




1 Key Research Laboratory of Germplasm Resources and Standardized Planting of Genuine Regional Medicinal Materials Produced in Hunan Province, School of Pharmacy, Hunan University of Chinese Medicine, 410208 Changsha, Hunan, China
2 Laboratory of Vascular Biology and translational medicine, School of Medicine, Hunan University of Chinese Medicine, 410208 Changsha, Hunan, China
3 Department of Science and Education, The first Hospital of Hunan University of Chinese Medicine, 410007 Changsha, Hunan, China
Abstract
Vascular diseases are amongst the most serious diseases affecting human life and health globally. Energy metabolism plays a crucial role in multiple vascular diseases, and the imbalance of energy metabolism in cells from the blood vessel wall can cause various vascular diseases. Energy metabolism studies have often focused on atherosclerosis (AS) and pulmonary hypertension (PH). However, the roles of energy metabolism in the development of other vascular diseases is becoming increasingly appreciated as both dynamic and essential. This review summarizes the role of energy metabolism in various vascular diseases, including AS, hemangioma, aortic dissection, PH, vascular aging, and arterial embolism. It also discusses how energy metabolism participates in the pathophysiological processes of vascular diseases and potential drugs that may interfere with energy metabolism. This review presents suggestions for the clinical prevention and treatment of vascular diseases from the perspective of energy metabolism.
Keywords
- glycolysis
- mitochondria
- atherosclerosis
- hemangioma
- aortic dissection
- pulmonary hypertension
Various pathologies associated with the vasculature, including hypertension, atherosclerosis (AS), aortic aneurysm, aortic dissection, pulmonary hypertension (PH), and stenosis, are considered to be some of the most serious diseases affecting human life and health worldwide [1]. Furthermore, energy metabolism is known to play a crucial role in the etiology of a number of vascular diseases. An imbalance of energy metabolism has been closely associated with vascular diseases, such as AS [2], abdominal aortic aneurysm [3], PH [4], aortic dissection [5], and vascular aging [6]. Pathological changes in specific vascular diseases usually cause changes in the energy metabolism of cells in the vascular wall, including vascular endothelial cells (VECs), vascular smooth muscle cells (VSMCs), and macrophages [7, 8, 9].
Energy metabolism is critical for maintaining normal cell function and life. In
eukaryotes, nutrients are converted by glycolysis to pyruvate, which then enters
the mitochondria, where it is further decarboxylated to acetyl coenzyme A
(acetyl-CoA). Under aerobic conditions, a acetyl-CoA enters the tricarboxylic
acid (TCA) cycle where it is reduced through the oxidative respiratory chain
pathway to produce nicotinamide adenine dinucleotide (NADH), with the generation
of H
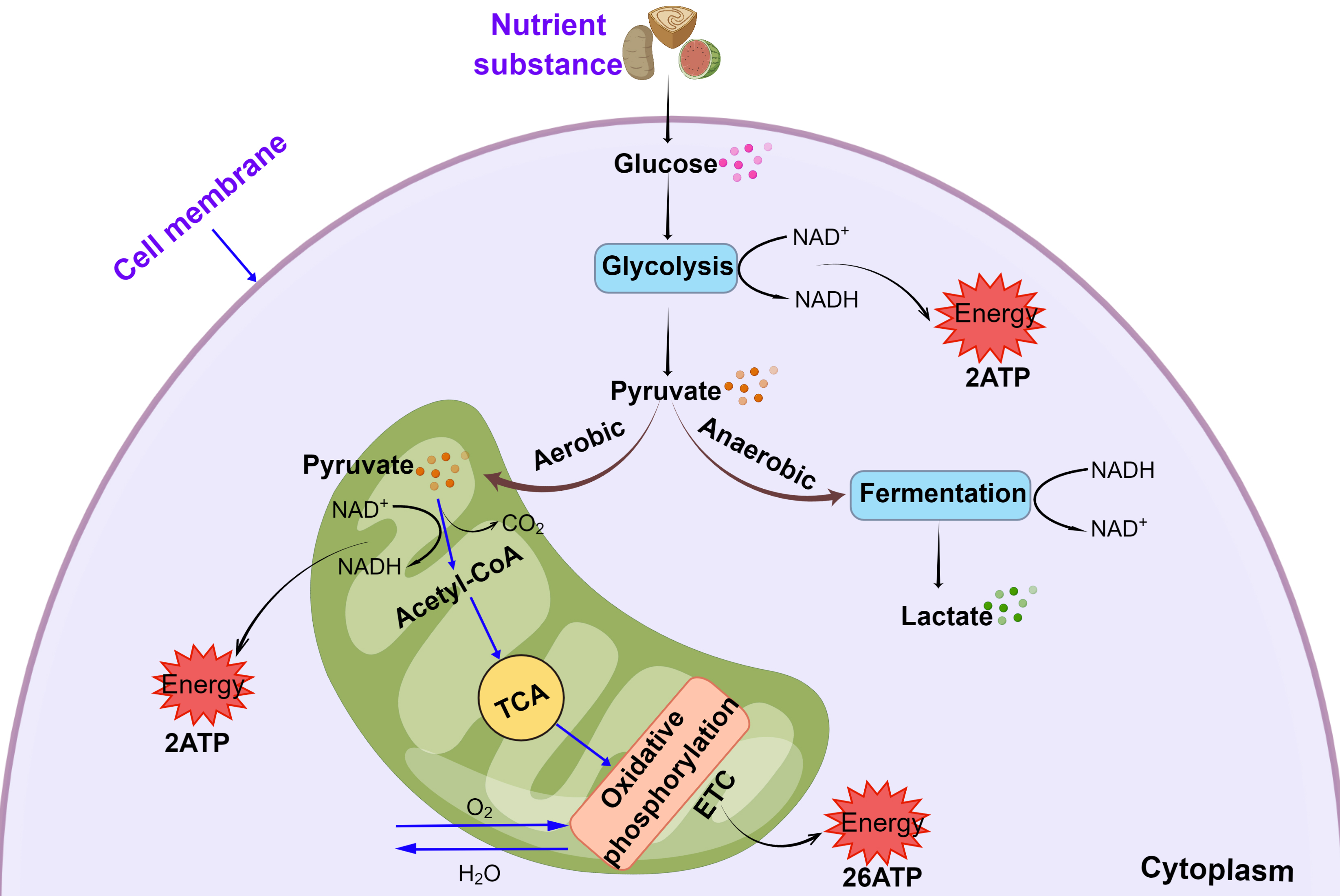 Fig. 1.
Fig. 1.Mitochondrial oxidative phosphorylation and glycolytic pathways.
Abbreviations: ATP, adenosine triphosphate; NAD
Mitochondrial respiration and glycolysis are the major energy metabolism pathways in cells. However, they also regulate various cellular processes, including differentiation, proliferation, apoptosis, immune response, cell death, and the transmission of secondary signals to the nucleus, all of which play important roles in a variety of vascular diseases [16, 17, 18]. It is therefore imperative to study the role of energy metabolism in vascular diseases. This review will be elaborated the regulatory role and mechanism of energy metabolism in vascular diseases in two part. The first part of this review will summarize mechanisms responsible for the regulatory of energy metabolism in vascular diseases, such as AS, hemangioma, aortic dissection, PH, vascular aging, and arterial embolism. The second part will discuss drugs and clinical treatment strategies, which might help to improve energy metabolism function in vascular diseases.
AS is a chronic inflammatory vascular disease driven by abnormal lipids and
characterized by progressive thickening and the gradual formation of plaques on
the blood vessel wall [19]. Cell of the arterial wall are very vulnerable to
mitochondrial dysfunction due to their barrier and metabolic functions.
Mitochondrial dysfunction in AS causes alteration in cellular metabolism and
respiration, leading to the production of excessive amounts of reactive oxygen
species (ROS) and resulting in oxidative stress [20]. Mitochondrial DNA (mtDNA)
is more susceptible to oxidative stress, making it vulnerable to oxidation.
Induction of mtDNA damage is a critical mechanism in the development of AS [21, 22]. Damaged mtDNA is usually accompanied by a change in the number of abnormal
mitochondria due to alterations in mitochondrial fusion and fission, thereby
leading to abnormal energy metabolism in AS [23]. Mutations in mtDNA also lead to
an increased number of in pro-inflammatory macrophages. These phagocytose large
amounts of lipid to form foam cells, resulting in cell death and necrotic plaque
cores. The destruction of mitochondrial membranes, inhibition of the respiratory
chain, and the reduction in mitochondrial enzyme activity, can also affect energy
metabolism in AS [24]. Yu E, et al. [25] confirmed that mtDNA damage is
seen early in AS in Apolipoprotein E knockout (ApoE
Besides mitochondrial metabolism, the glycolytic process is the chief source of rapid energy production in response to environmental changes, with the rate of glycolysis playing a critical role in maintaining cellular homeostasis. Intermediates produced during glycolysis can affect cell viability and thus increase the risk of AS [26, 27]. Moreover, abnormalities in mitochondrial and glycolytic energy metabolism can affect the function of VECs, the proliferation of VSMCs, and the polarization of macrophages, which ultimately promote the progression of AS lesions (Fig. 2).
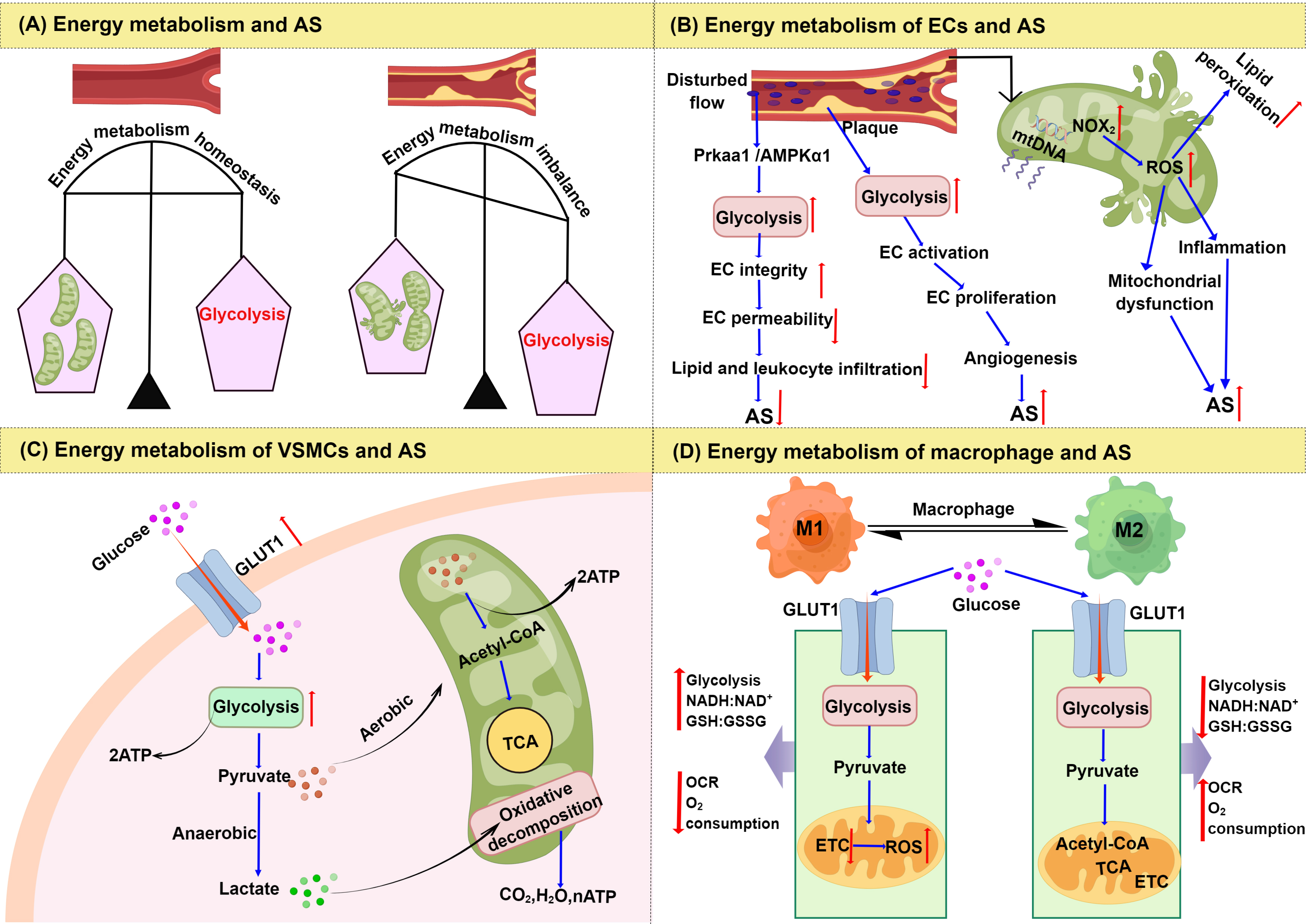 Fig. 2.
Fig. 2.Changes of energy metabolism in AS. (A) Energy metabolism
imbalance and AS. (B) Signaling regulation of glycolysis and mitochondrial
dysfunction in endothelial cells. (C) Signaling regulation of the glycolysis
pathway in vascular smooth muscle cells. (D) Comparison of energy metabolism
between M1 and M2 macrophages. Abbreviations: AS, atherosclerosis; ECs,
endothelial cells; NOX
Endothelial dysfunction is a key early event in the pathogenesis of AS. Unlike other cell types, the energy required for the physiological function of VECs primarily derives from glycolysis [28]. Even when oxygen is sufficient, VECs preferentially catabolize glucose through aerobic glycolysis rather than OXPHOS [29, 30]. Therefore, aerobic glycolysis provides various substrates for cell proliferation and DNA synthesis.
VECs that are exposed to disturbed flow patterns in areas of arterial
bifurcation or curvature are more vulnerable to injury and loss of integrity, and
show increased glycolysis both in vitro and in vivo [31].
Multiple mechanisms are involved in this process, protein kinase AMP-activated
catalytic subunit alpha 1 (PRKAA1)/AMP-activated protein kinase
Although aerobic glycolysis is beneficial for VEC proliferation; pathological angiogenesis also increases [33]. Aerobic glycolysis can bypass OXPHOS, so that the rate of glucose hydrolysis is faster. This can produce large amounts of energy very rapidly for VECs, thus accelerating their migration and proliferation, and promoting the formation of AS plaques [34]. The new vessels developed as a consequence of aerobic glycolysis are immature and have structural and functional abnormalities, thereby leading to plaque hemorrhage which is a major risk factor for the development of AS [34, 35]. Thus, aerobic glycolysis in VECs can have both beneficial and harmful roles in the development of AS.
Mitochondrial respiration is not the main energy metabolism pathway in VECs, but
the disruption of mitochondrial redox homeostasis in VECs can cause chronic
inflammation, which then contributes substantially to AS development [36].
Recently, the disruption of mitochondrial redox homeostasis in VECs was shown to
be a key factor in triggering inflammation. Reducing or neutralizing
mitochondrial ROS levels may therefore be an effective strategy to mitigate
inflammation, promote endothelial repair, and inhibit AS development [37]. The
primary enzymatic source of ROS during cardiovascular pathogenesis is the
nicotinamide adenine dinucleotide phosphate oxidase (NOX) family of proteins.
These are mainly expressed in VECs and provoke oxidative inactivation of nitric
oxide (NO) and the consequent uncoupling of endothelial nitric-oxide synthase
(eNOS), leading to sustained oxidative stress [38] (Fig. 2B). NOX has become a
target for drug therapy in AS, Tanshinone IIA, for example, can attenuates
oxidative stress by decreasing NOX2 and NOX4 levels and downregulating the
nuclear factor-
Abnormal proliferation of VSMCs promotes plaque formation in the early stage of AS lesions, but the apoptosis and senescence of VSMCs and VSMC-derived macrophage-like cells may promote inflammation and plaque rupture in the late stages of AS. Therefore, VSMCs are beneficial during the advanced stage of plaques, for example, by preventing the fibrous cap from rupturing [40, 41]. A major event leading to AS development is the change in VSMC phenotype from a physiological contractile state to an adverse proliferative or synthetic state, accompanied by enhanced aerobic glycolysis [42].
Aerobic glycolysis is the main energy metabolism pathway in VSMCs. Even though
approximately 90% of glucose is used in aerobic glycolysis, this pathway only
produces about 30% of the ATP is produced [42]. Currently there are two
hypotheses to explain this: The first is that the ATP produced by glycolysis and
glucose oxidation provides energy for different physiological processes. For
example, ATP produced by glycolysis is used to supply energy for sodium and
potassium transport, whereas ATP produced by OXPHOS supplies energy for other
physiological processes [43]. The second is that the large amount of lactate
produced by glycolysis can enhance the reserve capacity of mitochondria. The
lactate can then be converted to pyruvate by lactate dehydrogenase after entering
the mitochondria and being completely oxidized to produce CO
The uptake of carbohydrates in VSMCs is determined by glucose transporter 1 (GLUT1), which can further affect energy metabolism and especially the glycolytic pathway [47, 48]. The GLUT1-mediated glycolytic pathway in VSMCs has been closely associated with AS [49]. In one study, GLUT1 overexpression led to a 44% increase in intracellular glucose concentration in rat aortic smooth muscle cell lines (A7r5) and in human VSMC lines. An increase in the glucose flux through glycolysis and the TCA cycle further promotes the proliferation of VSMCs [42] (Fig. 2C). Overexpression of GLUT1 can also inhibit VSMC apoptosis by inhibiting caspase-8 activation in mouse primary VSMCs, thereby promoting cell proliferation [50]. Metabolic reprogramming may be involved in VSMC phenotypic transition. Prohibitin 2 is a pleiotropic scaffold protein involved in the regulation of energy metabolism and proliferation, which maintains VSMC contractile phenotype by interacting with heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) to counteract hnRNPA1-mediated pyruvate kinase isoform M2 (PKM2) alternative splicing and glucose metabolic reprogramming [45].
Furthermore, VSMCs were recently found to rely on a combination of mitochondrial and glycolytic metabolism, with the energy switch occurring at different stages in the AS disease process. Mitochondrial dysfunction and DNA damage are defining traits of aged VSMCs in AS [51]. Klotho is an anti-aging protein that could play a significant role in improving the VSMC phenotype. Targeting Klotho during early stages of AS could induce energy switching and improve VSMC survival in the plaque [51]. A follow-up study demonstrated that phosphatase and tensin homolog deleted on chromosome ten (PTEN) induced putative kinase 1 (Pink1) kinase can regulate mitochondrial homeostasis and the recycling of dysfunctional organelles that are critical for maintaining energetic homeostasis [52]. Energy switching between oxidative and glycolytic metabolism were found to be regulated by Pink1.
Macrophages exist in two distinct subsets: the classically activated M1
macrophages, and the alternatively activated M2 macrophages [53]. Glycolysis is
the main energy pathway in M1 macrophages, and the TCA cycle and OXPHOS are
impaired in these cells. In contrast, M2 macrophages are more dependent on
mitochondrial OXPHOS [54]. GLUT1 is a rate-limiting glucose transporter in
pro-inflammatory polarized macrophages. Overexpression of GLUT1 in
macrophage/monocyte-like leukemia cells (RAW264.7) resulted in a significant
enhancement of glucose uptake and metabolism, and increased production of pentose
phosphate pathway intermediates [55]. With increased the enhancement of
glycolytic metabolism, the proportion of M1 macrophages also increases, followed
by reduced lipid oxidative metabolism and lipid accumulation [56]. The
polarization of M1 macrophages induces AS by increasing ROS levels, which then
cause OXPHOS dysfunction. This in turn increases the rate of glycolysis and
stimulates pro-inflammatory reactions and the synthesis of inflammatory factors
[57]. As a consequence, glycolysis is increased in M1 macrophages together with
the levels of NADH:NAD
Recent studies highlight the importance of mitochondria in the metabolic and signaling pathways necessary to maintain macrophage effector functions [60]. Lipoprotein, oxidized phospholipid, and oxidized low-density lipoprotein (ox-LDL) present within AS plaques can cause oxidative stress-induced damage to macrophage mitochondria [61]. Mild oxidative stress damages the mitochondrial respiratory chain, stimulates mtDNA replication and transcription, and increases the mtDNA copy number. However, under severe oxidative stress the damaged mitochondria gradually lose the ability to self-regulate, which then leads to decreased rates of replication and transcription and a decrease in mtDNA copy number. This ultimately affects mitochondrial function and leads to abnormal energy metabolism [62]. Concurrently, oxidative stress damage to mitochondria in macrophages causes dysfunction of the mitochondrial electron transport chain (ETC), leading to stalled electron transport and increased ROS production [63]. Macrophage polarization is activated by increased ROS through a variety of signaling pathways. For example, activation of the mitogen-activated protein kinase (MAPK)-dependent nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signaling pathways by ROS-induced cytotoxicity ultimately increases the transcription of pro-inflammatory cytokines and other inflammatory mediators that promote polarization of M1 macrophages in RAW264.7 cells [64]. Consequently, ROS is generated in AS plaques as a result of damaged ETC function and further induces the polarization of M1 macrophages. Once polarized, the energy metabolism of macrophages relies mainly on glycolysis, meaning that fat metabolism is reduced and leading to further lipid accumulation and the deterioration of AS plaques [65, 66].
In addition to oxidative stress damage to mitochondria in macrophages,
HIF-1
Hemangioma is the most common benign tumor of infancy and is caused by dysfunction during embryogenesis and vasculogenesis. Hemangiomas progress during fetal life and manifest fully at birth. They arise in the skin and soft tissues and can occur at birth or shortly after birth [69]. The primary characteristics of hemangioma are a high incidence rate and strong destructiveness. Hypoxia and abnormal glucose metabolism are considered to be key factors in the pathogenesis of this disease [70].
Under normoxic and hypoxic conditions, glucose consumption and ATP production
occur at higher levels in hemangioma-derived endothelial cells compared to human
umbilical vein endothelial cells [70]. In addition, several glycolysis-related
genes are expressed at higher levels, including GLUT1, hexokinase 2 (HK2),
6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), PKM2, and lactate
dehydrogenase A (LDHA). Inhibition of glycolytic-related genes in
hemangioma-derived ECs can reduce the proliferation, migration, and angiogenesis
of these cells. The above results suggest that increased glycolytic metabolism is
an important cause of infantile hemangioma [70, 71]. Some genes related to
glycolysis are also highly expressed in infantile hemangioma. For example, the
expression of LncRNA MCM3AP-AS1 is significantly upregulated in infantile
hemangiomas and also promotes their progression by increasing glycolysis via
regulation of the miR-138-5p/HIF-1
Although tumor cells rely on glycolysis for energy supply, mitochondrial transformations in bioenergetics, metabolism, and fission-fusion dynamics are also likely to be important. This is especially true for rapidly growing tumor cells that rely on mitochondrial function for their sustenance [73]. However, some studies have identified tumorigenic cell populations that rely more on mitochondrial respiration than on glycolysis [74, 75, 76]. Mouse hemangioendothelioma ECs have been used to study the mechanism of hemangioendothelioma, which depend heavily on mitochondria to thrive. The targeting of mitochondria may therefore be an effective strategy for the management of hemangiomas [77].
Aortic dissection is a life-threatening condition in which blood in the aortic cavity enters the aortic medial membrane from a tear in the aortic intima, resulting in separation of the different layers of the aortic wall. Although the incidence of aortic dissection is low, it has a major clinical impact due to its high mortality and poor prognosis [78]. The potential mechanisms underlying energy metabolism-induced aortic dissection primarily involve two aspects: ROS and inflammation [79, 80].
Fan LM, et al. [81] investigated the special role of endothelial ROS in the development of aortic dissection by establishing a mouse model with endothelium-specific Nox2 overexpression. These workers then determined the susceptibility of the Nox2 over-expressing mice to aortic dissection after infusion with angiotensin II (Ang II). Compared to wild-type mice, a specific increase in endothelial ROS production in the Nox2 transgenic mice was sufficient to cause Ang II-mediated aortic dissection. Increased expression of Nox2 in VECs can lead to increased ROS production, upregulated expression of vascular cell adhesion molecule-1 (VCAM-1), enhanced matrix metalloproteinase activity, and accelerated CD45 inflammatory cell infiltration. Compared to controls, ECs from Nox2-overexpressing mice grown in conditioned medium induced greater phosphorylation of extracellular-regulated kinase 1/2 (ERK1/2) in VSMCs by secreting cyclophilin A (CypA). These findings suggest that VEC-derived ROS plays a critical role in determining the susceptibility of the aortic wall to dissection mediated by Ang II. The ROS-dependent secretion of CypA by VECs is an important signaling mechanism that regulates the susceptibility of aortic wall structural components to aortic dissection. These results suggest that endothelial ROS and CypA are potential therapeutic targets for the prevention of aortic dissection [81].
Aortic dissection is a severe inflammatory vascular disease associated with the
metabolic reprogramming of macrophages. The effect of macrophage metabolic
reprogramming has been studied in a mouse model by inducing aortic dissection
with
Pulmonary Hypertension (PH) is a pulmonary vascular disease with complex etiology. It is characterized by the remodeling and occlusion of small peripheral pulmonary vessels, and a progressive increase in pulmonary vascular resistance. This eventually leads to hypertrophy of the right ventricle and right heart failure [82]. The pathological characteristics of PH are excessive proliferation and apoptosis tolerance of VSMCs, adventitial fibroblasts and VECs, eventually leading to continuous pulmonary vascular wall thickening and pulmonary vascular remodeling [83]. Changes in metabolism and bioenergetics are increasingly recognized as universal hallmarks of PH in patients and in animal models of this disease [84].
Excessive proliferation of pulmonary artery smooth muscle cells (PASMCs) is the
main cause of hypoxic PH. Molecules that induce abnormal proliferation of VSMCs
may trigger PH [85]. For example, the increase in the main end products of
intracellular lipid peroxidation, 4-HNE, leads to abnormal proliferation of
VSMCs, thus promoting the development of PH. Therefore, inhibiting the
intracellular increase of 4-HNE can prevent the occurrence and progression of PH.
Aldehyde dehydrogenase 2 (ALDH2) is a detoxifying enzyme for 4-HNE, which can
regulate mitochondrial fission and VSMC proliferation through the
4-HNE/HIF-1
An emerging metabolic theory for PH suggests that cellular and mitochondrial metabolic dysfunction underly the pathology of this disease [89]. Zhang et al. [89] showed that high expression of polypyrimidine tract-binding protein 1 (PTBP1), controlled by decreased expression of miR-124, underlies the elevated PKM2/PKM1 ratio and subsequent metabolic reprograming of pulmonary adventitial fibroblasts isolated from calves and humans with severe PH. These findings are similar to those made in cancer cells and support the idea that pyruvate metabolism can be exquisitely controlled by miR-124 and by PTBP1 [89].
Metabolic changes together with endothelial dysfunction that favor increased
glycolysis are important in the pathophysiology of PH [90]. Lung ECs express high
levels of glucose metabolic enzymes, such as PFKFB3, and consequently produce
large amounts of glucose metabolites. Low levels of HIF-1
Vascular aging involves a deterioration in the normal function of vessels, resulting in increased arterial stiffness and pulse wave propagation rate and accumulation of oxidative damage. Vascular aging also greatly increases the incidence of AS and PH [92, 93]. Mitochondrial dysfunction plays a critical role in disease progression. For example, mitochondria-targeted esculetin alleviates AS in the setting of aging by delaying vascular senescence and pro-inflammatory processes, and by improving mitochondrial biogenesis and function [92].
Senescence appears to be closely linked with changes in cell metabolism. Replicative senescent VECs show upregulation of glycolytic and oxidative glucose metabolism compared to young VECs. This was demonstrated by metabolic profiling and glucose flux measurements, and by analyzing the expression of key metabolic enzymes. Senescent cells exhibit higher glycolytic activity and lactate production, enhanced expression of LDHA, and increased activity of the TCA cycle and mitochondrial respiration [6].
Age-related endothelial dysfunction is mediated primarily by the reduced bioavailability of NO. Impaired NO-mediated endothelial function is largely due to excessive production of ROS, which reacts with NO to reduce its bioavailability. The resulting arterial oxidative stress leads in turn to vascular aging and dysfunction [94]. The effects of oxidative stress on vascular aging also correlate with endothelium-dependent dilation (EDD), which is a marker of endothelial dysfunction. Impaired or reduced EDD can promote vascular aging and the stiffening of arteries by increasing the tension in arterial activity. Increased expression of NADPH oxidase and abnormal mitochondrial function lead to increased ROS production, decreased NO bioavailability, damaged EDD, and age-related endothelial dysfunction [95]. Furthermore, the expression of serine biosynthetic enzyme phosphoglycerate dehydrogenase (PHGDH) is significantly reduced due to decreased transcription of the activating transcription factor 4 (ATF4) during senescence, which leads to reduction of intracellular serine. Mechanistically, PHGDH interacts with PKM2, which prevents p300/CBP-associated factor (PCAF)-catalyzed PKM2 K305 acetylation and subsequent degradation by autophagy. In addition, PHGDH facilitates p300-catalyzed PKM2 K433 acetylation, which promotes PKM2 nuclear translocation and stimulates its activity to phosphorylate H3T11 and regulate the transcription of senescence-associated genes. Vascular endothelium-targeted expression of PHGDH and PKM2 ameliorates ageing in mice. This findings reveal that enhancing serine biosynthesis could become a therapy to promote healthy ageing [96].
Arterial embolism involves the narrowing or occlusion of the lumen due to AS, thus making it difficult to transport nutrients in the blood to the limbs and resulting in ischemia and serious tissue necrosis [97]. The occurrence of arterial embolism causes changes in energy metabolism. Insufficient blood supply and hypoxia not only lead to changes in energy metabolism, but also decrease mitochondrial activity and increase the carbon dioxide level, which may cause hypercapnia [98]. However, most of research on the relationship between energy metabolism and arterial embolism was carried out in the 1990s, with almost no recent research. The reason may be that the inducing factors for arterial embolism are related to hypertension, hyperlipidemia, AS, etc. Therefore, study of the inducing factors for arterial embolism and energy metabolism may be more important for the prevention of arterial embolism. Here, we review the research progress on arterial embolism and energy metabolism during the 1990s.
In one study, cerebral ischemia was induced in rats by administering microspheres into the right carotid canal, and the time course of changes in brain energy metabolism was examined on day 7 post-surgery. Nearly 50% of the operated rats showed the typical symptoms of stroke, i.e., cerebral ATP and creatine phosphate (PCr) were significantly decreased in the right hemisphere of microsphere-induced cerebral ischemia from day-1 to day-7, while tissue lactate levels were increased on day-1, -3, and -5 after embolism [99]. These results indicate that ischemia alters the energy metabolism of rat cerebra. Subsequently, Takeo et al. [98] studied the sustained damage to cerebral regions of rat brain induced by cerebral ischemia. These workers examined animal behavior and cerebral energy metabolism after microsphere embolization. On day-1 of post-microsphere embolization, the lactate content increased to its peak, and the ATP and PCr contents decreased to their lowest level in these cerebral regions in the right hemisphere. The glucose and glycogen content in these regions increased to their peak on day-3. Although these markers returned to control levels over time, metabolic changes in all of these areas persisted for up to 28 days post-surgery. The behavioral scoring as a result of the temporal changes after microsphere embolization showed stroke-like symptoms on day-1 after embolization, with the symptoms disappearing relatively quickly with the passage of time. These observations indicate that microsphere embolization in brain regions can cause extensive and sustained damage to energy metabolism [98].
Hepatic energy metabolism and oxidative attack were also studied following transcatheter arterial embolization (TAE) and chemoembolization (TAC) in the left and median lobes of the liver in a rat model with thioacetamide-induced liver cirrhosis. The energy charge of the embolized lobes of the lungs decreased from 0.86 to 0.78 and 0.74 one hour after the TAE and TAC processes, respectively, but recovered after 3 h. The ATP and total adenine nucleotide content of embolized and non-embolized lobes were also temporarily reduced. Total hepatic blood flow in the catheter-embolized and chemo-embolized lobes decreased by nearly 50% and returned to normal after 1 week. After chemoembolization for 3 h, the total liver glutathione content decreased from 7.02 µmol/g to about 4.5 µmol/g, while after chemoembolization for 6 h the malondialdehyde content increased from 196.94 nmol/g to more than 300 nmol/g. However, these parameters did not change in the catheter-embolized liver. The above results suggest that hepatic energy metabolism is temporarily altered by ischemia after treatment with TAE and TAC [100].
The regulation of energy metabolism in various tissues and cells helps the body
adapt to environmental changes. These regulatory disturbances are closely related
to the molecular pathology of many vascular diseases, including AS, PH and
hemangioma [70, 84, 101]. Under specific pathological conditions, glycolysis and
the mitochondrial energy pathway can affect VEC function, macrophage
polarization, and VSMC proliferation [51, 102, 103]. Abnormal glycolysis
metabolism in VECs, VSMCs, and macrophages is related to AS, infantile
hemangioma, and PH [70, 84, 101]. In addition to glycolysis, mitochondrial
metabolism is also crucial for vascular diseases such as AS, aortic dissection,
PH, vascular aging, and arterial embolism [20, 90, 104]. In vitro and
in vivo studies indicate that regulation of energy metabolism-related
molecules and pathways is used to maintain cellular metabolic homeostasis and to
prevent vascular diseases. The mechanisms for preventing vascular diseases
include multiple genes and signaling pathways involved in energy metabolism, such
as PRKAA1/AMPK
Through literature analysis, GLUT1 is closely associated with the energy
metabolism of various vascular diseases, which overexpressed in high glycolytic
flux, resulting in the overproduction of pyruvate, alanine, and lactate [106].
Concurrently, VECs and VSMCs display characteristics of cell proliferation and
migration, while the polarization of M1 macrophages also increases [48, 107, 108]. NOX2, ROS, Pink1 and NF-
Treatment strategies for vascular diseases that target energy metabolism have attracted much attention, especially the use of drugs. Pharmacotherapies that target endothelial dysfunction caused by energy metabolism disorders can potentially be used to treat AS and many other vascular diseases [111]. For example, the small molecule 3PO (3-[3-pyridyl]-1-[4-pyridyl]-2-propen-1-ketone) may exert beneficial effects against plaque formation by partially inhibiting the glycolysis-mediated impairment of new angiogenesis without affecting plaque composition, as well as by increasing the number of autophagosomes in VECs [112]. 3PO is known to be an inhibitor of PFKFB3. Heterozygous global deficiency of Pfkfb3 was shown to protect mice from developing hypoxia-induced PH, while administration of 3PO almost completely prevented PH in rats treated with Sugen 5416/hypoxia [7]. Tetrahydrobiopterin (BH4) has been shown to reduce endothelial NO synthase uncoupling, reverse oxidative stress-mediated inhibition of EDD, and improve EDD in the elderly, thereby alleviating vascular aging [95]. Furthermore, natural products may be a good primary source of drugs for the treatment of vascular diseases. Extracts of Rhodiola and Salidroside have been suggested to provide beneficial effects for metabolic disorders and AS. The postulated mechanisms for Rhodiola extracts and Salidroside in the treatment of AS involve energy metabolism and include oxidative stress, inflammation, mitochondria, and cell death [113]. Apocynin, extracted from the roots of Apocynum cannabinum and Picrorhiza kurroa, is an inhibitor of NADPH oxidase and could therefore restore EDD and NO levels in vitro and alleviate vascular aging [95, 114]. Oral apigenin supplementation can reverse two clinically important and age-related indicators of arterial dysfunction, namely vascular endothelial dysfunction and large elastic artery stiffening [115]. This novel work also showed that apigenin prevents foam cell formation in an established cell culture model of early AS. The above preclinical findings provide an experimental basis for future translational studies to assess the potential of natural products in treating arterial dysfunction and reducing vascular disease risk. In addition, the non-selective-adrenergic receptor antagonist propranolol is already in clinical use and is becoming the first line therapy as an approved treatment for infantile hemangioma. The latest evidence showed that propranolol-induced apoptosis in VECs may occur via mitochondrial stress [116]. Thus, it would be advantageous to discover the mechanisms related to energy metabolism for drugs that are already used in clinical practice.
In terms of clinical research, a mitochondrial-targeted antioxidant (MitoQ) has been shown to improve vascular mitochondrial function. Acute oral administration of the MitoQ (80 mg) can effectively improve endothelial function and superoxide dismutase in patients with peripheral artery disease (PAD). In addition, acute intake of MitoQ has also been found to be effective in improving maximum walking capacity and delaying claudication onset in PAD patients [117]. Pyruvate dehydrogenase kinase (PDK), an inhibitor of the mitochondrial enzyme pyruvate dehydrogenase in the pulmonary arteries of human PH compared to healthy lungs. In a 4-month, open-label study, DCA (PDK inhibitor dichloroacetate, 3 to 6.25 mg/kg b.i.d.) administered to patients with idiopathic PH already on approved idiopathic PH therapies led to reduction in mean pulmonary artery pressure and pulmonary vascular resistance and improvement in functional capacity [118].
Besides medication, there may be other potential therapeutic methods for the treatment of vascular diseases, such as cytotherapy. ALDHs are a superfamily that includes 19 subtypes in humans. ALDH2 is the mitochondrial isoform of ALDH and a key metabolizer of acetaldehyde. It is known to be a protective factor for vascular diseases such as AS, aortic dissection, aortic aneurysm, and PH by clearing toxic aldehydes generated from lipid peroxidation [86, 119, 120]. A Bayesian network meta-analysis of randomized controlled trials showed that intra-arterial injection of large amounts of ALDH-positive cells could improve the activity of daily living in ischemic stroke patients [121]. Therefore, we speculate that cytotherapy is a potential treatment for vascular disease.
Cellular energy metabolism is a crucial factor in vascular disease, with a number of studies having been conducted over the years to investigate its role. The vascular disease types related to energy metabolism include AS, hemangioma, aortic dissection, PH, vascular aging, and arterial embolism. Some drugs can directly or indirectly affect the expression of key rate-limiting enzymes in glycolysis, or regulate the mitochondrial energy metabolism pathway. In summary, this review should increase our understanding of energy metabolism in various vascular diseases. It may also help to guide future research efforts aimed at exploring the pathogenesis of vascular diseases and clinical treatment methods.
Acetyl-CoA, acetyl coenzyme A; ADP, adenosine diphosphate; ALDH1A3, aldehyde
dehydrogenase 1A3; ALDH2, aldehyde dehydrogenase 2; AMPK
QT designed the study. TS, WY, YW and DL performed the literature research. TS and WY wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Figures were created by Figdraw (https://www.figdraw.com/#/).
This study was supported by the Key Research and Development Projects of Hunan Provincial Science and Technology Department (No. 2022SK2011), the Science and Technology Innovation team of Hunan Province (No. 2021RC4064), the Key Projects of Hunan Provincial Department of Education (No. 20A379) and Graduate Innovation Project of Hunan University of Chinese Medicine (No. 2022CX15). 2023 Undergraduate Innovation and Entrepreneurship Training Program of Hunan University of Chinese Medicine (No. S202310541108).
The authors declare no conflicts of interest for this work.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.