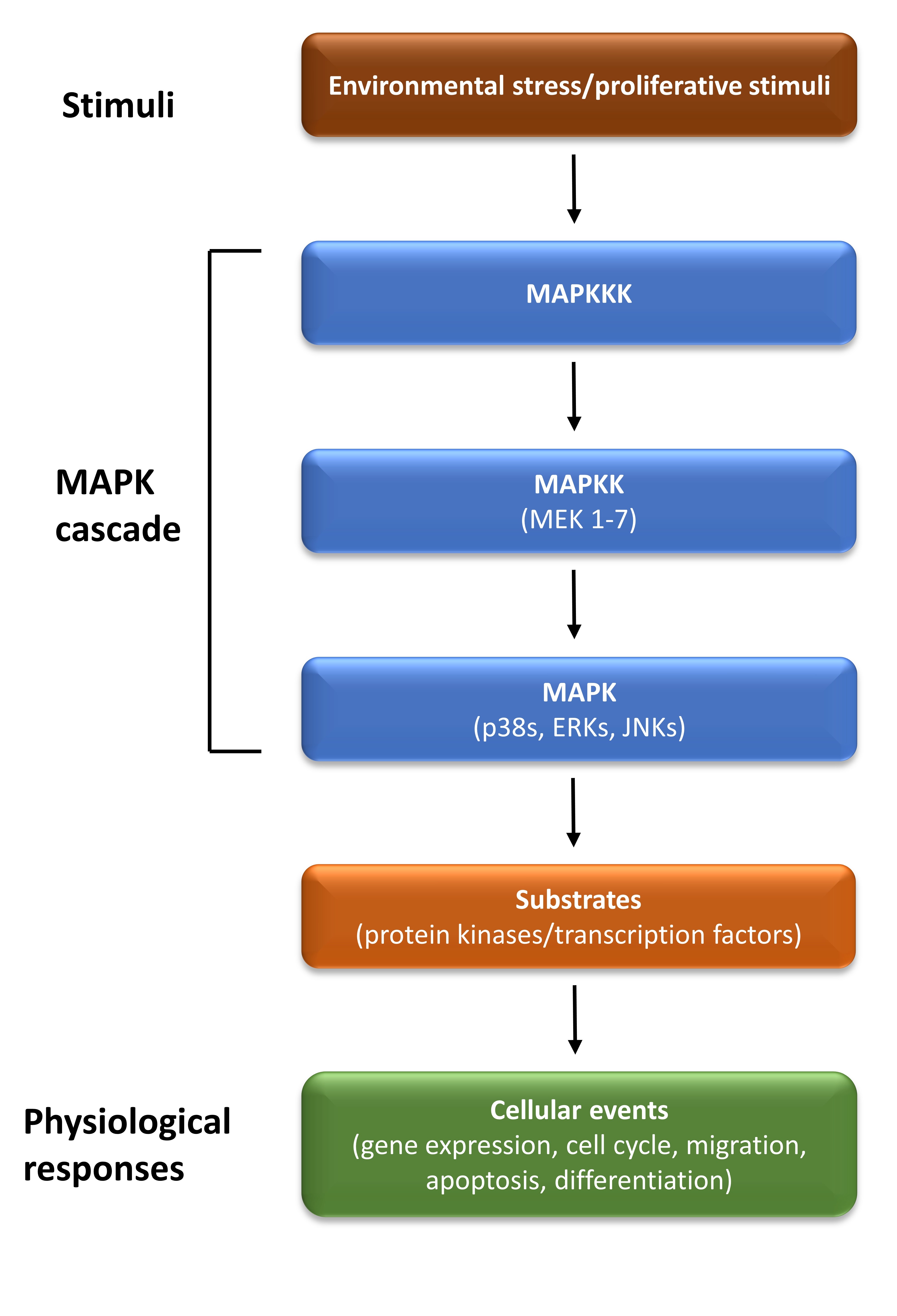 Fig. 2.
Fig. 2.The MAPK signalling pathway. The mitogen-activated protein kinase (MAPK) cascade consists of three sequentially acting kinases: a MAPK, a MAPK kinase (MAP2K), and a MAPKK kinase (MAP3K). It is activated by a variety of stimuli, and it phosphorylates various substrates, to regulate diverse cellular events. ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase.
Stress-activated protein kinases/mitogen-activated protein kinases (SAPK/MAPK)
signalling pathways are activated by pro-inflammatory cytokines in RA [42, 53]
and are considered to be key proteins for targeted therapies [54]. In injured
joint tissues, MAPKs are implicated in the regulation of proinflammatory
cytokines in response to IL-1, IL-17, and TNF receptors [55]. ERK1/2 also
regulates IL-6, IL-12, IL-23 and TNF production in response to lipopolysaccharide
(LPS)-stimulated macrophages [56]. Epidermal growth factor (EGF), which is
released from FLS, induces ERK1/2 to control the production of
cyclooxygenase-2-dependent prostaglandin E2 [57]. A major role of JNK in RA
development is the control it exerts on MMP levels and the extent of cartilage
destruction. IL-1
PI3K/Akt/mTOR is an important intracellular signalling network in mammalian cells involved in different cellular functions such as cell proliferation, survival, angiogenesis and differentiation [61]. The pathway is also activated to regulate autophagy during fasting or starvation. Elevated levels of PI3K/Akt/mTOR are reported in several types of cancers and autoimmune diseases, due to specific mutations and essential proteins such as phosphatase, tensin homologue deleted on chromosome 10 (PTEN), and mTOR [62, 63]. During the initiation phase of RA, NGF, PDGF, VEGF, FGF, and cytokines induce PI3K/Akt/mTOR signalling [64, 65]. As a result, increased levels of neutrophils, macrophages, eosinophil chemotaxis, mast cell degranulation, and both T- and B-cell infiltration, allied to the proliferation of RA-FLS and increased IL-17 production by CD4+ T-cells may occur. The PI-3K/AKT/mTOR pathway may also inhibit immune-cell proliferation by reducing the activity of forkhead box protein O (FOXO) transcription factor [42], which prevent Fas ligand-induced neutrophil apoptosis in inflammatory arthritis [66].
Type I interferons are activated as the first line of the immune response
against viruses and microbial products, such as LPS [67]. Theoretically,
regulation of the expression of type I interferon might be of therapeutic value,
as has been demonstrated for RA [68]. For example, most immune cells, and
particularly plasmacytoid dendritic cells (pDCs) release IFN
The endoplasmic reticulum (ER), as the largest intracellular membrane-bound
organelle in eukaryotic cells, has essential responsibilities in many important
cellular functions and interactions, including biosynthesis of lipids,
carbohydrate metabolism, Ca
All proteins that fail to fold properly are retained and removed from the system as they’re targeted for degradation by the ER-associated degradation (ERAD) pathway, by a process of ubiquitylation. Chaperones that reside in the ER assist in folding of the polypeptides. For instance, Hsp70-type chaperone/BiP (binding-immunoglobulin protein, GRP-78) interacts with intact polypeptide chains to prevent their aggregation and facilitates the exquisite protein folding into the correct conformation for their functional structure [77].
Any problems that may arise during protein folding, such as an imbalance between protein synthesis and protein demand, accumulation of misfolded proteins, lack of nutrients, failure of post-translational modification, irregularities of calcium storage and oxygen levels, defects in ERAD and the autophagy pathway may all lead to ER stress (ERS) [78]. The chief mechanism employed by the cells to cope with these various stresses (stressors) is known as the unfolded protein responses (UPR). During an UPR there is upregulation of those proteins which are necessary for protein folding or inhibition of protein synthesis [79].
The UPR is initiated by three ER transmembrane proteins, each having its own
signalling pathway, but also with an inherent capacity for crosstalk between
these 3 proteins. These three sensors, individually known as inositol requiring
enzyme-1 (IRE1, both
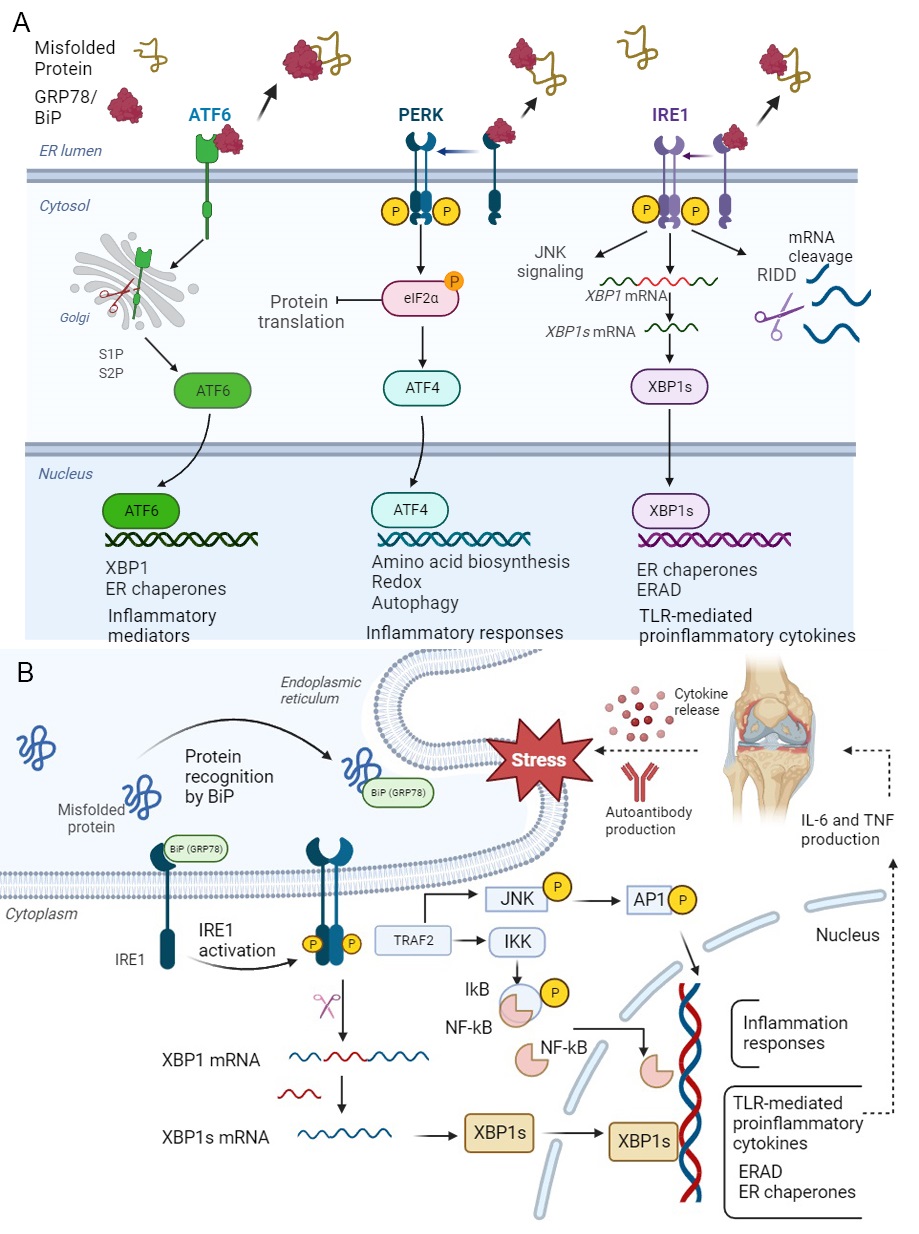 Fig. 3.
Fig. 3.Unfolded Protein Response (UPR) and IRE1
The PERK sensor is the first to be activated during UPR with dimerisation,
oligomerisation, and autophosphorylation. Upon activation, the PERK protein
phosphorylates the eukaryotic initiation factor 2 subunit-
IRE1, as a transmembrane protein in the ER, has two domains: the N-terminal, ER
luminal domain, senses unfolded proteins, and the C-terminal cytoplasmic region
initiates the UPR via serine/threonine kinase and endoribonuclease (RNase)
domains. IRE1 then dissociates from the GRP78 and binds to mal-folded proteins to
transmit the signal to the nucleus (Fig. 3A). IRE1 has two isoforms in mammals;
IRE1
ATF6 acts as both monomer and oligomer to form intra- and inter-disulfide bridges within the ER luminal. Under ER stress, the full format of ATF6 (ATF6p90) moves to the Golgi and is cleaved by two kinds of proteases called site-1 protease (S1P) and site-2 protease (S2P), then is converted to a basic leucine zipper (bZIP) translation figure, named ‘ATF6p50’. The spliced ATF6 form can translocate to the nucleus to induce the expression of genes such as ER chaperones and ER protein translocation, as well as proteins involved in folding, maturation and secretion, in addition to proteins involved in the degradation of misfolded proteins [90]. ATF6p50, like XBP1s, enhances the expression of secretory proteins from the ER and Golgi under ER stress [91] (Fig. 3A).
Although the most significant function of IRE1
In RA the presence of acute or chronic inflammation, the secretion of excessive
amounts of inflammatory cytokines, matrix metalloproteinases (MMPs), and other
components of the immune response as well as hypoxia and low glucose levels
combine to increase levels of ER stress. To restore the homeostasis, both the
ER-associated degradation (ERAD) system and the UPR are activated for maintenance
and survival of the cells. In the ERAD pathway, accumulated misfolded/unfolded
proteins are transferred to the cytosol and ubiquitinated through E2
(ubiquitin-conjugating enzyme) and synoviolin (E3 ubiquitin ligase) in order to
be degraded by the cytosolic 26S proteasome system. In the UPR pathway, the three
separate branches, IRE1
Upregulation of ER stress signalling plays a critical role in several diseases such as RA which is activated in immune and somatic cells such as FLS, B cells, myocytes, myeloid cells and chondrocytes [99]. FLS demonstrate invasive features, like tumour cells, which lead to increased synovial proliferation, joint destruction and persistent inflammation [100]. Activation of ERS key factors such as GRP78, IRE1, XBP1s, and eIF2-P have been reported in macrophages and synovial tissues of patients with RA [101]. The upregulation of GRP78, IRE1 and XBP1s have also been observed in PBMC of patients with RA [88]. Indeed, the inflammatory responses in RA can induce the ERS pathway, and UPR activation may also stimulate the production of inflammatory cytokines, such as TNF [98, 102, 103].
One of the key markers in ERS pathway is GRP78, which plays a critical role in
the pathogenesis of RA [100]. GRP78 overexpression not only occurs in the
synovium of patients but may also act as autoantibody in sera of patients with RA
[101, 104]. Anti-citrullinated BiP (citBiP) antibodies are also detectable in the
serum, FLS and macrophages of these patients. In addition to their role in
disease causation, extracellular GRP78 acts as an immunoregulatory marker and
induces a number of immunosuppressive cytokines such as IL-4 and IL-10 to prevent
RA development [105]. Moreover, BiP is also involved in other cellular functions
such as antibody production, T cell proliferation and proinflammatory cytokine
generation, and therefore has potential to be targeted or induced in the
treatment of RA [106]. Increased levels of IRE1 in FLS and macrophages of
patients with RA induce the production of TLR-mediated proinflammatory cytokines.
The increased expression of XBP1s in PBMC and FLS of active RA patients in
contrast to healthy controls or those in remission has also been observed and its
activation shown to be mediated through TLR2/4 signalling, leading to IL-6 and
TNF secretion. However, in active RA, the other ERS mediators such as ERN1,
HSPA5, or SYNV1 are decreased, suggesting the IRE1-XBP1s pathway act in a
TLR-dependent manner without any impact on ERS signalling [107]. The increased
level of PERK-eIF2 present in synovial tissues and macrophages of patients with
RA can activate the NF-
ATF6 is another branch of UPR that is increased in the macrophages of patients with RA. ATF6 may stimulate the inflammatory response and TNF may induce the cleavage of ATF6 in FLS; however, activation of ATF6 may be inhibited by use of proteasome inhibition or autophagic functions [109].
One of the major mechanisms of RA pathogenesis is dysregulated apoptosis mechanisms that cause abnormal expansion of FLS, the most common cell type contributing to joint destruction in RA at the pannus–cartilage junction [110]. Although apoptosis is activated by two major types of signal, either an extrinsic death receptor or an intrinsic mitochondrial death receptor, other models of cell death have also been proposed, including T-cell-mediated cytotoxicity and perforin-granzyme signalling [111, 112]. In the exogenous route, following the binding of Fas ligand (FasL, CD95L) to Fas (CD95) receptors, various proteins are activated that result in upregulation of both initiation and activator caspases, with subsequent activation of proteins involved in apoptosis. The endogenous pathway is activated by a range of intracellular signals such as increased oxidative stress or DNA damaging agents. These factors impact mitochondrial outer membrane permeabilisation (MOMP) and are associated with release of the apoptotic effectors. Apoptosis-dependent mitochondria are regulated by various members of the BCL-2 family, which have both anti- and pro-apoptotic functions [110, 113, 114].
Under severe ERS, when the cells are no longer able to establish and maintain
homeostasis, the three aforementioned UPR pathways trigger apoptosis pathway, via
different mechanisms. For example, permanent activation of PERK leads to
increased levels of CHOP transcription factor which regulate the expression of
many anti-apoptotic and pro-apoptotic genes, including genes encoding the
BCL2-family proteins. Furthermore, PERK protein enhances the expression of GADD34
that can dephosphorylate eIF2
CHOP transcription factor, which is regulated by ATF4, induces BIM (a
pro-apoptotic member of the BCL-2 family) expression to trigger cell death [115].
Indeed, ATF4 and CHOP increase the levels of reactive oxygen species (ROS)
production in stressed cells leading to cell death [116]. On the other hand,
severe ERS may cause hyperphosphorylation of IRE1
Autophagy is a self-eating process activated to remove the impaired organelles, unusual and inactive proteins, or other intracellular substances through lysosomal degradation. This recycling process is crucial for optimal cell life and function [122]. The various molecular mechanisms of autophagy are initiated by formation of autophagosome vacuoles to digest unwanted intracellular contents, which then fuse with lysosomes to form autophagolysosomes for selective degradation of specific cargos, such as organelles and proteins, with reuse of the primary materials. Each step in the process involves regulatory proteins, and mutation in these autophagy genes (ATGs) lead to failure of autophagic mechanisms at different steps in the pathway [123].
It has been reported that the expression levels of Beclin1, ATG5, and LC3, as autophagy-related proteins, are all raised in the synovial tissue of patients with RA [124]. In has also been documented that anti-TNF therapy and use of IL-6R inhibition bring about a reduction in the autophagic response [125]. Furthermore, the use of hydroxychloroquine and chloroquine as autophagy inhibitors drugs, in treating patients with RA, may decrease activation of immune systems and reduce cytokine production [111, 126]. 3-MA, as an autophagy inhibitor, also reduces citrullinated peptide levels in RA with induction of apoptosis pathways in RA-FLS compared to OA-FLS [127]. Autophagy can competitively inhibit apoptosis in proliferating FLS in RA [128]; the FLS induce autophagosome formation and enhance the expression of HMGB1 and Beclin-1 [129]. Autophagy is also activated under severe ERS to eliminate any unwanted or misfolded proteins by a mechanism referred to as ERphagy [77]. In addition, there is crosstalk between the ER stress and autophagy mechanisms. IRE1 activation leads to an increase in tumour necrosis factor receptor-associated factor 2 (TRAF2) and consequently apoptosis occurs via kinase-1 (ASK1) and c-JNK signalling. This process mediates the phosphorylation of Bcl-XL/Bcl-2 and enhances Beclin-1 expression [130]. PERK activation also regulates autophagy through ATF4 and CHOP transcription factors, which enhance the expression of autophagy-related proteins, including ATG-12 and ATG5 which bind to ATG16L to induce autophagosome formation [131, 132]. In RA, proinflammatory cytokines such as TNF are activated to induce autophagy via conversion of LC3-I to LC3-II [133, 134] which leads to apoptosis and drug resistance. Hence, autophagy inhibition, or the use of an anti-TNF treatment may aid in reactivation and restoration of apoptosis in RA [129, 135]. For instance, suppression of mTOR signalling in autophagy, in combination with MTX, is used to treat RA [136] and has shown a better response in RA patients than MTX alone; these findings suggest that autophagy modulators could be added to treatment protocols to increase therapeutic effectiveness. However, due to the dual role of autophagy in the pathogenesis of RA, autophagy induction in the early steps of RA may be used to increase apoptosis [129]. Autophagy exercises a dual role of both cytoprotection and cell death induction in RA-FLS. ER homeostasis is dependent on autophagy activation and a functioning ERAD pathway. Indeed, autophagy activation is correlated with CHOP reduction, with protection of FLS from apoptosis. The use of tapsigargin as an ERS inducer leads to independent autophagic cell death, whereas the use of MG132 as a proteasome inhibitor induces the cytoprotective effects of autophagy [102].
Reactive oxygen species (ROS), including superoxide anion, hydroxyl radical, hydroperoxyl, and hydrogen peroxide are all produced in the cells via endogenous or exogenous sources due to impaired oxygen reduction capacity [136]. Endogenous sources of ROS may include products of metabolism in intracellular organelles such as mitochondria, ER, peroxisomes, and macrophages [137]. Exogenous ROS may be derived from radiation, the use of certain drugs, smoking, and infection [138, 139]. The intracellular accumulation of ROS leads to oxidative stress [139], which may play a central role in the development of different autoimmune diseases, such as RA [140].
Changes in intracellular Ca
Furthermore, correct folding of proteins is completely dependent on redox status, as increased oxidative stress impairs the ER folding process which, in turn, may lead to the accumulation of unfolded proteins, UPR activation, ROS production, more inflammation, and ultimately apoptosis activation [143]. The use of ERS inhibitors or antioxidants reduces the levels of ROS and proinflammatory cytokines which, in turn, leads to inactivation of the UPR markers [137, 144].
The ER and mitochondria engage in crosstalk to maintain the cellular homeostasis
through redox signalling, Ca
There is significant crosstalk between the ERS and inflammatory pathways, as ERS
induces the production of the cytokines, with triggering all three ER stress
sensor proteins, IRE1, PERK and ATF6, which combine to inducing an inflammatory
response, through activation of NF-
One of the key features of cancer and inflammation is angiogenesis or activated vascular development, which is involved in tissue growth, dysregulated tissue perfusion, and increased responses to normal or pathological stimuli. The vascular endothelial growth factor (VEGF) superfamily and their receptors (VEGFRs) are important factors for the regulation of vascular permeability [151]. VEGF family members are found in both transmembrane and soluble format. VEGF and its receptors play the major role in synovial tissue pathology in adult RA. Seven isoforms of VEGF have been identified, including PlGF and snake venom VEGF. This growth factor is considered to be the most essential of the three different VEGF isoforms with at least 9 subtypes due to RNA splicing. Each subtype is believed to have a unique function in the development of blood vessels and arteries. VEGF signalling involves binding to tyrosine kinase receptors—VEGFR1 and VEGFR2. VEGFR2 promotes vascular growth, while VEGFR1 acts as a negative regulator during embryogenesis and inflammatory responses. VEGFR3, a receptor for lymphatic growth factors, regulates vascular and lymphatic endothelial function in development. Loss-of-function VEGFR3 variants have been found in lymphedema [64].
The presence of angiogenesis in the synovial lining is essential for RA pannus formation which relies on new blood vessels for growth. The initial discovery of highly vascularized pannus led to intensive investigations into angiogenesis factors in RA, with VEGF being the most potent in both adult RA and juvenile chronic arthritis (JCA) [152]. VEGF causes increased vessel permeability, which plays a critical role in development of chronic oedema and swelling in RA. It also generates chondrocyte and osteolytic fragments found in joint effusions. Although VEGF levels were higher in patients with inflammatory arthritis, there were no significant differences between the types of arthritis studied [64].
Spleen tyrosine kinase (SYK) is a key part of B-cell receptor (BCR) signalling and phosphorylated SYK levels are significantly higher in RA patient’s peripheral blood B-cells [153]. Bruton’s tyrosine kinase (BTK) is a crucial molecule in the Tec family of non-receptor tyrosine kinases. It is present in all hematopoietic cells except T-cells and NK cells [154]. BTK links BCR signalling, chemokine receptor and TLR signalling, and has important roles in B-cell regulation [154]. BTK is activated by SYK or PI3K in an antigen-dependent BCR signalling context and regulates B-cell survival and proliferation [155]. BTK can also interact with multiple molecules to promote antibody secretion, cell proliferation, generation of pro-inflammatory cytokines and regulation of B-cell migration [156]. B-cell dysregulation in RA leads to production of autoantibodies and cytokines, which promote disease progression. Moreover, increased levels of phosphorylated BTK in B cells of RA patients, lead to FLS proliferation. In RA patients who are RF positive, phosphorylated BTK is correlated with RF levels [157]. Since BTK regulates osteoclast proliferation and differentiation, this could be a viable target in treating RA [158].
Cancer and embryonic development, as well as RA, are characterised by abnormal
activation of the Wingless/Integrated (Wnt) signalling pathway. This pathway
plays a central role in synovial inflammation and bone metabolism in RA [159].
Two Wnt pathways, classical and non-classical, are involve in secretion of
proteins. The classical Wnt family is involved in secreted frizzled family
transmembrane receptor protein Dishevelled (Dsh), glycogen synthesis kinase 3
(GSK3),
Notch genes code for receptors that regulate cell development in organisms and
affect cell processes like proliferation, differentiation, apoptosis, and
boundary formation. The Notch pathway mainly consists of receptors, ligands, and
regulatory molecules. Mammals have 4 receptors and 5 ligands. Notch signalling
involves the interaction between adjacent cells via the Notch ligand and
receptor. Cleavage of Notch protein releases NICD into the cytoplasm, which then
enters the nucleus to form the NICD/CSL complex and activate its target genes.
Notch signalling induces inflammation in RA by stimulating various cells to
produce proinflammatory cytokines, while its inhibition of this pathway impairs
Th17 cell differentiation [163]. For example, Notch-1 binds to IL-17 and
ROR-
RA is characterized by synovial hyperplasia and increased proliferation of immune cells, including mainly B-cells, T-cells, dendritic cells, and macrophages which all play critical roles in RA pathogenesis. To adapt to these changes, the cells undergo metabolic reprogramming which creates a vicious cycle that further accelerates the degree of inflammation in the affected joint. Herein, the relationship between metabolites and RA is discussed (Fig. 4).
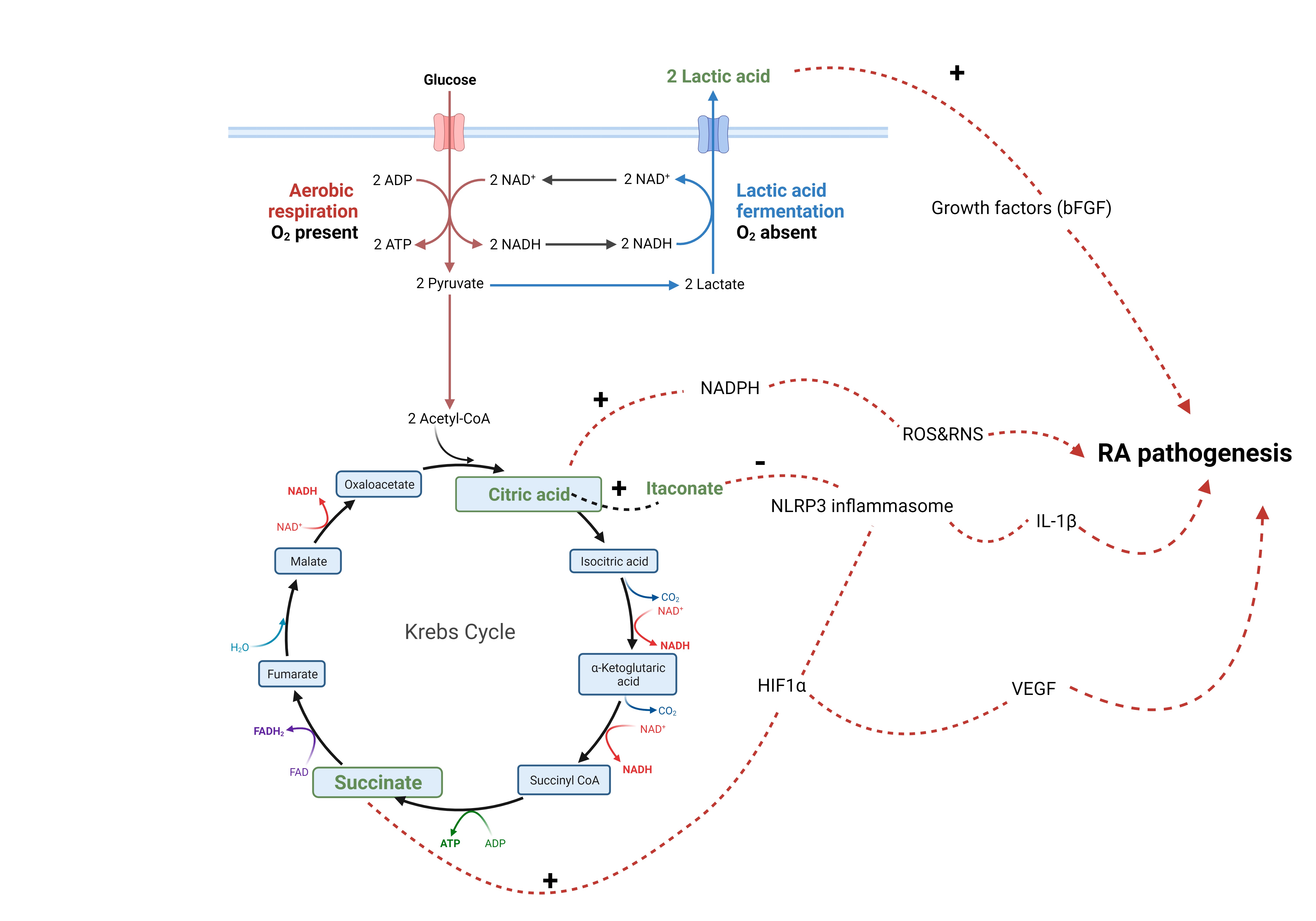 Fig. 4.
Fig. 4.Metabolites and RA pathogenesis. Lactic acid (lactate)
stimulates the release of growth factors from synovial fibroblasts. Citric acid
(citrate) provides a source for NADPH, a co factor for production of reactive
oxygen and nitrogen species (ROS and RNS). Citrate can be converted into
itaconate, which inhibits activation of NLRP3 inflammasome. Succinate activates
HIF1
6.6.5.1 Lactate (Lactic Acid)
Hypoxia is a basic metabolic change that occurs in almost all inflammatory disease states including RA. Hypoxia has been shown to cause mitochondrial dysfunction [165]; therefore, to meet the energy demands, cells must undergo a metabolic shift to anaerobic glycolysis. Decreased glucose levels increase intracellular lactate. The lactate transporters MCT-4 and SLC5a12 are upregulated to facilitate the transfer of lactate from the cytosol to the interstitial fluid within the synovial joint [166]. The increased lactate levels within the interstitial space activates quiescent synovial fibroblasts which lead to secretion of growth factors and consequently accelerates proliferation and migration of these cells as well as driving angiogenesis. Biniecka et al. (2016) [165] demonstrated an increase in the secretion of basic fibroblast growth factor (bFGF) as well as increased invasion of RA synovial fibroblasts, when cultured with increased lactic acid levels. This hostile microenvironment facilitates the recruitment of leukocytes that aggravate the inflammation within the synovial joint, leading to formation of a pannus which is the characteristic clinical manifestation of RA.
Lactic acidosis is also associated with alteration of the p53 suppression gene which increases cell survival by conferring survival advantages, such as evasion of apoptosis, and increased proliferation. This may result in a consequent exacerbation of inflammation. In fact, levels of lactate dehydrogenase (LDH) enzyme, which is responsible for the conversion of pyruvate into lactate are reported to be elevated within the synovium of patients with RA [167].
6.6.5.2 Succinate
Previous work has shown the significance of measuring levels of the
tricarboxylic acid (TCA) metabolite succinate in the diagnosis of RA. Kim
et al. (2014) [168] found succinate to be a potential biomarker for
RA (p
6.6.5.3 Citrate
Citrate plays a crucial role in cell metabolism and is the key regulator of various metabolic reactions that are the essential sources of energy production [166]. Citrate has also been shown to be a modulator of inflammation and provides a source of NADPH which is necessary to produce both reactive oxygen and nitrite species. Citrate can be converted to the anti-inflammatory metabolite, itaconate, via the enzyme aconitate decarboxylase 1 (ACOD1) [166, 170]. Citrate was found to be elevated in the synovial fluid of patients with RA, indicating its potential use as a biomarker of RA [168]. However, the precise mechanism(s) on how it affects the progression of RA remain to be explored.
6.6.5.4 Itaconate
Itaconate is a vital cell metabolite that has recently emerged in the
progression and diagnosis of RA. It is well known to possess anti-inflammatory
properties [171, 172, 173] and activates the key transcription factor Nrf2 which
downstream induces gene expression of antioxidant and anti-inflammatory
molecules [171]. Itaconate has also been shown to inhibit the
pro-inflammatory complex protein NLRP3 inflammasome [174] NLRP3 initiates a
cascade of events leading to production of inflammatory cytokines such as
IL-1
Peresolimab is a monoclonal antibody that stimulates human programmed cell death protein 1 (PD-1), which is predominantly expressed on activated T cells. Peresolimab has the capacity to induce physiological immune inhibitory pathways that restore immune homeostasis, but the exact function of PD-1 in Tregs remains unclear at this point. In a phase 2 trial of peresolimab in RA, the peresolimab arm of this study was shown to meet the primary endpoint for efficacy, with similar rates of adverse events between the drug and placebo arms [185]. It is quite possible that this drug, marketed by Eli Lilly, will be of significant therapeutic benefit in treating patients with RA, based on previous experience with the use of immune-checkpoint inhibitors in cancer immunotherapy [186].
Despite the wide-ranging targets and armamentarium of therapy available to treat RA, there are still patients with RA who unfortunately never quite achieve remission or even low disease activity states, the so-called difficult-to-treat RA [187]. These include drug resistant RA, recognised as refractory RA [188]. Drug resistance in RA is likely to be multifactorial, and can stem from several causes, such as the development of neutralising antibodies against biologics, alterations in drug metabolism, or shifts in the underlying disease mechanism [189]. There is also the recognition that the age of onset of RA can affect the response to treatment, with more drug resistance with increasing age of onset [190].
Personalised medicine and the ability to stratify and predict patients’ response to therapy may reduce the drug resistance in RA. Recently, it has been shown that an increase in dendritic cell precursors can predict the prognosis of RA [191]; such findings provide opportunities for novel targets for precision RA therapies. With improved understanding of the pathogenesis of RA comes advancement in the various facets of precision medicine in RA. Multidrug resistance is one of the most common reasons for refractory RA [192], but the ability to identify the right drug for a particular patient with a RA is somewhat lagging behind [193]. Real world registry studies may have a place in helping to close the gap in RA precision medicine, as we understand how patients respond to different therapies, particularly as patients with resistant RA are likely to have cycled through a number of different biologics and targeted synthetic DMARDs (tsDMARDS) [194].
Delineation of the intracellular signalling pathways activated in RA and development of novel targeted therapies are essential steps to improving patient outcome in this complex disease.
As befits the heterogeneity of drug resistant RA, there is a complex myriad of potential targets for therapy [195, 196]. Therefore, the knowledge gained in discovering the intracellular signalling pathways in RA will complement precision medicine strategies in providing difficult-to-treat RA, particularly those exhibiting drug resistance and may also offer the hope of achieving disease remission, or, at least, a less active disease state.
MR and MFM conceived the study and retrieved the literature; MPK, JL and ALT retrieved the literature. All the authors were involved in writing the paper, and all authors read and approved the final manuscript. All authors have participated sufficiently in the work and are accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.