
- Academic Editor
†These authors contributed equally.
Objective: This study investigated the role of long non-coding RNAs (lncRNAs) FTX in vascular endothelial cells (ECs). Methods: Transfection of FTX/Sh-FTX with lentivirus was used to construct gain and loss of function cell models in human umbilical vein endothelial cells (HUVECs). Liquid chromatography-mass spectrometry was used for quantitative proteomics analysis of differentially expressed proteins (DEPs). Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and protein interaction analysis were further conducted to investigate the key molecules and pathways that respond to lncRNA-FTX. Results: In the proteomics analysis, 3308 quantifiable proteins were identified, 64 proteins were upregulated and 103 were downregulated when lncRNA FTX was overexpressed. Additionally, 100 proteins were upregulated and 147 were downregulated when lncRNA FTX was knocked down. Functional clustering analysis of DEPs demonstrated that lncRNA FTX was involved in multiple biological processes. Among them, the expression of complement 3 (C3), cartilage oligomeric matrix protein (COMP), faciogenital dysplasia 6 (FGD6), and tissue inhibitor of metalloproteinase 1 (TIMP1) was significantly upregulated when lncRNA FTX was knocked down, and significantly downregulated when lncRNA FTX was overexpressed. They are associated with inflammation, collagen deposition, angiogenesis, and regulation of liver stem cell differentiation, which may be associated with the occurrence and development of liver fibrosis. Conclusions: The study demonstrated that lncRNA FTX might play a potential role in ECs and contribute to the development of liver fibrosis. Thus, FTX may be a promising target for the prevention or reversal of liver fibrosis.
Endothelial cells (ECs) lining the heart, blood vessels, and lymphatic vessels serve as a crucial interface between the vascular wall and blood. ECs lining the walls of blood vessels can regulate blood flow and play pivotal roles in inflammatory responses through the expression of adhesion molecules, recruitment of inflammatory cells, and neovascularization [1]. Moreover, ECs can secrete an array of proteins that can lead to coagulation disorders. Additionally, they are able to regulate vascular smooth muscle cells (VSMCs) and help inhibit thrombosis by secreting cytokines [2]. Under normal physiological conditions, the proliferation and apoptosis of endothelial cells are in balance, enabling the normal function of blood vessels [3]. Endothelial dysfunction is associated with the development of various diseases such as coronary heart disease [4], stroke [5], diabetes [6], pulmonary fibrosis [7], and coronavirus disease 2019 [8]. Drugs that target endothelial dysfunction possess therapeutic potential in numerous diseases [9].
Many factors have been shown to regulate the function of ECs, including the long non-coding RNAs (lncRNAs), which participate in epigenetic modifications. LncRNAs such as antisense non-coding RNA in the INK4 locus (ANRIL) induce the expression of inflammation-related genes and alternative splicing in human umbilical vein endothelial cells (HUVECs) when overexpressed [10], and CA7-4 promotes autophagy or apoptosis by sponging microRNA 877-3p (miR-877-3p) and miR-5680 in high glucose-induced ECs [11]. Moreover, lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) inhibits ECs proliferation or angiogenesis while promoting invasion and apoptosis [12], indicating that they may function as regulatory factors in the ECs dysfunction-related diseases.
FTX is a non-coding gene on the X chromosome in humans and was initially discovered and named lncRNA FTX in human embryonic stem cells. Abnormal expression of FTX is related to various congenital diseases and acquired diseases such as tumors or fibrosis [13, 14]. For example, lncRNA-FTX upregulates the fragile X mental retardation gene 1 (Fmr1) by sponging miR-410-3p to regulate myocardial cell injury induced by hypoxia/reoxygenation [15]. However, the role of FTX in ECs remains uncertain.
In our study, the intervention of lncRNA FTX expression FTX in HUVECs was performed in vitro and its influence on downstream protein composition was analyzed. We identified the biological effects of lncRNA FTX in ECs.
The HUVECs were obtained from the Chinese Academy of Sciences Cell Bank
(Shanghai, China) and were cultured in Dulbecco’s modified Eagle medium (Hyclone,
MA, USA) at 37 °C with 95% air and 5% CO
According to the manufacturer’s instructions, lncRNA FTX overexpression (FTX) and its mock control (FTX-negative control [NC]), as well as lncRNA FTX RNA interference (single hairpin [Sh]-FTX) and its mock NC (Sh-NC), were transfected using lentivirus (Shanghai Geochemistry, Shanghai, China). Puromycin (2 g/mL) was used to select transfected cells, and the stability of transfection was verified by quantitative polymerase chain reaction (qPCR).
Total RNA was extracted from HUVECs and subjected to qPCR. Beta-actin (ACTB) was
selected as the internal reference, and the
2
| Primer | Sequences (5′-3′) |
| lncRNA FTX Forward Primer | GAATGTCCTTGTGAGGCAGTTG |
| lncRNA FTX Reverse Primer | TGGTCACTCACATGGATGATCTG |
| ACTB Forward Primer | GGCACCACACCTTCTACAATG |
| ACTB Reverse Primer | TAGCACAGCCTGGATAGCAAC |
Samples removed from the –80 °C freezer were treated with lysis buffer (8 M urea, 1% protease inhibitor), followed by sonication to ensure complete lysis. After centrifugation of the solution at 4 °C and 12,000 g for 10 minutes to eliminate cellular debris, and the supernatant was transferred to a new centrifuge tube. The protein concentration was determined using a bicinchoninic acid kit. Equivalent amounts of protein were obtained from every sample to digest, with the proper amount of standard protein added during volume adjustment with the lysis buffer. Subsequently, dithiothreitol (DTT) was added into the solution to a final concentration of 5 mM and incubated at 56 °C for 30 minutes, followed by the addition of iodoacetamide (IAA) to a final concentration of 11 mM and incubation for 15 minutes at room temperature in the dark. The alkylated samples were transferred to an ultrafiltration tube, centrifuged at room temperature for 20 minutes at 12,000 g, washed three times with 8 M urea, and washed three times with a replacement buffer. Trypsin was added to the samples at a ratio of 1:50 (protease: protein, m/m) and allowed to enzymatically digest the proteins overnight. The peptide fragments were recovered by centrifugation at 12,000 g for 10 minutes at room temperature, followed by recovery using ultra-pure water and a combination of the peptide solution.
Tryptic peptides were first dissolved in 0.5 M TEAB. Each sample of 50 µg peptide was labeled with their respective TMT reagent (ThermoFisher Scientific, MA, USA), and incubated for 2 hours at room temperature. The labeling information is shown in Table 2. Five microliters of each sample were pooled, desalted, and analyzed by MS to check labeling efficiency. After the labeling efficiency check, samples were quenched by adding 5% hydroxylamine. The pooled samples were then desalted with Strata X SPE column (Phenomenex) and dried by vacuum centrifugation.
| Sample Groups | Labeling information |
| H_OE_C | 126 |
| H_OE | 128 |
| H_KD | 129 |
| H_KD_C | 130 |
H_OE_C, HUVEC cells transfected with FTX-NC as a control; H_OE, HUVEC cells overexpressing FTX; H_KD, HUVEC cells knocked down for FTX; H_KD_C, HUVEC cells transfected with shRNA-NC as a control.
The sample (200 µg labeled peptide) was fractionated into fractions by high pH reverse-phase high-performance liquid chromatography (HPLC) using Agilent 300 Extend C18 column (10 µm particles, 4.6 mm ID, 250 mm length). Briefly, peptides were separated with a gradient of 8% to 32% acetonitrile in 10 mM ammonium bicarbonate pH 9 over 54 min into 54 fractions. The 54 peptide fractions were combined into 9 fractions and dried by vacuum centrifuging.
After dissolving the peptides in mobile phase A (the water-based solution
including 2% acetonitrile and 0.1% formic acid), separation was carried out on
the EASY-nLC 1200 ultra-HPLC (UHPLC) system. Meanwhile, mobile phase B
(water-based solution including 90% acetonitrile and 0.1% formic acid) was
utilized as well. The liquid gradient was set as follows: 0–26 min, 5–22% B;
26–34 min, 22–32% B; 34–37 min, 32–80% B; 37–40 min, 80% B, with a flow
rate maintained at 450.00 nL/min. The peptides were ionized through a nanospray
ionization source and analyzed with the HF-X Mass Spectrometer after separation
using the UHPLC system. An ion source voltage of 2.2 kV was utilized, and the
detection and analysis of parent ions and secondary fragments were performed by
high-resolution Orbitrap MS. The first mass scan was conducted between 400 and
1600 m/z, with a resolution set at 120,000.00, and the second mass scan was
initiated at 100 m/z with a resolution of 15,000.00. A data-dependent acquisition
(DDA) program was used for data acquisition mode, where the top 25.00 peptide
parent ions with the highest signal intensity were sequentially sent to the pool
for high energy collision dissociation (HCD) and then subjected to secondary mass
spectrometry (MS) analysis after being fragmented with 28% collision energy. To
enhance MS efficiency, the automatic gain control (AGC) was set at 5
The resulting MS/MS data were processed using the MaxQuant search engine
(v.1.6.15.0, https://www.maxquant.org/). Tandem mass spectra were searched against the human SwissProt
database (20,366 entries) concatenated with the reverse decoy database. Trypsin/P
was specified as a cleavage enzyme allowing up to 2 missing cleavages. The mass
tolerance for precursor ions was set as 20 ppm in the First search and 4.5 ppm in
the Main search, and the mass tolerance for fragment ions was set as 0.02 Da.
Carbamidomethyl on Cys was specified as a fixed modification. Acetylation on
protein N-terminal, oxidation on Met, and deamidation (NQ) were specified as
variable modifications. TMT-6plex quantification was performed. False discovery
rate (FDR) was adjusted to
Identified proteins were enriched for protein function, structural domains, and
biological pathways via annotation with the UniProt-Gene Ontology Annotation
(GOA) database (http://www.ebi.ac.uk/GOA/), InterPro structural domain database
(http://www.ebi.ac.uk/interpro/) and Kyoto Encyclopedia of Genes and Genomes
database (http://www.genome.jp/kaas-bin/kaas_main). Wolfpsort software
(https://www.genscript.com/wolf-psort.html) was used for annotating the
subcellular localization of submitted proteins. Differential expressed protein
(DEPs) database numbers, or protein sequences from different comparison groups,
were compared using the STRING (v10.5; http://cn.string-db.org) protein network
interaction database to extract protein interaction relationships that possessed
a confidence score
Statistical analyses were performed using SPSS software (v 25.0; IBM, Chicago,
IL, USA) and the GraphPad Prism 5 software (GraphPad, San Diego, CA, USA).
Protein expression was compared using the two-sample, two-tailed t-test,
with significance set at p
HUVECs were transfected with the FTX overexpression lentivirus, its NC lentivirus, the shRNA lentivirus, and its NC lentivirus. Total RNA extracted from each cell group was collected, and qPCR was used to confirm the changes in FTX expression in HUVECs after successful transfection with overexpression and shRNA lentivirus (Fig. 1).
 Fig. 1.
Fig. 1.The validation of lentiviral transfection efficiency by real time quantitative polymerase chain reaction (RT-qPCR). The results of RT-qPCR were normalized to Beta-actin (ACTB).
To study the effects of lncRNA FTX on ECs, we performed quantitative proteomics analysis to analyze DEPs in HUVECs with functional loss or gain of lncRNA FTX. A total of 4348 proteins were identified by high-resolution LC-MS/MS analysis, among which 3308 proteins were quantifiable. A total of 64 significantly upregulated proteins and 103 significantly downregulated proteins were identified in the FTX group compared with the FTX-NC group. A total of 36 significantly upregulated proteins and 44 significantly downregulated proteins were identified in the FTX-Sh group compared with the Sh-NC group. Logarithmic transformation of the fold changes of all proteins with the base two generated the volcano plot (Fig. 2). The top 10 significantly upregulated and downregulated proteins in each group are shown in Tables 3,4, respectively. These data indicate that the intervention of lncRNA FTX expression could lead to changes in the proteomics profile of HUVECs, suggesting its potential involvement in the regulation of EC function.
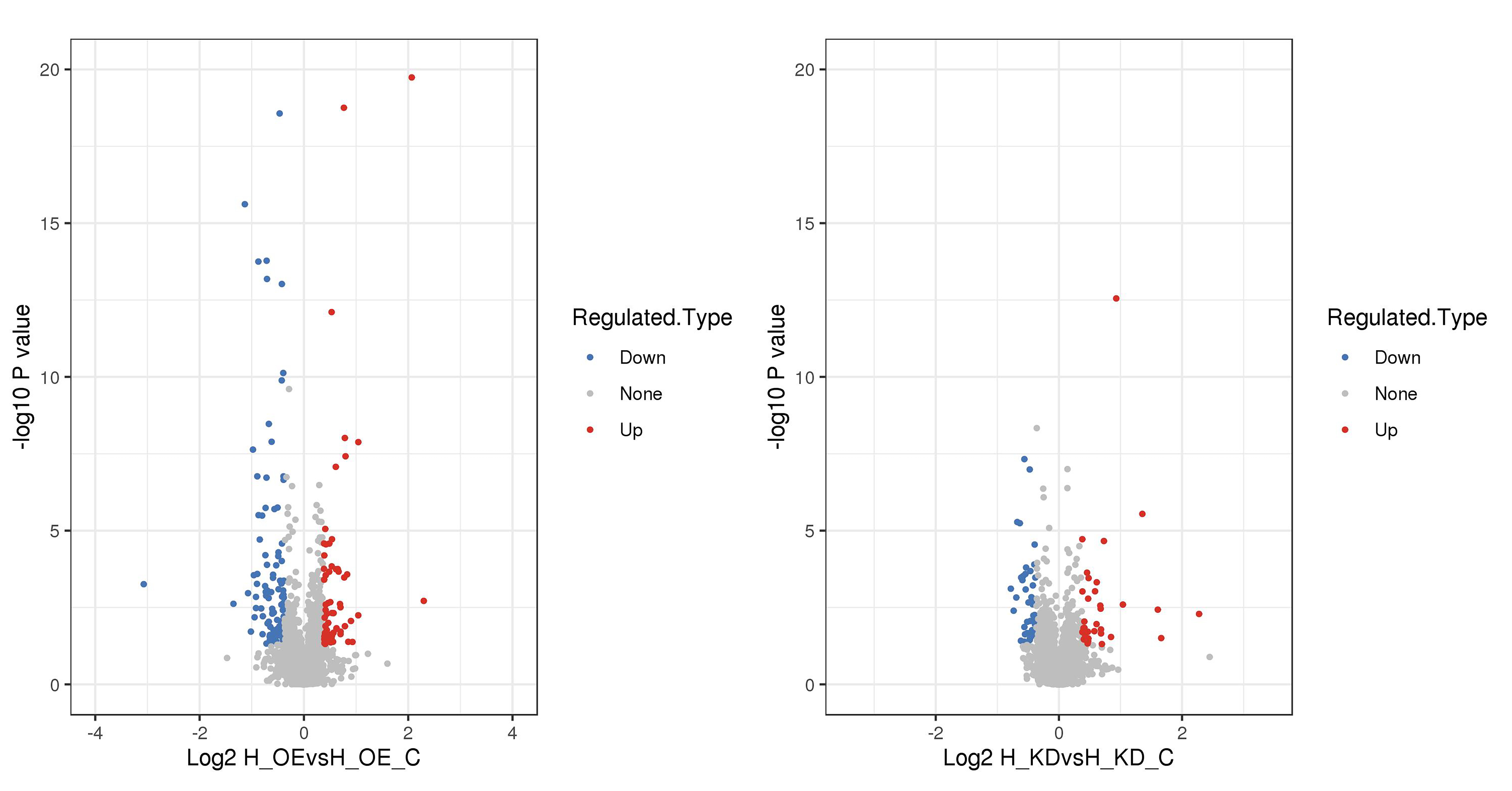 Fig. 2.
Fig. 2.Volcano plot of Differentially expressed proteins (DEPs) quantification.
| Upregulated protein | Protein description | Downregulated protein | Protein description |
| P04264|KRT1 | Keratin, type II cytoskeletal 1 | Q9NRR5|UBQLN4 | Ubiquilin-4 |
| P0DMV9|HSPA1B | Heat shock 70 kDa protein 1B | P67936|TPM4 | Tropomyosin alpha-4 chain |
| P31689|DNAJA1 | DnaJ homolog subfamily A member 1 | P80297|MT1X | Metallothionein-1X |
| P07099|EPHX1 | Epoxide hydrolase 1 | P05783|KRT18 | Keratin, type I cytoskeletal 18 |
| Q9H9Y6|POLR1B | DNA-directed RNA polymerase I subunit RPA2 | Q06323|PSME1 | Proteasome activator complex subunit 1 |
| Q5BJF2|TMEM97 | Sigma intracellular receptor 2 | P55327|TPD52 | Tumor protein D52 |
| P56182|RRP1 | Ribosomal RNA processing protein 1 homolog A | P62328|TMSB4X | Thymosin beta-4 |
| P50995|ANXA11 | Annexin A11 | O60664|PLIN3 | Perilipin-3 |
| P50454|SERPINH1 | Serpin H1 | Q9UK76|JPT1 | Jupiter microtubule associated homolog 1 |
| Q96QD8|SLC38A2 | Sodium-coupled neutral amino acid transporter 2 | Q9Y2U5|MAP3K2 | Mitogen-activated protein kinase kinase kinase 2 |
| Upregulated protein | Protein description | Downregulated protein | Protein description |
| Q68D86|CCDC102B | Coiled-coil domain-containing protein 102B | O75131|CPNE3 | Copine-3 |
| Q6ZV73|FGD6 | FYVE, RhoGEF and PH domain-containing protein 6 | O94925|GLS | Glutaminase kidney isoform, mitochondrial |
| P49747|COMP | Cartilage oligomeric matrix protein | P62917|RPL8 | 60S ribosomal protein L8 |
| P04114|APOB | Apolipoprotein B-100 | P62851|RPS25 | 40S ribosomal protein S25 |
| P07148|FABP1 | Fatty acid-binding protein, liver | P10412|H1-4 | Histone H1.4 |
| O43175|PHGDH | D-3-phosphoglycerate dehydrogenase | O15231|ZNF185 | Zinc finger protein 185 |
| Q8IZ83|ALDH16A1 | Aldehyde dehydrogenase family 16 member A1 | P62424|RPL7A | 60S ribosomal protein L7a |
| P35237|SERPINB6 | Serpin B6 | P55263|ADK | Adenosine kinase |
| P01033|TIMP1 | Metalloproteinase inhibitor 1 | P62987|UBA52 | Ubiquitin-60S ribosomal protein L40 |
| Q01581|HMGCS1 | Hydroxymethylglutaryl-CoA synthase, cytoplasmic | Q92804|TAF15 | TATA-binding protein-associated factor 2N |
Enrichment and clustering analyses were conducted to evaluate the properties and functions of DEPs in response to the overexpression or knockdown of lncRNA FTX. GO functional enrichment analysis showed that the DEPs after knockdown or overexpression of lncRNA FTX mainly existed in the cell, organelle, and membrane-enclosed lumen, performing molecular functions such as binding and catalytic activity, which are primarily involved in cellular processes, biological regulation, metabolic process, and response to the stimuli. Based on GO enrichment clustering analysis (Fig. 3), it was further shown that the DEPs related to the endoplasmic reticulum (ER) lumen and extracellular region part were upregulated after lncRNA FTX knockdown, while those related to the rough ER, rough ER membrane, cytosolic large ribosomal subunit, and large ribosomal subunit were downregulated. The upregulated proteins after overexpression of lncRNA FTX were enriched in cellular components such as the cytosolic small ribosomal subunit, small ribosomal subunit, and ribonucleoprotein complex, whereas the downregulated proteins were enriched in the actin cytoskeleton and extracellular region part. In terms of biological processes, lncRNA FTX knockdown led to the upregulation of proteins involved in the biosynthetic process of alpha-amino acid, whereas the downregulated proteins participated in processes such as amide biosynthetic process, protein localization to the membrane and ER, protein targeting to the membrane, translation, peptide biosynthetic process, cotranslation protein targeting to the membrane, and protein targeting to the estrogen receptor. At the same time, after overexpression of lncRNA FTX, the upregulated proteins were mainly associated with the regulation of cellular response to heat, ncRNA, and ribosomal RNA metabolic process, whereas the downregulated proteins were associated with actin filament bundle organization. Regarding molecular function, the structural constituent of the structural molecule activity and ribosome was enriched in downregulated proteins after lncRNA FTX knockdown. The upregulated proteins included heat shock protein and C3HC3-type RING finger domain binding when lncRNA FTX was overexpressed.
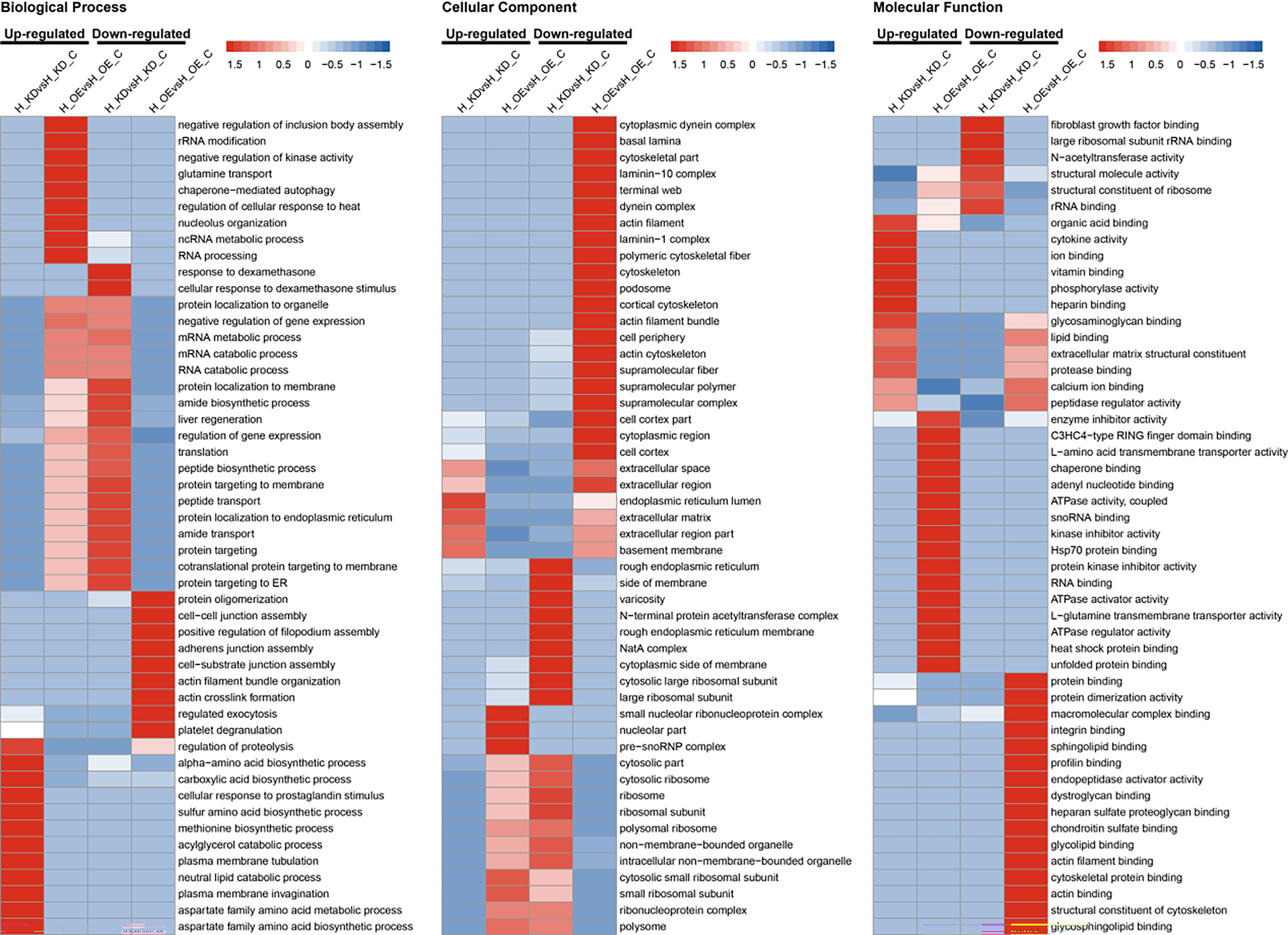 Fig. 3.
Fig. 3.Gene Ontology (GO) enrichment clustering analysis of DEPs. (A) Cellular Component. (B) Biological Process. (C) Molecular Function. H_OE, HUVEC cells overexpressing FTX; H_OE_C, HUVEC cells transfected with FTX-NC as a control; H_KDsH, HUVEC cells knocked down for FTX; H_KD_C, HUVEC cells transfected with shRNA-NC as a control.
Next, we further used the Kyoto Encyclopedia of Genes and Genomes analysis (KEGG) to perform pathway enrichment-based clustering analysis of proteins responding to the changes in lncRNA FTX (Fig. 4). After the knockdown of lncRNA FTX, the peroxisome proliferator-activated receptor (PPAR) signaling pathway, leucine, valine, and isoleucine degradation, pyrimidine metabolism, fat digestion and absorption, cysteine and methionine metabolism, drug metabolism-other enzymes, and biosynthesis of amino acids were enriched by upregulated proteins, whereas leukocyte transendothelial migration, malaria, viral myocarditis, and the Rap1 signaling pathway were enriched by downregulated proteins. Overexpression of lncRNA FTX led to the enrichment of upregulated proteins in legionellosis, protein processing in the ER, longevity regulating pathway-multiple species, antigen processing and presentation, and the estrogen signaling pathway, whereas folate biosynthesis, tyrosine metabolism, hypertrophic cardiomyopathy (HCM), phenylalanine metabolism, dilated cardiomyopathy (DCM), vasopressin-regulated water reabsorption, shigellosis, phagosome, and focal adhesion pathways were enriched by downregulated proteins.
 Fig. 4.
Fig. 4.Kyoto Encyclopedia of Genes and Genomes analysis (KEGG) pathway clustering analysis of DEPs. H_OE, HUVEC cells overexpressing FTX; H_OE_C, HUVEC cells transfected with FTX-NC as a control; H_KDsH, HUVEC cells knocked down for FTX; H_KD_C, HUVEC cells transfected with shRNA-NC as a control.
Lastly, we further performed domain enrichment analysis to determine the domain
of DEPs after lncRNA FTX knockdown and overexpression (Fig. 5). The upregulated
proteins after lncRNA FTX knockdown contained the SH3 domain, whereas
downregulated proteins contained the thioredoxin-like domain. When the lncRNA FTX
was overexpressed, the upregulated proteins contained the ribosomal protein
L7Ae/L30e/S12e/Gadd45 family, DnaJC terminal domain, NOP5NT (NUC127) domain, and
DnaJ domain structures, whereas the downregulated proteins contained laminin
EGF-like (domains III and V), spectrin repeat, FYVE zinc finger, EF-hand domain
pair, kazal-type serine protease inhibitor domain, S-100/intestinal
calcium-binding protein (ICaBP) type calcium-binding domain, calcium-insensitive
EF-hand, and inosine-5
 Fig. 5.
Fig. 5.Protein domain clustering analysis of DEPs. H_OE, HUVEC cells overexpressing FTX; H_OE_C, HUVEC cells transfected with FTX-NC as a control; H_KDsH, HUVEC cells knocked down for FTX; H_KD_C, HUVEC cells transfected with shRNA-NC as a control.
The DEP interaction network was visualized using the STRING database, and the core modules identified by the MCODE plugin and the top 10 hub genes recognized by the MCC algorithm of the CytoHubba plugin are presented in Fig. 6. The core module in the overexpression lncRNA FTX group included eukaryotic translation initiation factor-5, ribosomal protein S18 (RPS18), RPL7A, nucleolar protein 56, mRNA turnover 4 homolog, SMG1, and serpine1 mRNA binding protein 1. The core module in the knockdown lncRNA FTX group included ubiquitin A-52, RPL8, RPL28, RPL7A, RPS25, RPL14, and RPS19.
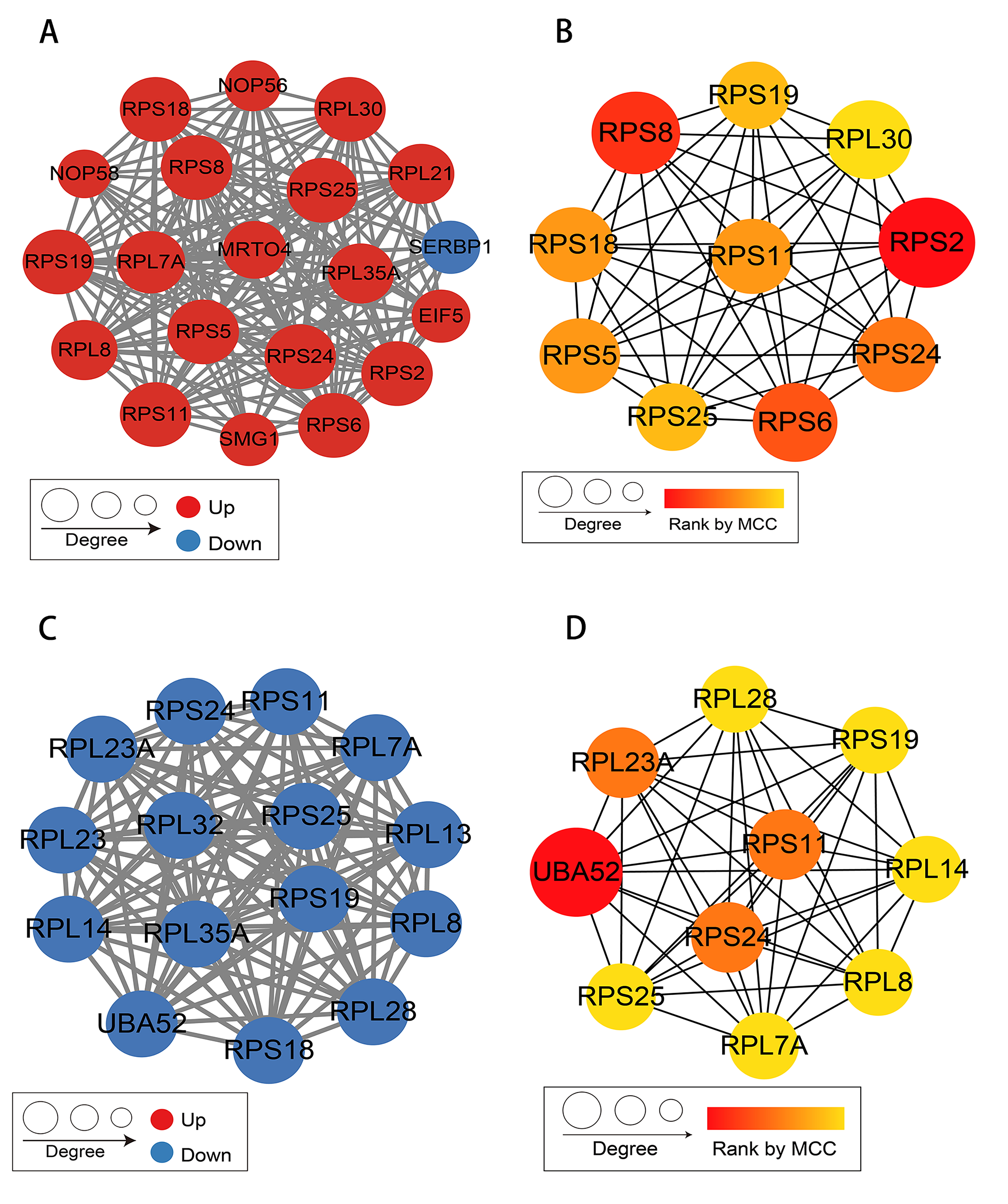 Fig. 6.
Fig. 6.Protein-protein interaction network analysis. (A) Core module of protein interaction in the H_OE vs H_OE_C group. (B) Top 10 hub genes identified by Matthews correlation coefficient (MCC) algorithm in the H_OE vs H_OE_C group. (C) Core module of protein interaction in the H_KDsH vs H_KD_C group. (D) Top 10 hub genes identified by MCC algorithm in the H_KDsH vs H_KD_C group.
In normal conditions, ECs remain in a dormant state and play crucial roles in upholding the vascular structure’s integrity, regulating vascular tension and permeability, and participating in immune responses [16]. However, during ischemia or tissue damage, ECs may become activated to proliferate significantly and facilitate angiogenesis. Meanwhile, they can also lead to fibrosis in several organs through processes such as inflammation, endothelial–mesenchymal transition, secreting exosomes, and regulating capillary rarefaction and vascular aging [17].
LncRNAs are abundant RNAs of over 200 nucleotides without protein-coding potential [18]. Studies have reported the significant roles played by lncRNAs in the regulation of various biological processes including gene expression regulation, chromatin remodeling, and epigenetic modifications [19, 20]. In the vascular system, lncRNAs expressed by ECs (such as MALAT1) can regulate vascular growth and function [21]. LncRNAs also participate in the fibrosis of various organs [22], including the liver [23, 24], heart [25], and kidney [26, 27]. As an increasing number of lncRNAs have been identified, they have become promising targets for antifibrotic therapy.
The FTX gene is located on the human X chromosome, which encodes nine introns, seven of which are transcribed into RNA fragments and linked to the lncRNA FTX [28]. LncRNA FTX is composed of about 2300 nucleotides. Some studies have found that lncRNA FTX can promote the progression of colon, liver, and kidney cancer [29, 30, 31], other studies have suggested that it can inhibit the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma (HCC) by regulating the M1/M2 polarization of Kupffer cells [32]. Despite these findings, the role of FTX in ECs remains unclear. Thus, the current study used quantitative proteomics and bioinformatics to systematically determine its molecular mechanisms and biological functions.
We analyzed the DEPs, with a focus on those whose expressions was increased in the lncRNA FTX knockdown group and decreased in the lncRNA FTX overexpression group. These proteins were encoded by genes including C3, chromobox protein homolog 1, coiled-coil domain containing 102B, cartilage oligomeric matrix protein (COMP), D-dopachrome tautomerase, faciogenital dysplasia 6 (FGD6), serpin family B member 6, and tissue inhibitor of metalloproteinase (TIMP). Among them, C3, COMP, FGD6, and TIMP1 are related to inflammation, collagen deposition, angiogenesis, and liver stem cell differentiation regulation, respectively, which may be involved in the development of liver fibrosis.
The C3 gene encodes the complement C3 protein. The complement system functions as a bridge between innate and adaptive immunity, and comprises a complex network of proteins. It can modulate mediator release, chemotactic effects, and vascular permeability. Additionally, it can enhance the phagocytosis of microbes through opsonization and participate in inflammatory responses [33]. The C3 protein is a crucial component in the complement system activation pathway. C3 can regulate liver inflammation through Kupffer/macrophages [34]. When the complement cascade enters an overactivated state, excessive C3 molecules are cleaved to release C3a [35], triggering the activation of Kupffer/macrophages in the liver via specific interaction with C3a/C3aR. This leads to the secretion of inflammatory chemokines and the recruitment of large numbers of bone marrow-derived mononuclear cells/macrophages to the liver, thereby amplifying the inflammatory response [36, 37, 38, 39]. Additionally, C5 molecules activated by the C3 cascade may cause liver fibrosis through mediating inflammatory, chemotaxis responses, and anaphylatoxin recognition in the innate and adaptive immune system [40] or regulating the activation and migration of hepatic stellate cells (HSCs) [41, 42]. Studies suggest that blocking the C3aR/C5aR and downstream Wnt/b-catenin signaling pathway could prevent endothelial-to-mesenchymal transition (EndMT) and alleviate glomerular fibrosis, indicating the potential role of C3 in the development of fibrosis [43].
COMP is a glycoprotein that primarily exists in the extracellular matrix (ECM)
of synovium, ligaments, cartilage, and tendons [44]. COMP promotes tumor
progression by increasing invasiveness, tumor cell vitality, and metabolic
transformation in breast and colon cancer [45, 46]. These findings suggest that
COMP might serve as a novel prognostic indicator and therapeutic target for
breast and colon cancers. Furthermore, studies have demonstrated that COMP levels
are markedly elevated in the serum and tissues of patients with HCC, thus serving
as a non-invasive tool for assessing HCC [47, 48, 49]. Besides its role in promoting
tumor progression, the potential for COMP to induce fibrosis in multiple visceral
organs has been extensively researched [50]. For example, COMP can serve as a
transforming growth factor beta 1 (TGF-
The FGD6 gene, which encodes guanine nucleotide exchange factors (GEFs) and features an FYVE-type zinc finger structure, a Dbl homology domain, and two pleckstrin homology domains, is part of the FGD family consisting of six members (FGD1-6) [53]. FGD6 regulates bone degradation by osteoclasts [54] and increases the risk of polypoid choroidal vascular disease [55]. Furthermore, FGD6 is significantly overexpressed in gastric cancer (GC) tissues and its overexpression is correlated with high T classification, histological grade, and a poor prognosis, making it an independent prognostic factor for GC patient survival [56]. FDG6 also regulates the angiogenic effects of vascular endothelial growth factor (VEGF) in ECs, including permeability, network formation, orientation, and proliferation [57]. Moreover, studies have demonstrated that inhibiting FGD6 gene expression in embryonic liver stem cells promotes their differentiation into liver cells, suggesting that the FGD6 gene might play a crucial role in regulating liver stem cell differentiation [58]. All of these mechanisms may provide potential targets for preventing or reversing liver fibrosis.
TIMP is an inhibitor of the matrix metalloproteinase (MMP), consisting of 184-194 amino acids and weighing 25–31 kD. TIMP encompasses four subtypes: TIMP-1, TIMP-2, TIMP-3, and TIMP-4 [59]. TIMP-1 primarily inhibits the activity of collagenases such as MMP-1, MMP-8, and MMP-13 [60]. MMPs are calcium-dependent zinc-containing peptidases that can degrade excessive ECM, whereas TIMPs can inhibit MMP activity appropriately without damaging normal liver tissue, thereby preserving microenvironment homeostasis of the liver [61]. ECM deposition is the primary pathological process of fibrosis, characterized by an imbalance between TIMP and MMP expression [62]. Thus, targeting MMP-mediated ECM degradation presents significant potential in treating fibrosis [63].
The excess proliferation of myofibroblasts and the deposition of ECM for tissue repair form the biological basis of fibrosis [9]. Although myofibroblasts are considered to be effector cells of fibrosis, the contribution of ECs to the development of fibrosis has also garnered increasing attention [64, 65]. Research findings indicate that environmental stimuli activate ECs, which potentially trigger and aggravate organ fibrosis via diverse mechanisms [66]. EndMT-mediated myofibroblast conversion is among the direct effects [67, 68, 69]. Furthermore, ECs contribute to fibrotic progression indirectly by driving inflammatory reactions and exosome secretion [70], through participation in regulating capillary rarefaction [71] and vascular aging [72]. The microenvironment of hepatic fibrosis comprises immune cells, macrophages, hepatic sinusoidal (LSECs), HSCs, extracellular matrix (ECM), and other cell types. This unique, complex, and highly dynamic region is rich in growth or signaling factors [73]. These different cell types and ECM proteins can coordinate liver remodeling, hematopoiesis, immune function, and tissue regeneration, which may be potential targets for the treatment of liver fibrosis [74, 75].
Previous studies by our research team identified that FTX enhances T-cell immunoglobulin and mucin domain 3 expressions through miR-545 inhibition, leading to the decreased production of inflammatory cytokines - interleukin 6 (IL-6), tumor necrosis factor-alpha, nuclear factor kappa B, and IL-1b, which delays the progression of cirrhosis [76]. In this study, we constructed HUVESs with the gain and loss of lncRNA FTX function. Application of quantitative proteomics and bioinformatics analyses enabled us to present a detailed overview of quantitative proteome maps in HUVEC cells upon knockdown and overexpression of lncRNA FTX. Further, protein annotation, enrichment, and clustering analyses facilitated the identification of DEPs. Intriguingly, our results showed that lncRNA FTX markedly decreased the expression of C3, COMP, FGD6, and TIMP in HUVEC cells. This observation indicates that these genes may serve as valuable therapeutic targets for the clinical prevention or reversal of hepatic fibrosis through vascular ECs. However, the specific regulatory mechanisms need to be further verified in cell experiments and in vivo experiments to evaluate its possibility.
The results of this study demonstrated that lncRNA FTX might play a potential role in ECs and contribute to the development of liver fibrosis, thus providing a promising target for the prevention or reversal of liver fibrosis.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
QZ and XFS designed the research study. YDL and XJS performed the research and analyzed the data. YDL wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the Natural Science Foundation of Shandong Province (grant no. ZR2021MH028), and the Medical Science and Technology Development Plan of Shandong Province (grant no. 202003031362).
QiZhao and Xiangfei Sun are the postdoctoral researchers in the Shandong Booke Biotechnology Co., Ltd, but there are no conflicts of interest for all authors
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.