








 , Shahab Uddin 1,2,8,*
, Shahab Uddin 1,2,8,*
1 Translational Research Institute, Academic Health System, Hamad Medical Corporation, 2030 Doha, Qatar
2 Dermatology Institute, Academic Health System, Hamad Medical Corporation, 2030 Doha, Qatar
3 Department of Human Genetics-Precision Medicine in Diabetes, Obesity and Cancer Research Program, Sidra Medicine, 26999 Doha, Qatar
4 Department of Dermatology and Venereology, Rumailah Hospital, Hamad Medical Corporation, 3050 Doha, Qatar
5 Department of Medicine, Weill Cornell Medicine Qatar, Qatar Foundation-Education City, 24144 Doha, Qatar
6 Department of Medicine, Weill Cornell Medicine Qatar, New York, NY 10065, USA
7 College of Medicine, Qatar University, 2713 Doha, Qatar
8 Laboratory of Animal Research Center, Qatar University, 2713 Doha, Qatar
Abstract
Background: The benzophenanthridine Sanguinarine (Sng) is one of the
most abundant root alkaloids with a long history of investigation and
pharmaceutical applications. The cytotoxicity of Sng against various tumor cells
is well-established; however, its antiproliferative and apoptotic potential
against the cutaneous squamous cell carcinoma (cSCC) cells remains unknown. In
the present study, we investigated the anti-cancer potential of Sng against cSCC
cells and elucidated the underlying mechanisms relevant to the drug action.
Methods: The inhibitory effect of Sng on cSCC cells was evaluated by
analyzing cell viability, colony-forming ability and multi-caspase activity.
Apoptosis was quantified through Annexin-V/Propidium iodide flow cytometric assay
and antagonized by pan-caspase inhibitor z-VAD-FMK. Mitochondrial membrane
potential (
Keywords
- cutaneous squamous cell carcinoma
- Sanguinarine
- apoptosis
- reactive oxygen species
- JNK signaling pathway
Squamous cell carcinoma of the skin or cutaneous squamous cell carcinoma (cSCC) is an epidermal keratinocyte carcinoma that falls under the umbrella of non-melanoma skin cancer (NMSC). It is one of the most frequent neoplasms affecting humans, especially the fair-skinned population, and second only to basal cell carcinoma in incidence [1]. Several population-based surveys have demonstrated a steady rise in the incidence of cSCC over the last three decades [2, 3, 4, 5]. The Australian population has recorded the highest burden of cSCC [6]. cSCC onset generally correlates with cumulative sun exposure (primarily UV-B radiation) and indoor tanning [7], regardless of its multifactorial etiology. Although cSCC is typically benign and indolent, it can present as a spectrum of progressively advanced stages ranging from precursor actinic keratosis (AK) to invasive and, in rare cases, metastatic cSCC [8, 9].
Surgical excision is the mainstay of treatment for common primary cSCC, with a high rate of complete clinical and microscopic resection [10]. However, the management of advanced cSCCs, that encompass unresectable and metastatic disease, has proven challenging causing significant morbidity and mortality. Moreover, the complex molecular landscape and high mutational burden of cSCCs have confounded advances in precision medicine approaches [11]. Nevertheless, recent scientific advances and clinical trials testing new management approaches have significantly improved the National Comprehensive Cancer Network (NCCN) guidelines for cSCC prophylaxis and regional disease treatment with systemic cytotoxic drugs, immune checkpoint inhibitors, and targeted therapies. In addition, many clinical trials have investigated the efficacy of a multimodal approach involving adjuvant systemic therapy, neoadjuvant therapy, or both combined with surgery and/or radiation therapy [12]. Nonetheless, the development of effective drugs against aggressive cSCCs, characterized by high rates of tumor recurrence and the occurrence of secondary primary tumors, is gaining high priority.
In recent years, alternative medicines employing ‘natural agents’, principally phytochemicals, have garnered considerable interest for their potential preventive and therapeutic applications against cSCC [13]. Many preclinical studies have demonstrated the intriguing advantages of these phytochemicals over synthetic chemotherapeutic drugs, attributable to their diverse pharmacological properties, multidimensional targeting, favorable pharmacokinetics and broad safety profiles. For example, topical ingenol mebutate, obtained from the sap of Euphorbia peplus, was reported to be an effective and safe therapy for AK [14]. At the molecular level, ingenol mebutate acted as a pleiotropic effector, inducing cell death and manipulating immune-mediated responses [15]. Curcumin, a broad-spectrum phenolic compound extracted from turmeric (Curcuma longa) rhizome, has also been shown to inhibit cSCC growth and tumor progression in vivo by inhibiting the mTOR pathway [16].
Sanguinarine (Sng), derived from plants such as Sanguinaria canadensis
and Macleaya cordata, belongs to the group of benzylisoquinolines and
has been classified as an alkaloid antibiotic and botanical antifungal agent
[17]. The anti-cancer potential of Sng has also been established both in
vitro and in vivo against various human malignancies, such as
colorectal cancer [18], head and neck cancer [19] and drug-resistant
non-small-cell lung cancer [20]. Sng has been shown to induce apoptosis
(extrinsic and intrinsic) in cancer cells through several mechanisms, including
reactive oxygen species (ROS) generation and activation of c-Jun-N-terminal
kinase (JNK) and nuclear factor-kappa B (NF-
In the present study, we adopted an in vitro approach to investigate Sng’s anti-cancer and therapeutic potential in cSCC. Herein, we provide experimental evidence of Sng-mediated suppression of cSCC cell growth and stemness potential via apoptosis through ROS-dependent activation of the JNK signaling pathway.
Human cSCC epithelial cells A431 (primary; source: male) and A388 (metastatic;
source: female) were obtained from American Type Culture Collection (ATCC,
Manassas, VA, USA). The cell lines were routinely tested to ensure they were free
of microbial contamination. Cells were cultured in DMEM (Gibco, ThermoFisher
Scientific, MA, USA) supplemented with 10% (v/v) fetal bovine
serum (FBS) and 1% (v/v) penicillin-streptomycin (Gibco,
ThermoFisher Scientific, MA, USA) and maintained in a humidified 5%
CO
All the cell lines utilized in this study were procured from the ATCC,
accompanied by a Certificate of Analysis confirming their authentication through
STR profiling and their mycoplasma-free status. Upon receipt, the cell lines were
immediately thawed and cultured according to standard protocols in a humidified
incubator maintained at 37 °C and 5% CO
The in vitro cellular cytotoxicity of Sng alone and in combination with
inhibitors against cSCC cells was evaluated using the Cell Counting Kit-8 (CCK-8)
assay, as described previously [26]. Firstly, Sng
(Sigma-Aldrich, St. Louis, MO, USA) was dissolved in DMSO (Sigma-Aldrich, St.
Louis, MO, USA) to make a stock solution and further diluted with the
corresponding culture fluid to make a series of drug solutions. cSCC (10
The time- and dose-dependent cellular response profiles of Sng treatment were
measured using the xCELLigence Real-Time Cell Analysis high-throughput detection
platform. After setting up the program, 50 µL of cell culture medium was
added to each well of an E-plate 16 (ACEA Biosciences, San Diego, CA, USA). After
equilibration at 37 °C for 30 min, the E-plates were inserted into the
xCELLigence RTCA station (ACEA Biosciences, San Diego, CA, USA) to check for
appropriate electrical connections and gather the background impedance of the
cell culture medium. A431 and A388 cells were resuspended in cell culture medium,
seeded at the required cell density into each well of the E-plate, and incubated
for 24 h at 37 °C with 5% CO
The cellular response to pharmacological inhibitors, with or without Sng, was assessed via the trypan blue dye exclusion assay [27]. Briefly, A388 cells were seeded in 60-mm culture plates and maintained for 24 h in complete DMEM medium. Post incubation, cells were pretreated with SB203580 (a p38 inhibitor; 20 µM) or U0126 (an ERK1/2 inhibitor; 20 µM) for 2 h followed by the treatment with Sng (4 µM) for 6 h. Following treatment, the cells were harvested, centrifuged, and resuspended in 1 mL PBS. Cells were then stained with 0.4% trypan blue solution (1:1; Sigma-Aldrich, St. Louis, MO, USA), and 10 µL of cell suspension was applied to the edge of the dual-chamber cell counting slide (Bio-Rad, Hercules, CA, USA). Finally, the slide was inserted into the TC20™ automated cell counter (Bio-Rad, Hercules, CA, USA), and the cell viability was estimated as the percentage of live cells in the sample via trypan blue exclusion.
The LIVE/DEAD® Viability Kit (Life Technologies Corporation,
Carlsbad, CA, USA) was used according to the manufacturer’s instructions and as
previously described [28] to visualize the distribution of viable and non-viable
A431 and A388 cells 24 h post-treatment with Sng alone and in combination with
inhibitors. Briefly, A388 and A431 cells were seeded at a density of 1
The colony-forming assay was performed as described previously [29] to evaluate
the proliferative and colony-forming abilities of A388 and A431 cells treated
with Sng alone and in combination with inhibitors. Following optimization of the
number of cells required to obtain the best size and distribution of colonies,
A431 and A388 cells were seeded at a density of 10
The quantification of apoptosis and necrosis after treatment with Sng alone and
in combination with inhibitors (NAC, 10 mM; z-VAD-FMK, 50 µM; and SP600125,
30 µM) was performed using conventional Annexin V-FITC and Propidium iodide
(PI) staining (Annexin V-FITC apoptosis detection kit, BD Biosciences, San Jose,
CA, USA) followed by flow cytometric analysis. Approximately 1
To evaluate the capacity of Sng to modulate the mitochondrial membrane potential
(
First, A388 and A431 cells were treated with Sng alone or in combination with
z-VAD-FMK (50 µM) for 24 h. After treatment, cells were harvested, washed
twice in PBS, and centrifuged. The cells were then fixed and permeabilized using
a BD Cytofix/Cytoperm Plus Fixation kit (BD Biosciences, San Jose, CA, USA)
according to the manufacturer’s protocol. Approximately 1
To investigate Sng-induced oxidative stress, we pursued the generation of ROS in A388 and A431 cells treated with Sng alone or in combination with NAC (10 mM) or z-VAD-FMK (50 µM). 24 h post-treatment, the cells were harvested, washed using Hanks’ balanced salt solution (HBSS), and stained with CellROX™ Green Reagent (10 µM; to detect intracellular ROS) and MitoSOX Red reagent (5 µM; to detect mitochondrial superoxide) for 30 min at 37 °C. ROS levels were quantified using flow cytometry (BD LSR Fortessa analyzer, BD Biosciences, USA).
Sng-induced DNA double-strand breaks (DSBs) were analyzed by detecting
phosphorylated histone H2AX Ser139 (p-H2AX
The effect of Sng on cell cycle distribution was analyzed as previously described [26]. A388 and A431 cells were seeded in 100-mm culture dishes and treated with gradient concentrations of Sng alone and in combination with NAC (10 mM) or z-VAD-FMK (50 µM) for 24 h. After treatment, cells were harvested, centrifuged, and fixed in ethanol (70%) for 2 h. For analysis, cells were resuspended in PBS, incubated with RNAse A solution (0.04 µg/mL) for 30 min at 37 °C, and then treated with PI DNA cell-cycle stain (40 µg/µL) for 30 min at 4 °C. By gating the forward and side scatter profiles of the samples, doublets, disintegrated nuclei, and other cell debris were excluded from the analysis. The gates were uniformly maintained across all the samples in each cycle. After that, the relative DNA content was determined, and the percentages of cells in the sub-G0/G1, G0/G1, S, and G2/M phases were quantified by flow cytometry (BD LSR Fortessa analyzer, BD Biosciences, USA) based on the amount of PI fluorescence.
Tumor-derived spheroids/tumorspheres from A388 cells were generated in
non-adsorbent, ultra-low attachment, 6-well plates (Corning, USA). The
two-dimensional cell culture was maintained for 24 h under standard culture
conditions (37 °C with 5% CO
Following cell lysis with a 2X Laemmli sample buffer (Bio-Rad, Hercules, CA,
USA), the total protein from cells was extracted and quantified using a Nanodrop
ND-100 spectrophotometer (Thermo Fisher Scientific). Approximately 30–100
µg of protein from each sample was loaded and resolved by SDS-PAGE
(10–12%), transferred to a polyvinylidene difluoride (PVDF) membrane, and
blotted with primary antibodies against p-p38 (Thr180/Tyr182), p38, p-p44/42
(Thr202/Tyr204), p44/42, p27, p21, CDK6, caspase-3 , caspase-9, cleaved
caspase-8, PARP, BH3 interacting-domain death agonist (Bid), growth arrest-and
DNA damage-induced 45 A (GADD45A), and p-H2AX
The statistical significance between the treated and untreated samples was
analyzed by one-way ANOVA followed by Tukey’s multiple comparison test using
GraphPad Prism. All error bars represent the mean
Several toxicological studies have demonstrated the anti-cancer potential of Sng in both in vitro and in vivo tumor models [31, 32, 33, 34, 35]. To obtain an accurate assessment of the Sng dose-response in cSCC cells, cell viability was assessed upon screening with escalating concentrations of Sng (0.5–16 µM) for 24 h. Sng began to show a response at 2 µM, evident from the significant decrease in the number of viable cells in both A388 and A431 cells (Fig. 1A,B). The RTCA system was also used to evaluate the cytotoxic actions of Sng. Being a high-throughput and quantitative technique, RTCA enables continuous, label-free and real-time monitoring of dose-dependent cellular responses such as cell proliferation and morphological alterations [36]. Dynamic real-time monitoring of cSCC cells treated with Sng confirmed the antiproliferative effect of the compound, and results were comparable to those obtained from the CCK-8 assay (Fig. 1C,D). Because the reduction in the number of viable cells obtained from the CCK-8 assay could have been caused either by cytotoxicity or inhibited cell proliferation, we performed a LIVE/DEAD® cell viability assay, which detects live and dead cells from the fluorescence emitted by calcein AM and EthD-1, respectively. The live/dead assay also confirmed the effective cytotoxicity exerted by Sng on A388 and A431 cells, as evidenced by the significant reduction in the number of viable cells (reflected by the green fluorescent signal) after Sng treatment compared to the control group. Moreover, it provided proof of apoptosis, as indicated by an increase in the relative number of dead cells (reflected by the red fluorescent signal) (Fig. 1E,F). To assess the effect of Sng on reproductive cell survival and growth in vitro, we used a conventional 2D clonogenic or colony formation assay. Fig. 1G,H show whole-well images of colony formation across all treatment groups and reflect the capacity of Sng to influence and antagonize the extent of clonogenic growth (reflected by the number and size of tumor cell colonies) of both primary and metastatic cSCC cells.
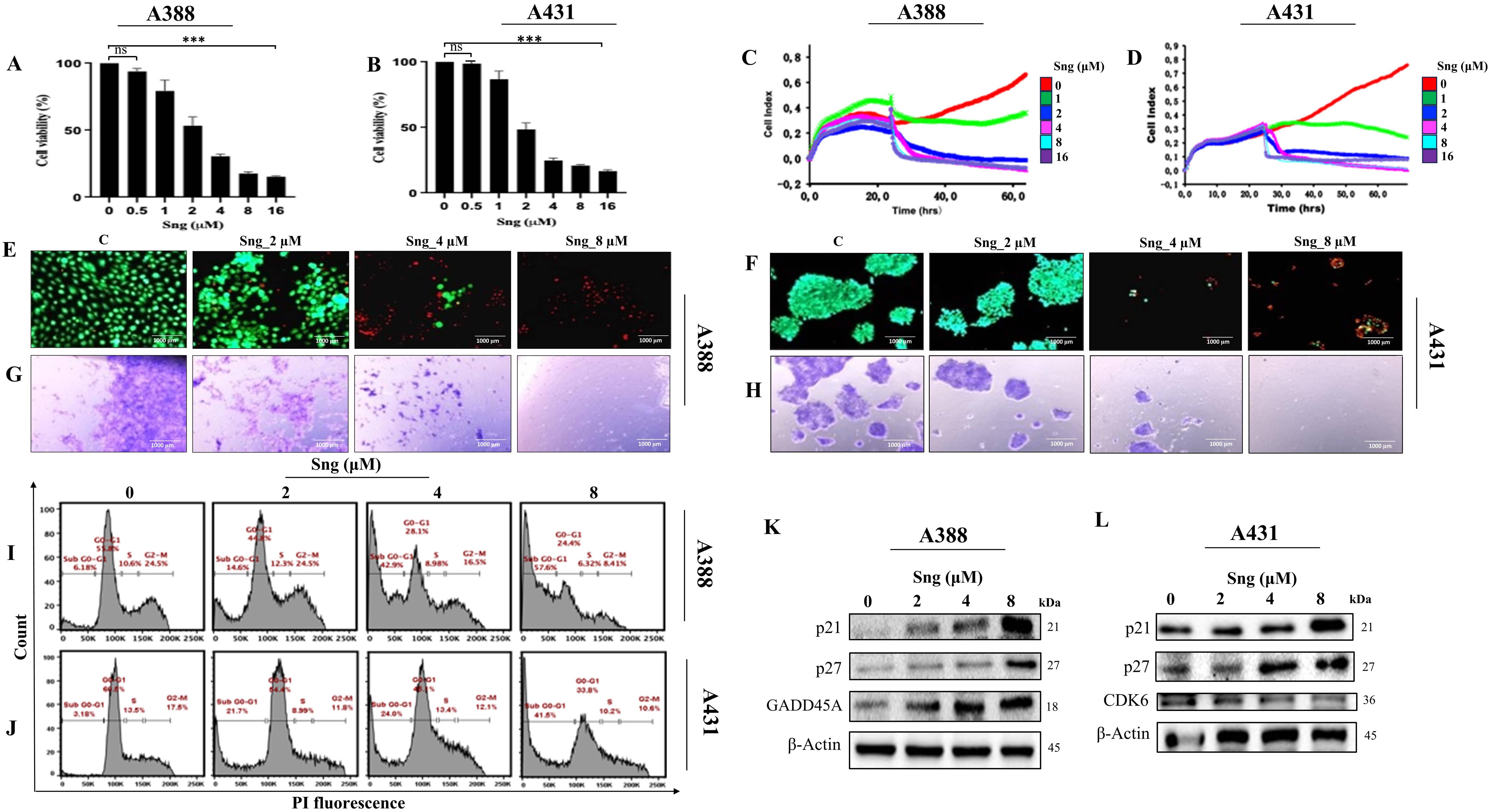 Fig. 1.
Fig. 1.Sanguinarine (Sng) inhibits proliferation, impairs viability,
and induces cell-cycle perturbations in cutaneous squamous cell carcinoma (cSCC)
cells. (A–D) Metastatic (A388) and primary (A431) cSCC cells were treated with
various concentrations of Sng (0.5–16 µM) for
24 h. The viability of the cells was determined by the CCK-8 assay
(A,B) while the Real-Time Cell Analysis (RTCA) system was used to record the cell
growth curves in response to different concentrations of Sng
(1–16 µM) (C,D) Data represented as mean
In addition to the drug dose-response, it is crucial to quantitatively assess the growth-inhibitory effect of the drug as a function of exposure time. To track the cellular response to Sng across time, A388 and A431 cells were exposed to 2 µM Sng at different time points (0–48 h) and subjected to toxicity and viability measurements. Time-dependent analysis of cellular response from 0 to 48 h indicated a decline in cell viability (Supplementary Fig. 1a,b), as well as morphological and biochemical changes consistent with apoptosis, including activation of caspase-3, -8 and DNA fragmentation (Supplementary Fig. 1c–f). These results implied the dose- and time-dependent cytotoxic effects of Sng in cSCC cells.
Concerning the cytostatic assessment of Sng via fluorescent PI-based
quantification of cell-cycle distribution, we determined that Sng alters the cell
cycle machinery by markedly increasing the accumulation of the pro-apoptotic
sub-G0/G1 fraction in both A431 and A388 cells in a
concentration-dependent manner (Fig. 1I,J). Subsequently, western blot analysis
was performed to delineate the molecular mechanisms underlying Sng-induced
cell-cycle arrest. The analysis showed that Sng modulates cell cycle regulatory
proteins, manifested by the upregulated expression of cyclin-dependent kinase
inhibitors p21
Focusing on the mechanistic aspects of cytotoxicity induced by Sng, we sought to unveil and characterize the major regulated cell death (RCD) modality [37] initiated by perturbations in the intracellular or extracellular microenvironments of cSCC cells. Since apoptosis has been reported to be the most profound RCD program triggered by Sng [26, 38], we assessed concentration-dependent morphological response that is characteristic of apoptosis in cSCC cells following exposure to Sng for 24 h. As shown in Fig. 2A,B, the morphological hallmarks of apoptosis, including cell shrinkage, membrane blebbing, and membrane-bound apoptotic bodies, were observed. To quantify apoptotic rates and segregate the apoptotic population, cSCC cells were subjected to Annexin V/PI staining. As shown in Fig. 2C,D, the majority of the Sng-treated cell population was in a late apoptotic state, although there was a significant increase in the percentages of both early and late apoptotic A388 and A431 cells associated with increasing concentrations of Sng. Induction of apoptotic cell death typically occurs via two major pathways: an extrinsic death receptor pathway and an intrinsic/mitochondrial pathway. Each of these pathways requires specific initiating stimuli to activate specific initiator caspases [39] and, in turn, executioner caspase-3. To ascertain whether the extrinsic pathway was associated with Sng-induced apoptosis, we assessed the expression of caspase-8 and caspase-3 in A388 and A431 cells treated with increasing concentrations of Sng. Treatment with Sng increased the expression of both caspase-3 (Fig. 2E,F) and cleaved caspase-8 (Fig. 2G,H) in the A388 and A431 cells in a concentration-dependent manner. Given that PARP is the preferred cellular substrate for caspases, we analyzed the cleavage of PARP in response to caspase-3 activation induced by Sng. As shown in Fig. 2G,H, and Supplementary Fig. 2a,b, there was an obvious increase in the activity of PARP, represented by its cleaved form, in both A388 and A431 cells exposed to increasing concentrations of Sng. DNA fragmentation is a biochemical hallmark of apoptosis, which can be detected through the phosphorylation status and foci formation of H2AX in cells [40]. Exposure to Sng triggered a concentration-dependent increase in Ser139 phosphorylation of H2AX in A388 and A431 cells, indicating the significant genotoxic effect of Sng (Fig. 2G,H). These findings suggest the execution of a caspase-dependent extrinsic signaling pathway following the cell death stimulus generated by Sng.
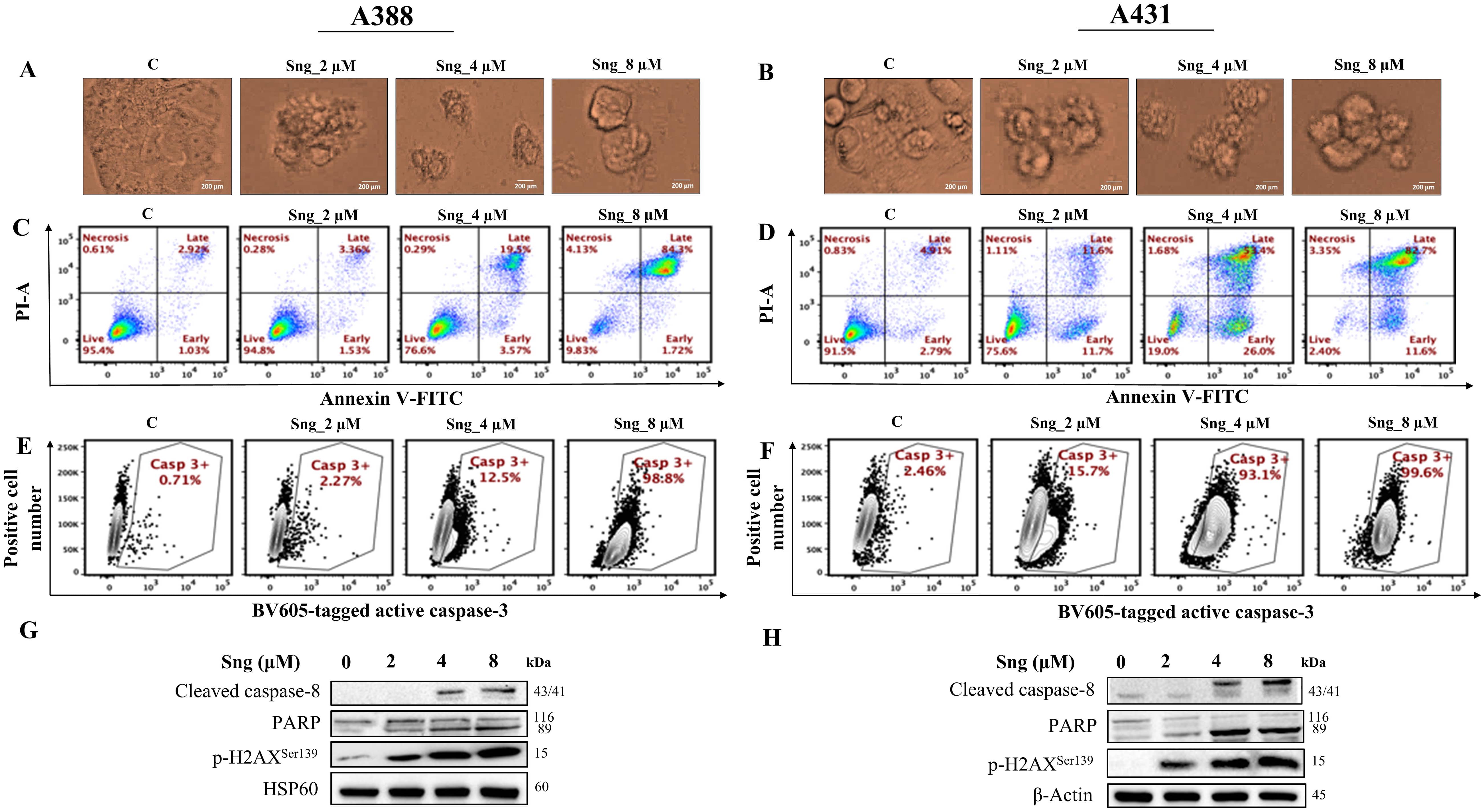 Fig. 2.
Fig. 2.Sanguinarine induces apoptosis in cSCC cells. (A,B) Microscopic images of cSCC cells showing morphological abnormalities induced by
Sng (2 µM, 4 µM, and 8 µM) (magnification 20
To substantiate the role of caspases in Sng-induced cellular apoptosis, A388 and A431 cells were preincubated with the pan-caspase inhibitor z-VAD-FMK (50 µM), followed by treatment with Sng (4 µM). z-VAD-FMK is a cell-permeable, irreversible fluoromethyl ketone (FMK)-derivatized peptide that acts as an efficient caspase inhibitor [41]. Applying z-VAD-FMK significantly restored the viability of the Sng-treated A388 and A431 cells (Fig. 3A,B). Evaluation of specific cell fractions by Annexin V/PI staining revealed that preventing the cleavage of caspases with z-VAD-FMK remarkably attenuated the accumulation of apoptotic cells relative to cells treated with Sng alone (Fig. 3C,D). The percentage of apoptotic (early and late) cells appeared as a readout of enhanced apoptosis in Sng-treated cells but was minimally detectable in z-VAD-FMK and Sng co-treated cells. Similarly, the Live/Dead assay also revealed an increase in the density of live cells in the z-VAD-FMK-treated group (Fig. 3E,F), corroborating the flow cytometry results. The clonogenic abilities of A388 and A431 cells were also restored upon pretreatment with z-VAD-FMK (Fig. 3G,H). Furthermore, co-treatment of cells with z-VAD-FMK and Sng significantly antagonized the expression of Sng-induced cleaved caspase-8 and PARP (Fig. 3I,J). These findings suggest that Sng induces caspase-dependent apoptosis in cSCC cells.
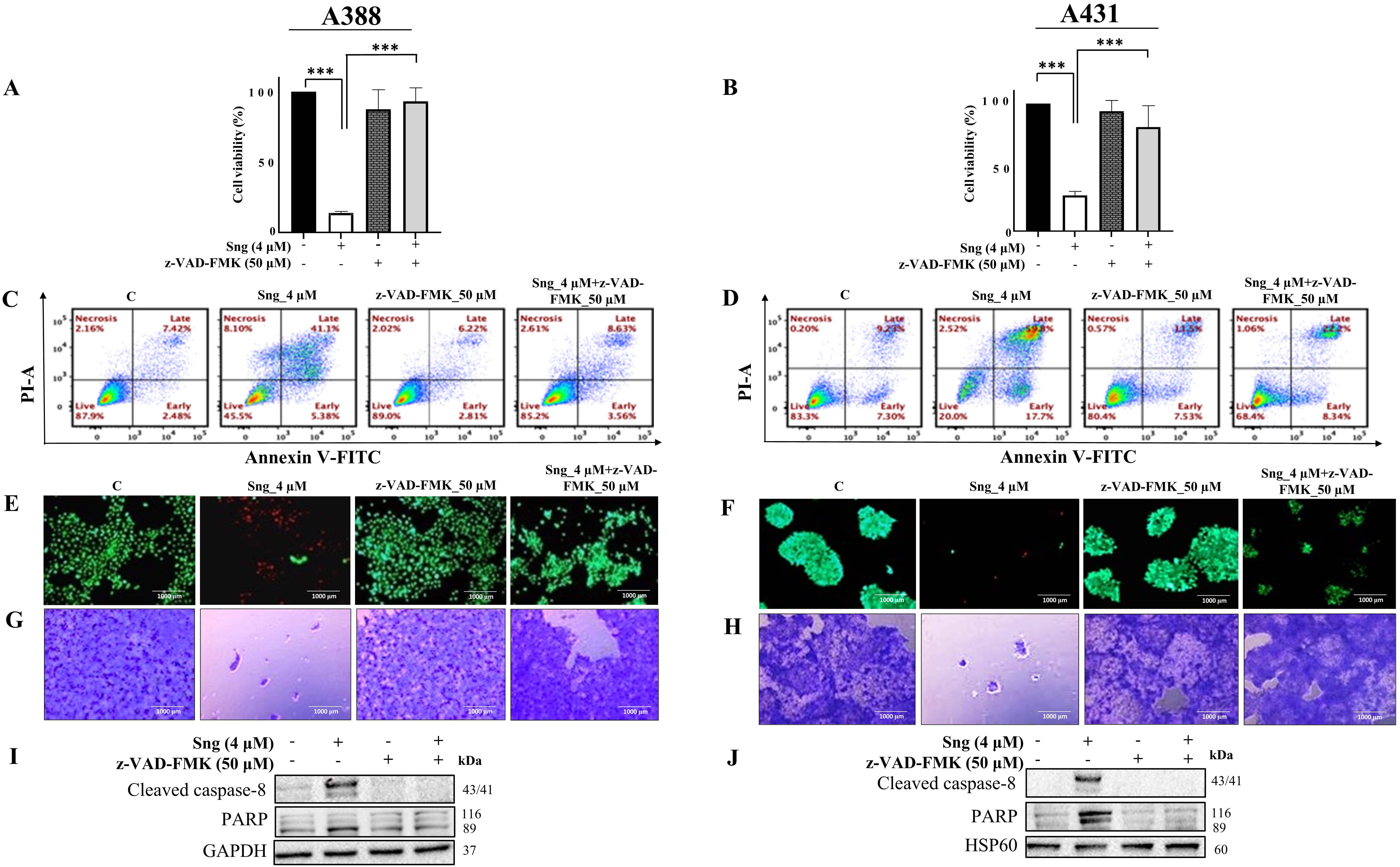 Fig. 3.
Fig. 3.Sanguinarine induces caspase-mediated apoptosis in cSCC cells.
(A,B) Pretreatment with the general caspase inhibitor z-VAD-FMK restores cell
viability in A388 and A431 cells following exposure to Sng. A388 and A431 cells
were pretreated with 50 µM z-VAD-FMK for 1 h
before being challenged with 4 µM Sng. Cell viability was
determined by CCK-8 analysis. Data presented as the mean
Delving deeper into the activity of z-VAD-FMK, we assessed whether z-VAD-FMK can
reverse Sng-induced cell-cycle defects and found that z-VAD-FMK prevented
the accumulation of sub-G0/G1 fraction in A388 cells
(Supplementary Fig. 3c). Because Sng-treated cells expressed active
caspase-3, we sought to determine its expression in response to pretreatment with
z-VAD-FMK. z-VAD-FMK reduced the expression of active caspase-3 compared to that
in the Sng-treated group (Supplementary Fig. 3e). Moreover, we
quantified p-H2AX
ROS, a class of highly reactive molecules, are critical regulators of apoptosis. Considering the bidirectional nature of ROS [42], we determined the ability of Sng to manipulate levels of ROS in cSCC cells. Using CellROX and MITOSOX fluorogenic probes, we showed that Sng markedly increased intracellular (Fig. 4A,B) and mitochondrial (Supplementary Fig. 2c,d) ROS production in a concentration-dependent manner in both A388 and A431 cells. This ROS production was blocked by NAC (10 mM), a universal cytoprotective antioxidant that serves as a ROS scavenger and precursor of glutathione (GSH) synthesis in Sng-treated A388 (Fig. 4C; Supplementary Fig. 4a) and A431 cells (Fig. 4D). NAC also restored GSH levels (Supplementary Fig. 4d) that were elevated in A388 and A431 cells exposed to Sng (Supplementary Fig. 2e,f).
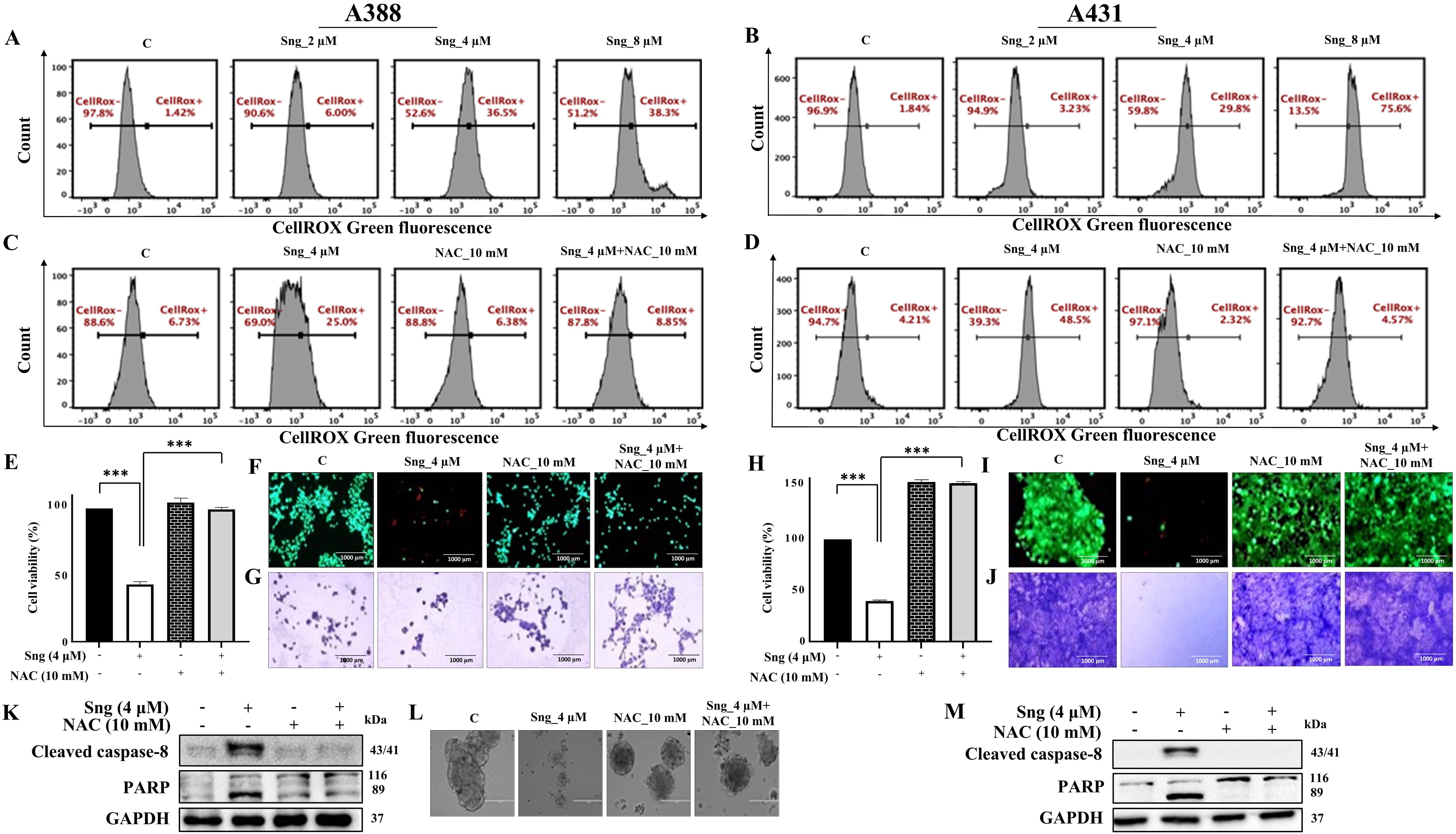 Fig. 4.
Fig. 4.Sanguinarine induces reactive
oxygen species (ROS)-dependent apoptosis in cSCC cells.
(A,B) Frequency histograms showing the generation of intracellular ROS in A388 and A431 cells treated with increasing
concentrations of Sng for 24 h. (C,D) Frequency histograms showing the effect on
intracellular ROS in A388 and A431 cells following pretreatment with
N-acetylcysteine (NAC) (10 mM) and then by Sng (4 µM) for 24 h. (E,H)
Pretreatment with the ROS scavenger NAC restores cell viability in A388 and A431
cells following exposure to Sng. A388 and A431 cells were pretreated with
10 mM NAC for 1 h before being challenged with
4 µM Sng. Cell viability was determined by CCK-8 analysis.
Data presented as the mean
Next, we investigated the role of ROS in the regulation of Sng-induced apoptosis. Pretreatment with NAC remarkably preserved cell viability and abrogated cytotoxicity in Sng-treated A388 (Fig. 4E,F) and A431 cells (Fig. 4H,I). The clonogenic assay in Fig. 4G,J further suggested that NAC restored cell survival and clonal expansion of A388 and A431 cells compared to cells treated with Sng alone. Furthermore, applying NAC decreased the sub-G0/G1 population (Supplementary Fig. 4b) of A388 cells. To determine whether ROS production precedes caspase-dependent apoptosis in cSCC cells, we determined cleaved caspase-8 and PARP expression levels in A388 and A431 cells cotreated with NAC and Sng. The expression of cleaved caspase-8 and PARP was markedly reduced upon pretreatment with NAC compared to cells treated with Sng alone (Fig. 4K,M). In 3D spheroid cultures, dissolution, the loss of well-circumscribed edges of normally compact cell aggregates, corresponds to the induction of apoptosis [43]. Interestingly, the dissolution of metastatic A388 cell-derived multicellular spheroids was also inhibited by NAC treatment alone or in combination with Sng (Fig. 4L).
To further investigate the role of ROS in caspase-mediated cell death, we analyzed changes in intracellular and mitochondrial ROS levels in A388 cells pretreated with z-VAD-FMK. Both intracellular and mitochondrial ROS production were significantly reduced upon pretreatment with z-VAD-FMK. In contrast, they remained more or less unchanged in cells treated with either z-VAD-FMK or Sng alone (Supplementary Fig. 3a,b). These findings emphasize the functional role of the caspase-dependent pathway in Sng-induced apoptosis, which is secondary to the modulation of ROS.
Sng has been previously shown to elevate ROS levels in neoplastic cells, thereby
disrupting redox homeostasis and inducing oxidative stress [21, 26]. Because
oxidative stress can occur at either a high
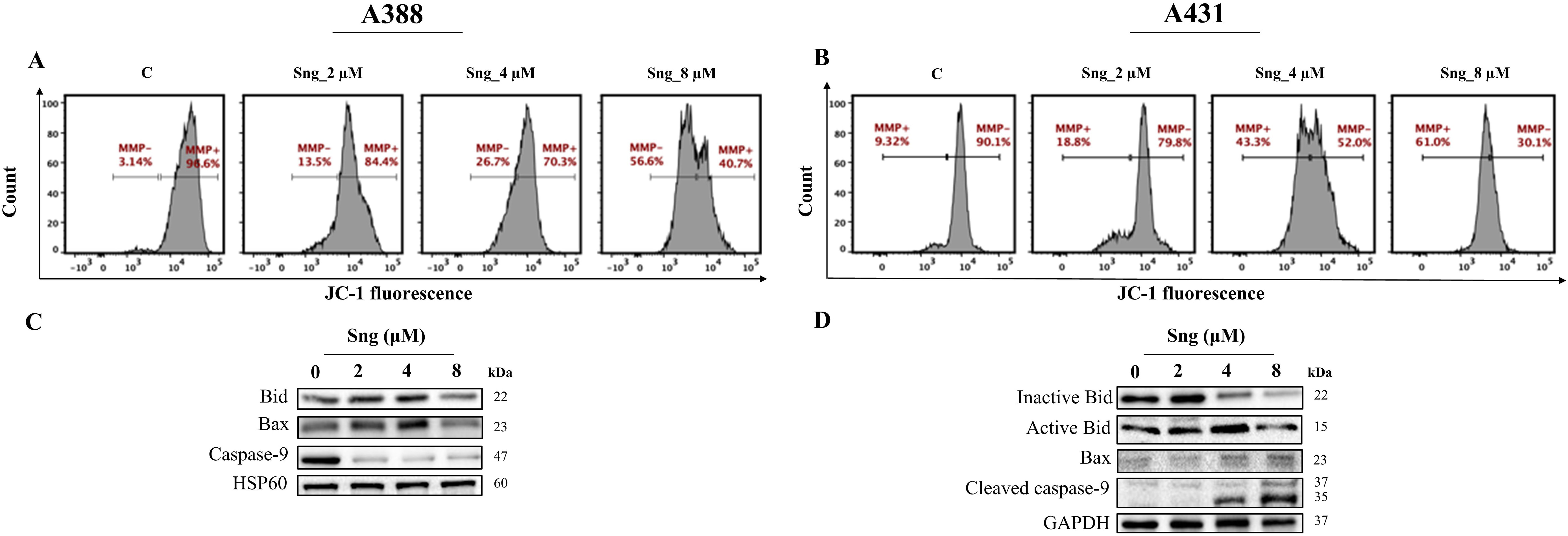 Fig. 5.
Fig. 5.Disruption of mitochondrial membrane potential in response to
Sanguinarine in cSCC cells. (A,B) Distribution plots representing the percentage
of cells exhibiting loss of
The fact that alterations in mitochondrial energization, particularly the loss
of
Substantial evidence has highlighted the pivotal role of MAPKs in regulating stimulus-specific cellular responses, including cell growth, migration, proliferation, differentiation, and apoptosis [48]. To elucidate the possible link between apoptosis and the MAPK pathway in cSCC cells, we investigated the influence of Sng on the MAPK signaling pathways in A388 cells. Indeed, Sng promoted the phosphorylation and activation of JNK, p38, and ERK (p44/42 MAPK), with maximal phosphorylation (specifically that of JNK and ERK) observed at 8 µM Sng (Fig. 6A), thus corroborating the involvement of MAPKs in Sng-induced apoptosis in A388 cells. The expression of JNK, p38, and ERK remained unchanged. Following this observation, we investigated the impact of SB203580 (a p38 inhibitor), U0126 (an ERK1/2 inhibitor), and SP600125 (a JNK inhibitor) on MAPK signaling-mediated caspase-dependent apoptosis in A388 cells. According to the trypan blue assay, the Sng-induced decrease in viable A388 cells was not restored upon the pretreatment with SB203580 and U0126 (Supplementary Fig. 5a,b). SB203580/Sng co-treatment inhibited the expression of cleaved caspase-8 and PARP, whereas U0126/Sng co-treatment failed to inhibit the Sng-induced activity of these proteins in A388 cells. Moreover, inhibition of p38 and ERK did not attenuate the Sng-induced H2AX phosphorylation at Ser139, indicating no reversibility of DNA damage prompted by Sng in A388 cells (Supplementary Fig. 5c,d).
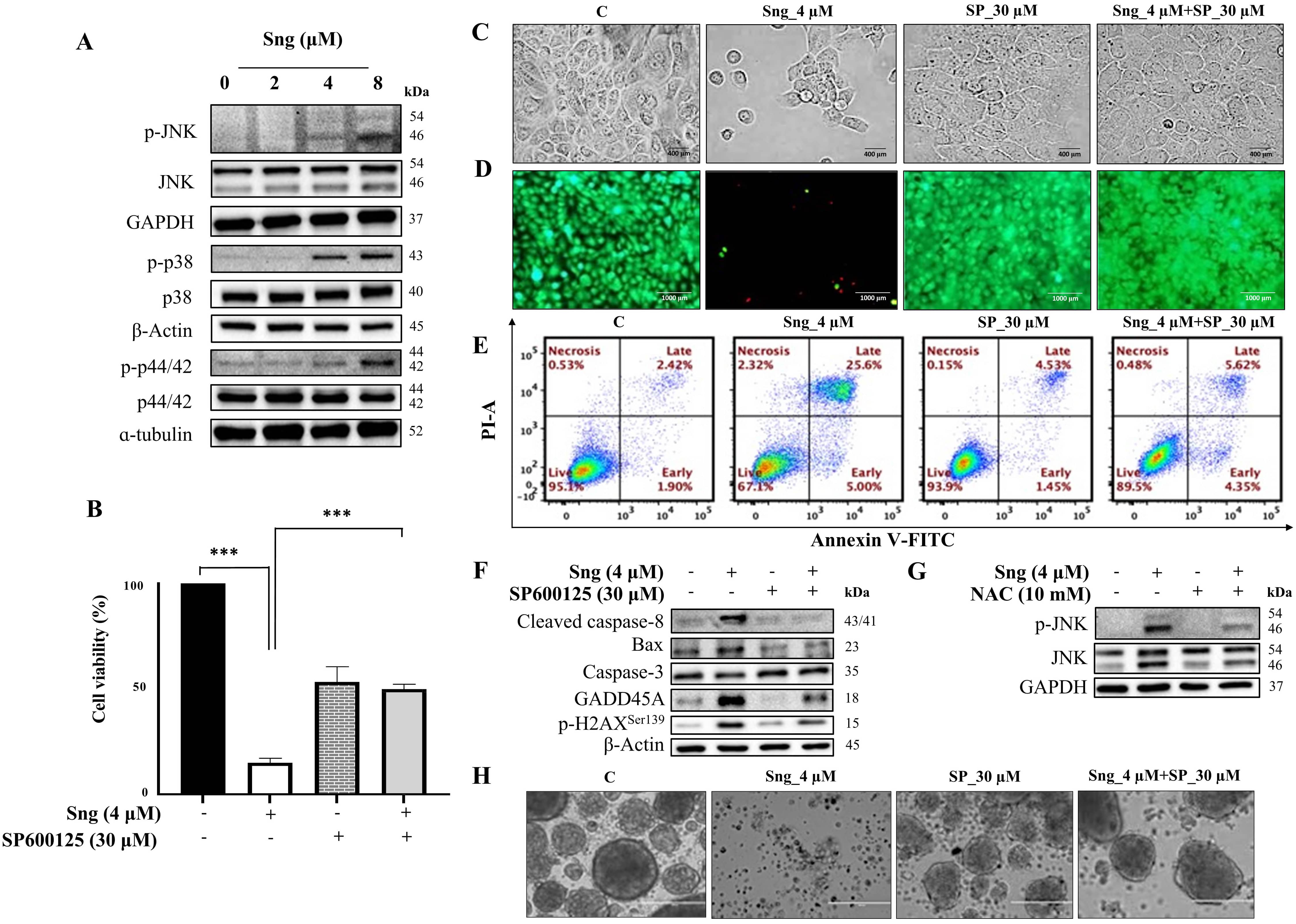 Fig. 6.
Fig. 6.Activation of the JNK signaling pathway by Sanguinarine in A388
monolayer cells and spheroids. (A) Western blot analysis of JNK, p38, and ERK
phosphorylation in A388 cells treated with indicated concentrations of Sng for 6
h. (B) Viability assessment of A388 cells treated with or without SP600125 (30
µM) by CCK-8 assay. Data presented as the mean
Earlier studies have delineated the capacity of Sng to induce ROS production and
effectively activate the JNK signaling pathway [49]. Therefore, we determined the
functional role of JNK signaling in Sng-induced apoptosis in cSCC cells using the
pharmacological JNK inhibitor SP600125. As shown in Fig. 6B, restoration of cell
viability was observed when Sng-treated A388 cells were pre-exposed to SP600125
for 2 h. Viability assessment following inhibition of JNK activity in A431 cells
also yielded similar results (data not shown). In A388 cells, pretreatment
preserved the morphological characteristics (Fig. 6C) and
markedly decreased the number of dead cells compared to the Sng-treated group
(Fig. 6D). As derived from flow cytometry analysis (Fig. 6E), SP600125
pretreatment significantly attenuated the accumulation of apoptotic cells
relative to cells treated with Sng alone. Furthermore, blocking JNK with SP600125
attenuated the Sng-induced expression of caspase-3, cleaved caspase-8, Bax,
GADD45A, and p-H2AX
To elucidate whether ROS are involved in Sng-mediated JNK activation, A388 cells were treated with Sng with or without NAC. Co-treatment of A388 cells with Sng and NAC for 6 h blocked JNK activation relative to those treated with Sng alone (Fig. 6G). These results confirmed the participation of ROS-dependent JNK signaling in the pro-apoptotic effects induced by Sng cSCC cells.
Compared to rescuing parental monolayer cells from the apoptotic effect of Sng, SP600125 also prevented the Sng-induced dissolution of intact spheroids. Both SP600125 and SP600125-Sng co-treated spheroids maintained well-circumscribed edges comparable to those of the control (Fig. 6H), indicating spheroid viability.
In vitro, 3D tumor spheroid models have emerged as viable tools for drug screening. To analyze the cell response to Sng in 3D culture, we assessed the dissolution of metastatic A388 cell-derived spheroids. The so-called tumor-derived spheroids were generated in a 3D scaffold-free culture system using 2D A388 adherent cells that were grown as floating spheres in an ultra-low attachment 6-well plate and sensitized to increasing concentrations of Sng for 7 days. A388 cells formed dense globe-like aggregates with prominent core formation (Fig. 7A). Following a 7-day treatment period, the dissolution of the spheroids in each well was assessed. At a concentration of 2 µM, Sng showed no apoptotic response, as the spheroids retained their well-circumscribed edges identical to the control. This contrasts the higher concentrations of Sng (4 and 8 µM), which showed a mixed apoptotic response, characterized by the co-existence of spheroids with significantly distorted edges and single-cell populations (Fig. 7A).
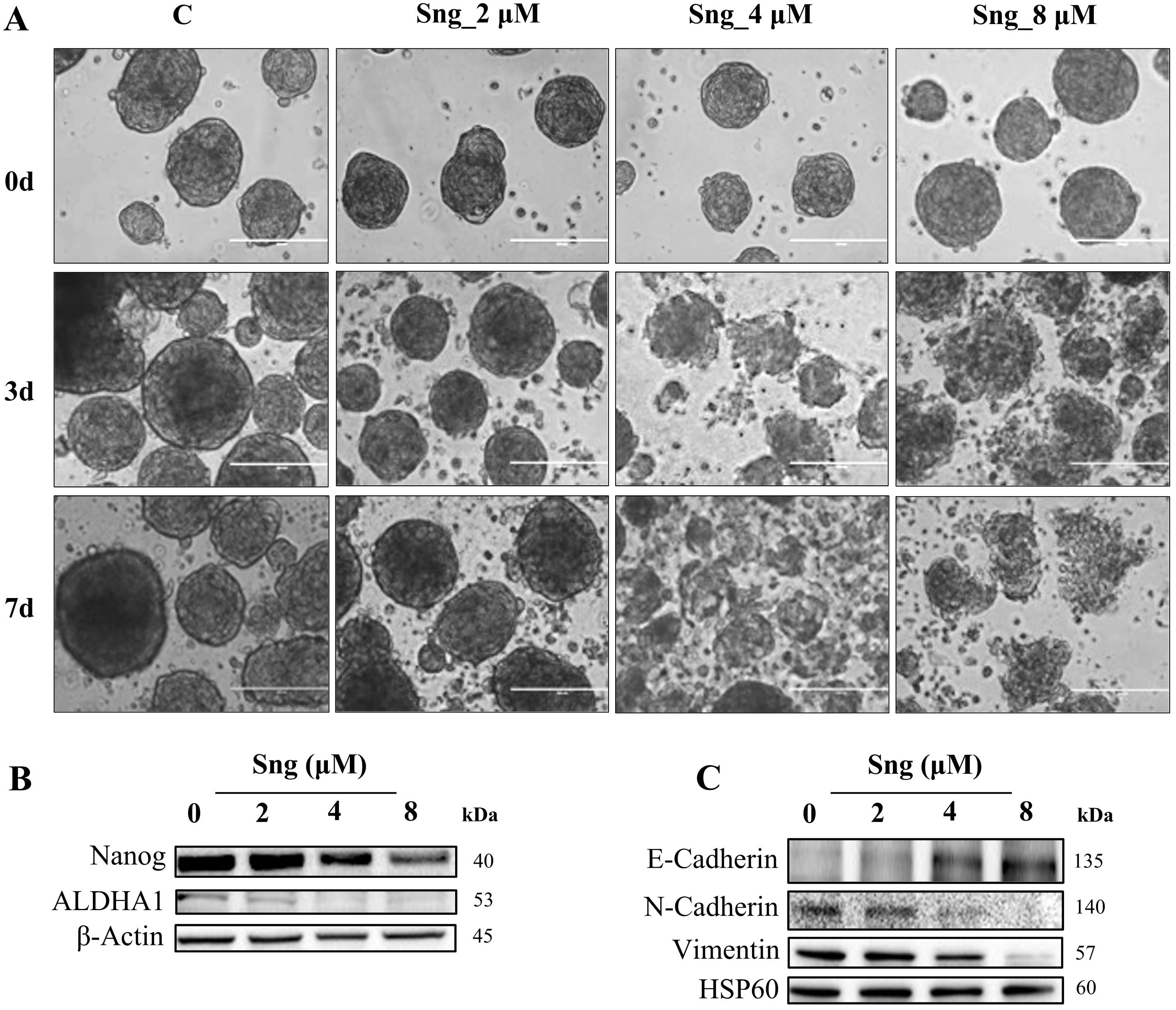 Fig. 7.
Fig. 7.Ablation of stemness potential by Sanguinarine in metastatic
cSCC cells. (A) Representative optical images of A388 cell-derived spheroids
that were systematically investigated for the effect of Sng on spheroidal
dissolution and apoptosis at 0, 3, and 7 cultivation days (magnification
10
To investigate the effect of Sng on cancer stem cells (CSCs) enriched in the formed spheroids, we assessed the expression of the key stemness gene Nanog and the cell surface marker aldehyde dehydrogenase A1 (ALDHA1). Sng-treated A388 spheroids displayed downregulation of both Nanog and ALDHA1 expression (Fig. 7B). Epithelial-mesenchymal transition (EMT) is a highly plastic, dynamic transitional process characterized by the loss of apical polarity and gain of mesenchymal properties, cell motility, and stemness in epithelial cells in a variety of cancerous tissues [50]. Considering that EMT confers stem cell-like traits to neoplastic cells, we evaluated the ability of Sng to abrogate the EMT phenotype in A388 spheroids. Western blot analysis demonstrated an increase in the expression levels of the epithelial marker E-cadherin, with concurrent downregulation in the levels of the mesenchymal markers N-cadherin and vimentin upon treatment with Sng (Fig. 7C).
Growing awareness of the complex dynamics of cSCC in recent decades has widened
the understanding of its molecular pathogenesis and, thus, the therapeutic
arsenal against this commonly occurring cutaneous malignancy. However, in
comparison to the common primary cSCCs that present indolent clinical behavior
and favorable prognosis, the burden of metastatic cSCCs portending poorer
prognosis and higher mortality rates (
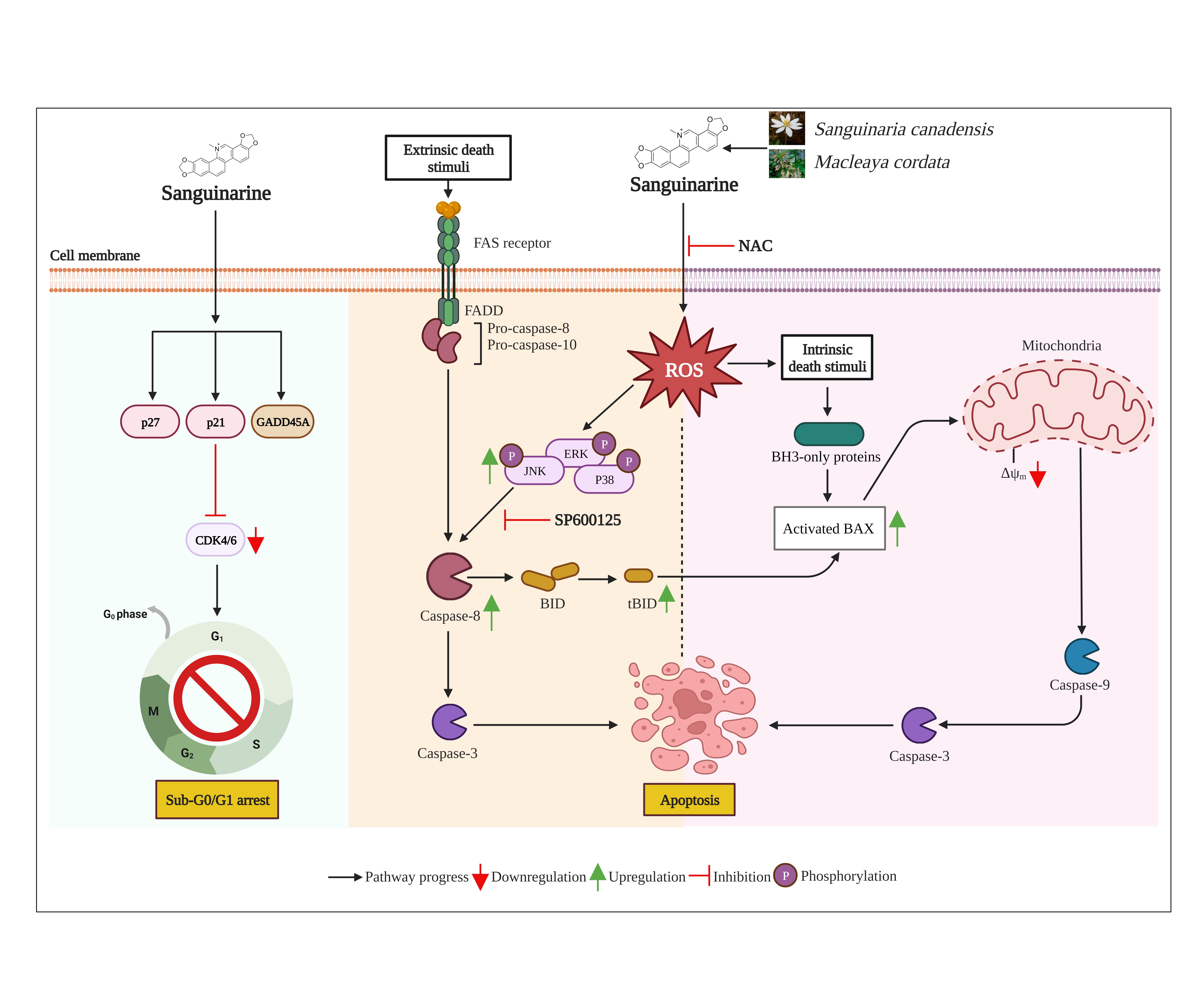 Fig. 8.
Fig. 8.Schematic diagram depicting the proposed molecular mechanisms that underscore the anti-cancer activity of Sanguinarine in cSCC cells. Sng induces sub-G0/G1 arrest and triggers apoptosis by activating the JNK pathway that is dependent on the ROS-mediated signaling in human cSCC cells. CDK, Cyclin-dependent kinase; FADD, Fas Associated Via Death Domain; GADD45A, growth arrest and DNA damage-inducible gene 45 alpha; tBID, truncated bid. Created with BioRender.com.
Drug toxicity can be defined as the combination of two processes: induction of
cell death (cytotoxic effect) and inhibition of proliferation and metabolism
(cytostatic effect). Consistent with the published literature [54], our study
suggests that Sng exerts both cytostatic and cytotoxic effects on cSCC cells.
According to the cytotoxicity assays, Sng decreased the number of viable cells
and dramatically reduced the clonogenic abilities of both primary and metastatic
cSCC cells. Previous experimental observations have demonstrated the preferential
cytotoxicity of Sng to A431 cells via apoptosis compared to normal human
epidermal keratinocytes [55], thus unraveling the differential growth-inhibitory
response of Sng in cancer cells versus normal cells. Cellular
homeostasis relies on the precise regulation of cellular signaling systems that
control the spatial and temporal balance of cell proliferation, growth arrest,
and apoptosis [56]. The hub of these signaling systems is the cell cycle, the
progression of which is regulated by sequential activation and inactivation of
different cyclin/cyclin-dependent kinase (CDK) regulatory complexes [57]. Our
study revealed that Sng induces significant sub-G0/G1 cell-cycle arrest in cSCC
cells, suggesting that Sng acts via a cell cycle-specific mechanism. Critical to
cell cycle exit is the activity of CDK inhibitors, including p21
In addition to growth arrest, negative growth rate control, such as apoptosis,
is critical in regulating aberrant development in cancer. Apoptosis is a
universal, highly complex RCD mode that occurs via two pathways: (a) the
extrinsic/death receptor pathway, initiated by perturbations of the extracellular
microenvironment that are detected by activation of cell surface death receptors,
propagated by apical caspase (-8), and precipitated by executioner caspases,
mainly caspase-3; and (b) the intrinsic/mitochondrial pathway, initiated by
perturbations of the intracellular or extracellular microenvironment, demarcated
by mitochondrial dysfunction, propagated by apical caspase (-9), and precipitated
by caspase-3 [37]. Both pathways converge on the activation of downstream
caspases, which are normally expressed as inactive zymogens (procaspases) until
stimulation. In the present study, we showed that initiator caspases-8 and -9 and
effector caspase-3 are concomitantly activated upon treatment with Sng,
suggesting the involvement of both the extrinsic and intrinsic pathways in
Sng-induced apoptosis. Caspase inhibition by z-VAD-FMK substantially inhibited
Sng-mediated inhibition of cell viability, clonogenic potential, and apoptosis,
thus implicating caspase cascades in Sng-mediated apoptosis of cSCC cells. The
morphological documentation also revealed the cellular changes typical of
apoptosis, which allied with the analysis from flow cytometric Annexin V/PI assay
and western blot, suggesting a majorly apoptotic mode of death. Progressive DNA
damage, reflected by upregulated p-H2AX
The complex relationship between ROS and cancer depends on the precise
fine-tuning of ROS production and scavenging. During carcinogenesis, ROS exhibits
a paradoxical role: ectopic ROS accumulation promotes the proliferation and
survival of cancer cells via the activation of redox-sensitive signaling
pathways, whereas excessive ROS levels promote cell death [59]. Given that
reprogramming of redox metabolism can counteract tumorigenesis, our study
demonstrated the potential of Sng to serve as a ROS-stimulating agent,
overwhelming redox adaptation and inducing oxidative stress in cSCC cells. The
loss of
Ser/Thr protein kinases, such as JNKs, are components of the “three-tiered” MAPK signaling that mediate an array of cellular responses, including cell survival and apoptosis, in response to a variety of abiotic and biotic stress insults, such as oxidative stress and DNA damage [61]. In this study, we demonstrated that JNK is an important mediator of cytotoxic damage induced by Sng in both monolayer and spheroid cultures of metastatic cSCC cells. The cytotoxic effect of Sng significantly increased the phosphorylation of JNK in metastatic cSCC cells. In contrast, the pharmacological inhibition of JNK protected the cells from Sng-induced cytotoxicity in vitro and attenuated the expression of caspase-3 and cleaved caspase-8. Inhibition of JNK activation also reversed the Sng-induced expression of p-H2AX and GADD45A, thus linking previously thought unrelated phenomena, that is, the DNA damage-induced expression of GADD45-like genes and the DNA damage-induced activation of the JNK pathway. In addition to cell cycle arrest, GADD45 proteins have been implicated in DNA damage and environmental stress-related responses via the activation of the MEKK4/p38/JNK signaling pathway [62]. Moreover, JNK inhibition downregulated the expression of Bax, thus connecting the stress-activated signaling pathway to the cell death machinery involved in mitochondrial apoptosis. Furthermore, the ROS scavenger NAC preserved Sng-induced JNK phosphorylation, suggesting the involvement of ROS/JNK pathway in Sng-induced apoptosis of metastatic cSCC cells.
Effect of Sng on spheroids revealed the possible potency of the drug in addition to traditional 2D monolayer cultures. Sng treatment elicited a mixed apoptotic response in 3D spheroids, as indicated by the co-existence of partially dissolute spheroids and single-cell populations. This response can be attributed to Sng targeting only proliferative cells predominantly residing in the peripheral zone of the spheroids. Future studies assessing the penetration of Sng into spheroids are required to determine whether it is one of the causal factors underlying the limited response of Sng. Nevertheless, Sng displayed considerable potency against cSCC cell-derived spheroids. Tumor-derived spheroids have been widely used to enrich putative CSCs and evaluate stem cell-like properties in vitro. Amidst the current focus on developing therapeutic strategies to target and eliminate CSCs to counteract drug resistance and tumor recurrence, our study revealed that the stemness-targeting ability of Sng was prompted by the downregulation of Nanog and ALDHA1. Moreover, considering that EMT and CSCs belong to the triad of major contributing factors of metastasis and are interlinked [63], we also demonstrated the capacity of Sng to target EMT effector molecules in tumorspheres obtained from metastatic cSCC cells.
In conclusion, our study explored the anti-cancer potential of Sng against cSCC cells, which is mediated through cell-cycle arrest leading to apoptosis via the ROS/JNK signaling pathway. In view of its chemosensitive abilities in vitro, the cytotoxic potential of Sng, at relatively lower dosages, requires further evaluation in animal efficacy models. Moreover, investigations into the pharmacologic synergy and efficacy between Sng and other phytochemicals and/or traditional chemotherapeutic drugs are warranted to identify, test, and develop an efficacious clinically relevant combinatorial drug regimen against cSCC.
This published article and its supplementary information files include all data generated and/or analysed during this study.
MS and SU designed and supervised the research and coordinated the project. KP conducted the main experiments. KP, SU and MS wrote the manuscript. AQK, FA, SK, RA, JMM and AAB helped in drafting the manuscript, critically reviewing it, data analyzing. AA, AAB and JB involved in designing, performing analysis of the data as well helped in writing of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was funded by Medical Research Center, Hamad Medical Corporation, under grant no. MRC-01-23-065.
Given his role as a guest editor, Dr. Shahab Uddin had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Prof. Amancio Carnero Moya. The other authors have no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.