



1 Department of Endocrinology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430000 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
Angiotensin-converting enzyme 2 (ACE2) is a member of the renin-angiotensin system (RAS), which was once considered a linear cascade. ACE2 mainly functions to convert AngiotensinⅡ (AngⅡ) to Angiotensin1-7 (A1-7). The biologically active product A1-7 then binds to the Mas receptor to form the ACE2/A1-7/Mas axis. In contrast to classic RAS, which plays a decisive role in regulation, the ACE2/A1-7/Mas axis effectively counteracts vasoconstriction, the inflammatory response, oxidative stress, and cell proliferation, and is thus a negative regulator of the RAS. ACE2 also functions as a chaperone to regulate intestinal amino acid uptake. It is widely expressed in the lungs, cardiovascular system, gastrointestinal tract, kidney, pancreas and adipose tissue. Previous studies have confirmed that ACE2 has a vital role in homeostasis. ACE2 also has a variety of other biological activities and plays a critical role in Type 2 diabetes (T2DM) and its complications, especially diabetic nephropathy, obesity, dyslipidemia and other diseases. In this review, we summarize the latest research on the regulation of glucose and lipid metabolism by ACE2 in different organs. Our focus was particularly on T2DM, with the aim of providing new clinical ideas for the use of ACE2 as an effective target in the prevention and treatment of metabolic diseases.
Keywords
- angiotensin-converting enzyme 2
- angiotensin 1-7
- Mas receptors
- type 2 diabetes
- glucose metabolism
- lipid metabolism
Angiotensin-converting enzyme 2 (ACE2) has two fundamental physiological functions. The first is a mono-carboxypeptidase that catalyzes a single residue from Angiotensin Ⅰ (Ang Ⅰ) and Ang Ⅱ to form Angiotensin1-9 (A1-9) and A1-7. The former can be converted to A1-7 by Angiotensin-converting enzyme (ACE) [1]. The second function of ACE2 is as a critical regulator of dietary amino acid uptake in the gastrointestinal tract. The generation of A1-7 is a vital mechanism of ACE2-mediated counteract regulator in renin-angiotensin system (RAS). Hypertension, Type 2 diabetes (T2DM), obesity and non-alcoholic fatty liver disease (NAFLD) are associated with disturbances in the RAS and changes in the expression level of ACE2 [2]. A local RAS occurs in the lung, liver, testis, cardiovascular system, gastrointestinal tract, kidney, pancreas and adipose tissue [3]. This affects metabolic organs and the progression of metabolic diseases. The current review describes the role of ACE2 in the regulation of glycolipid metabolism in different tissues, with the aim of finding effective therapeutic targets for metabolic diseases.
The RAS is a crucial homeostasis regulatory system in the human body that is closely associated with metabolic syndromes such as obesity, T2DM, and NAFLD. The RAS has two main axes: a classic ACE/Ang II/type 1 angiotensin receptor (AT1R) axis, and a new ACE2/A1-7/Mas axis (Fig. 1). The classic ACE/Ang II/AT1R axis contributes to vessel contraction, pro-inflammation, pro-oxidation, pro-fibrosis and pro-thrombus [4]. Dysregulated RAS accompanied by an increase in Ang Ⅱ results in a series of deleterious effects, including vasoconstriction, inflammation, cardiovascular damage, and increased oxidative stress. On the other hand, the ACE2/A1-7/Mas axis is thought to be a negative regulator of the classic RAS axis, with the ability to attenuate the majority of its deleterious effects. An imbalance between the two axes can damage the physiological functioning of different tissues and organs [5]. In 2000, Donoghue et al. [6] identified and isolated ACE2 from the left ventricular cDNA library of patients with heart failure. The ACE2 gene is located on the X chromosome and is the first human homologue of ACE. ACE2 and the ACE catalytic domains share 42% of their amino acid sequence [6, 7]. ACE2 is a chimeric molecule that includes a catalytic domain and a collectrin-like transmembrane domain. In renal tissue, collectrin stabilizes numerous single transporters that are needed for amino acid reabsorption [8]. The non-enzymatic collectrin domain of ACE2 functions as a chaperone protein for the amino acid transporter B0AT1. The ACE2-B0AT1 complex is involved in the absorption of neutral amino acids in the intestine [9, 10]. A1-7 is a bioactive, 7 amino acid peptide product of the RAS cascade that exerts its biological effects via the endogenous orphan receptor Mas [11, 12]. The ACE2/A1-7/Mas axis is therefore a fundamental component of RAS and has been confirmed to directly regulate metabolic homeostasis.
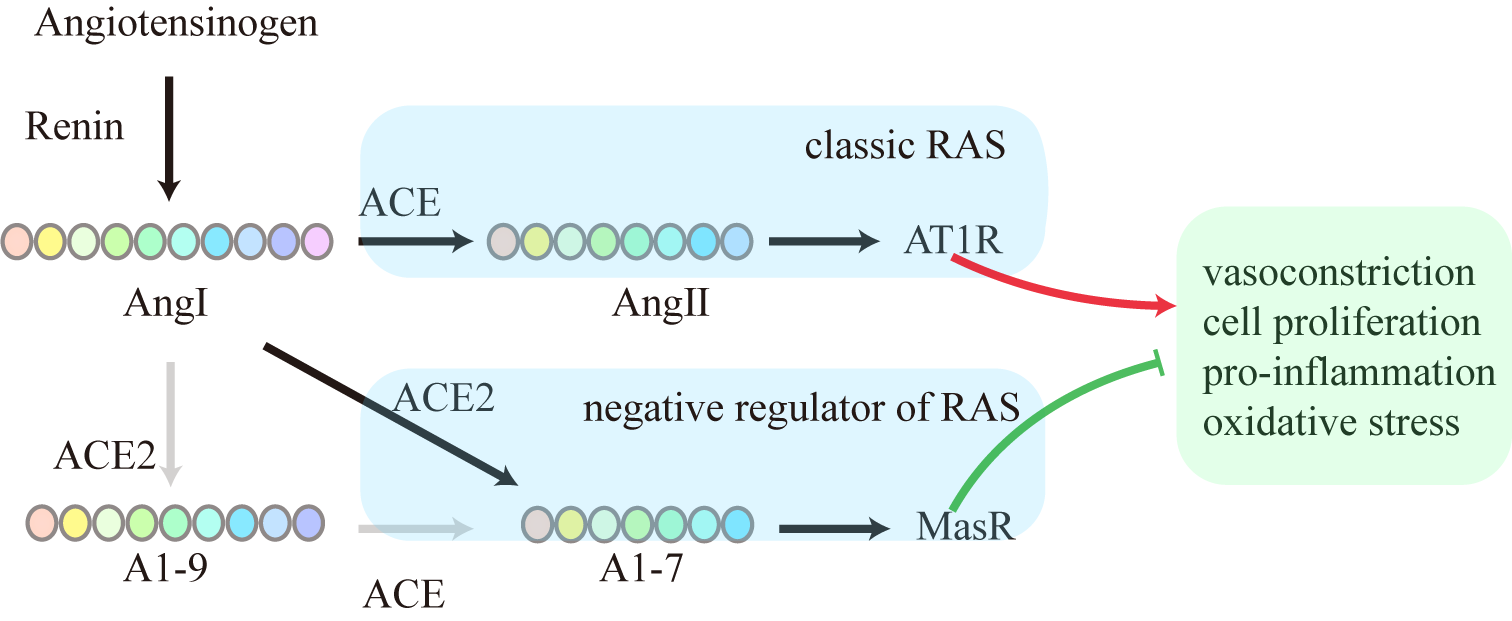 Fig. 1.
Fig. 1.Classic and negative regulator of RAS. ACE, angiotensin-converting enzyme; ACE2, angiotensin-converting enzyme 2; A1-9, angiotensin 1-9; A1-7, angiotensin 1-7; Ang I, angiotensin I; Ang II, angiotensin II; AT1R, type 1 angiotensin II receptor; RAS, renin-angiotensin system.
ACE2 is mostly expressed in the alpha cells of the mouse pancreas, whereas the A1-7 receptor Mas is mostly expressed in beta cells. This indicates the ACE2/A1-7/Mas axis may be important in regulating beta cell function [13]. Other research has shown that ACE2 is also expressed in pancreatic beta, acinar and ductal cells, as well as in the islet microvasculature [14, 15]. Through its catalytic product A1-7, ACE2 functions as a positive modulator of islet function and works through Mas receptors.
The primary pathophysiology of T2DM is relative insulin insufficiency and insulin resistance (IR), typically with developing beta cell dysfunction. The RAS is closely associated with pancreatic endocrine physiology and its pathophysiology. The classic RAS components are upregulated in T2DM, resulting in impaired islet blood flow and oxidative stress. During the compensatory period there are changes in islet morphology and beta cell hypertrophy, with an increased cell number [16, 17]. These changes eventually lead to the apoptosis and dedifferentiation of beta cells, thereby impairing islet function and exacerbating glucose metabolism disorders. The ACE2/A1-7/Mas axis exerts a positive role on pancreatic islet beta cell function by improving the microcirculation, regulating mitochondrial function and inhibiting oxidative stress, and reducing apoptosis and dedifferentiation.
The blood circulation of pancreatic islets comprises about 7–10% of the total pancreatic blood flow [18]. The high vascularization of islet is crucial for the survival and proper functioning of beta cells. In turn, beta cells secrete several substances that regulate vascular development, including vascular endothelial growth factor (VEGF). Interactions with islet microvascular endothelial cells can influence the function of beta cells. Impairment in the vascularization of pancreatic islets negatively effects glucose tolerance and insulin secretion. Under the dual action of glucose toxicity and lipid toxicity, dysfunction of microvascular endothelial cells is one of the important mechanisms leading to the failure of beta cells. Dysfunction of islet endothelial cells is thus a potential contributor to the pathophysiology of T2DM. Previous research reported the ACE2/A1-7/Mas axis performs an essential role in mediating the apoptosis of several cell types. Studies have provided evidence that A1-7 can prevent the apoptosis of alveolar epithelial cells induced by Ang II or endoplasmic reticulum stress [19]. Activation of the ACE2/A1-7/Mas axis could potentially improve islet blood flow and preserve beta cells by enhancing islet endothelial cells [20]. Our earlier research showed the ACE2/A1-7/Mas axis induced phosphorylation of endothelial nitric oxide synthase (eNOS) via activation of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathway, thereby promoting the production of NO [20]. Conversely, PI3K inhibitors and the Mas receptor-specific antagonist A779 can inhibit the phosphorylation of eNOS [21]. Upregulation of the ACE2/A1-7/Mas axis has been shown to reduce the apoptosis of islet endothelial cells by regulating phosphorylation levels of the p38 mitogen-activated protein kinase (MAPK) signaling pathway and of the c-Jun N-terminal kinase (JNK) signaling pathway [22]. Together, these findings demonstrate involvement of the AKT/eNOS/NO, JNK, and p38 MAPK signaling pathways in the anti-apoptosis effect of A1-7 on islet endothelial cells. Protecting the survival and function of islet endothelial cells is a major goal for reducing the risk of diabetes. Exogenous administration of A1-7 can also upregulate VEGF, while inhibition of inducible nitric oxide synthase (iNOS) may be one of the mechanisms that improves islet microcirculation, Elucidation of the specific mechanisms require further study [13], however. Based on the above findings, it can be concluded that ACE2 protects beta cells by reducing the dysfunction of islet endothelial cells and enhancing pancreatic microcirculation.
Pancreatic islets express lower levels of antioxidants compared to other tissues, making then more vulnerable to oxidative stress [23]. In T2DM, oxidative stress leads to beta cell failure [24], resulting in decreased insulin synthesis and secretion [25]. Insulin release from pancreatic beta cells is regulated by a complex system involving mitochondrial oxidative metabolism. the ACE2/A1-7/Mas axis was shown to have antioxidant effects on INS-beta cells, thereby restoring glucose-induced insulin secretion by reducing the generation of intracellular reactive oxygen species (ROS), suppressing mitochondrial membrane potential depolarization, and maintaining glucose sensitivity threshold of beta cells [26]. Activating the ACE2/A1-7/Mas axis can also regulate mitochondrial metabolism and calcium influx in islet beta cells, as well as affecting insulin secretion through the regulation of mitochondrial respiration and ATP production [27].
The ACE2/A1-7/Mas axis affects the proliferation and differentiation of pancreatic islet cells, which can regulate the development of embryonic pancreatic cells [28]. Apoptosis is thought to be the primary mechanism leading to beta cell dysfunction in T2DM patients. Treatment of T2DM rats with A1-7 markedly reduces the number of sensing beta cells in the pancreas, reduce the expression of iNOS, caspase-3, 8, 9, Bax and JNK in the pancreas islet, and increases expression of the anti-apoptotic factor Bcl-2 [29]. Loss of ACE2 impairs beta cell proliferation, resulting in a smaller islet area and reduced number of islet beta cells [30]. The main reason for reduced beta cell mass has been identified as an increase in the apoptosis of beta cells. However, recent studies have indicated that cell function is significantly influenced by cell dedifferentiation. Indeed, dedifferentiation could play a more important role in the etiology of beta cell failure than endocrine cell apoptosis [31]. Dedifferentiated beta cells can redifferentiate into mature islet beta cells to secrete insulin, therefore playing a more important role in T2DM [29]. Knockout of the ACE2 gene aggravated the dedifferentiation of beta cells under a high food diet (HFD), Intervention with A1-7 could reduce the degree of dedifferentiation of beta cells under metabolic stress, both in vitro and in vivo [13]. The above effects were mediated by improvement in islet cell microcirculation and a reduction in islet cell NOS [13, 21]. Gamma-aminobutyric acid (GABA), is a key inhibitory neurotransmitter in the mammalian central nervous system and is crucial for islet function and glucose homeostasis [32]. The GAD67/GABA signal is regulated by the ACE2/A1-7/Mas axis, which improves cell dysfunction. Up-regulation of the ACE2/A1-7/Mas axis and the GABA, either separately or together, can prevent the onset of obesity-induced diabetes and have additive effects on beta cell differentiation and maturation [33]. The GABA signaling system in islet is closely correlated with the ACE2/A1-7/Mas axis. In this network system, ACE2 is secreted by alpha cells and transforms Ang Ⅱ into A1-7, thereby activating Mas receptors in beta cells and promoting the synthesis of GABA. This indicate that local expression of ACE2 by alpha cells may be an important paracrine regulatory molecule between alpha cells and beta cells [34]. These findings offer a new theoretical foundation for the mechanisms of paracrine regulation of islet function, while suggesting new intervention targets for the management of T2DM.
The liver is essential for glycolipid metabolism and energy balance, and liver dysfunction results in various metabolic disorders, such as obesity, IR, NAFLD, and metabolic syndrome [35]. Numerous critical elements of the RAS have been identified in the liver, and an imbalance between the ACE/Ang II/AT1 and ACE2/A1-7/Mas axes results in metabolic dysfunction.
Metabolic abnormalities are often accompanied by IR, and insulin signaling is
impaired during the state of IR. The liver is one of the main target organs for
insulin and plays a crucial role in regulating glycolipid metabolism [36]. The
mechanism of IR is related to the negative regulation of several steps in the
insulin signaling cascade by Ang II. These include phosphorylation of the insulin
receptor and insulin receptor substrate-1 (IRS-1), and activation of the PI3K/Akt
pathway [37]. ACE2 can antagonize the above effects of Ang II. Previous studies
found that the ACE2/A1-7/Mas axis can improve IR and glycolipid metabolism. This
may be related to anti-oxidative stress and the regulation of insulin signaling
pathways [38]. Over-expressing of ACE2 in HepG2 cells inhibits JNK activation,
and decreases the expression of phosphoenolpyruvate carboxykinase (PEPCK) and
glucose-6-phosphate (G-6-P) through the IRS-1/PI3K/Akt pathway and attenuated
oxidative stress [39]. Additionally, high concentrations of A1-7 decrease PEPCK
expression by downregulating the transcription of hepatocyte nuclear factor
4
Overactivation and imbalance of RAS causes hepatic dysfunction and promotes
hepatic steatosis and lipid metabolism disorders [43]. ACE2 overexpression and
Ang II or ATR1 blockers can effectively prevent and treat metabolic diseases such
as T2DM, IR, hepatic steatosis, and NAFLD. Overexpression of ACE2 and of
recombinant human ACE2 significantly reduced fat accumulation and endoplasmic
reticulum stress levels in the liver of db/db mice [44]. Obvious hepatic
metabolic abnormalities have also been shown in ACE2KO mice. Moreover, the
positive effect of liraglutide on NAFLD is attenuated in ACE2KO mice, and
exogenous A1-7 intervention in a diabetes model of C57BL/6J mice can
significantly reduce NAFLD [43]. Furthermore, the ACE2/A1-7/Mas axis can improve
the glycolipid metabolism balance in HFD mice by regulating the liver GABA
signaling system, upregulating the PI3K/Akt signaling pathway, inhibiting the
expression of proteins involved in hepatic adipogenesis and gluconeogenesis,
promoting the expression of fatty acid beta oxidation-related genes, and reducing
macrophage infiltration [42]. In addition, oral administration of A1-7 can
ameliorate hepatic inflammation. This is accompanied by markedly reduced
expression of resistin, Toll-like receptor 4 (TLR4), interleukin-6
(IL-6) and tumor necrosis factor alpha, reduced phosphorylation of MAPK, and
increased expression of nuclear factor kappa B (NF-
The gastrointestinal tract is not only the major site of nutrient digestion and absorption, but also the largest endocrine organ in the human, and an active participant in the regulation of systemic metabolism. The “gut-islet axis” is a crutial endocrine signaling axis that modulates islet function through the communication between intestinal microbiota and endocrine metabolism [46]. ACE2 is highly expressed in the gastrointestinal tract, especially in the duodenum, jejunum, ileum, and colon [34]. This may partly explain why patients infected with COVID-19 have digestive symptoms. Intestinal dysbiosis is an important underlying inducer of metabolic diseases such as T2DM and obesity [47]. Studies have shown that ACE2 deficiency aggravates diabetes-induced dysbiosis, leading to worsening diabetes [48]. ACE2 is involved in the absorption of neutral amino acids by binding to B0AT1-specific receptors, thereby regulating intestinal nutrition, the inflammatory response, and gut microbiota [49]. Administration of the oral A1-7 to obese mice results in increased expression of neutral amino acid transporter (B0AT1) in the gut, possibly associated with the uptake of tryptophan (Trp). This was the first evidence of a link between A1-7 and regulation of the gut microbiota regulation [50]. Similarly, MasKO mice showed altered intestinal structure, which subsequently leads to dysbacteriosis [51]. Trp metabolites can improve islet function in db/db mice and may be act as signaling molecules for communication between the intestinal flora and pancreatic islets. In a recent study by our group, we found that intestinal ACE2 functions as an upstream regulator of the gut-islet axis. Gut ACE2 promoted islet beta-cell proliferation and insulin secretion by regulating intestinal Trp absorption [52, 53]. Furthermore, intestinal glucose transport depends on the sodium-glucose transport vehicle (SGLT) and glucose transport (GLUT) vehicles of intestinal mucosal epithelial cells. ACE2 is involved in regulating the expression level of SGLT and GLUT1, indicating the ACE2/A1-7/Mas axis may participate in the control of postprandial blood sugar levels [54, 55]. In conclusion, the ACE2/A1-7/Mas axis has a positive role in maintaining intestinal homeostasis, and in turn the regulation of glucose homeostasis. This suggests the gut-islet axis may be an important endocrine signaling pathway for intestinal microecology and intestinal endocrine metabolism in the regulate islet function
ACE2 is expressed on the brush border of proximal tubular cells and on podocytes, with glomerular endothelial and mesangial cells being only weakly positive or negative [56]. ACE2 may be an important target for the prevention and treatment of diabetic nephropathy. This is a progressive condition associated with chronic renal failure and characterized by mesangial matrix expansion, glomerular basement membrane thickening, and proteinuria. Changes in the expression of ACE2 in the kidney have been studied in animal models of T2DM. The expression of ACE2 increased in an early T2DM model, while the expression of ACE decreased [57]. However, in the diabetic nephropathy stage, ACE2 expression decreased while ACE increased [58]. Indeed, some studies have shown that ACE2 deficiency or inhibition can exacerbate diabetic nephropathy [59, 60, 61]. Treatment with ACE2 protects multiple organs and contributes to physiological metabolic homeostasis through its enzyme and amino acid transport functions has additional benefits for the progression of diabetic nephropathy due to its inhibition of renal RAS activation and reduction in glomerular fibrosis [62]. The localization of ACE2 in podocytes in the early stages of diabetes suggests that it may protect against the loss of podocytes, thereby preventing the glomerular damage from worsening [63]. Conversely, in streptozotocin-induced diabetes, A1-7 administration aggravates diabetic kidney injury, suggesting the protection provided by ACE2 may be due to the loss of Ang II rather than through A1-7 [64].
The two main types of adipose tissue are white adipose tissue (WAT) and brown
adipose tissue (BAT). WAT acts as an energy store, while BAT is responsible for
energy consumption. In addition to its energy storage and thermogenesis
functions, adipose tissue is a crucial endocrine organ that secrets a variety of
bioactive molecules (adipokines) such as leptin and adiponectin. These act on
different target organs, such as the brain, muscle, heart, vessels, liver and
others to regulate systemic metabolic homeostasis [65, 66]. The ACE2/A1-7/Mas
axis plays a key role in regulating glycolipid metabolism and energy metabolism.
A deficiency of Mas receptors in adipose tissue leads to reduced glucose
transport and decreased expression of adiponectin and GLUT4, which then manifests
as elevated serum triglyceride and cholesterol levels [41]. Over-activation of
the Ang II signaling cascade also exacerbates adipose IR, increases adipose
tissue inflammation and oxidative stress, and inhibits adiponectin secretion and
insulin signaling pathways [67, 68]. Conversely, activation of the ACE2/A1-7/Mas
axis reduces adipose tissue inflammation and oxidative stress, and increases
glucose uptake and adiponectin levels [69]. Moreover, exogenous treatment of
HFD-induced obese mice with A1-7 has been shown to increase the BAT and to
improve the insulin signaling pathway (PI3K/Akt/mTOR). Later studies confirmed
that recombinant human ACE2 can stimulate BAT, promote the differentiation and
proliferation of BAT, increase the energy expenditure in HFD mice, induced the
conversion of subcutaneous WAT to BAT, and finally improved IR [70]. Obesity is
an independent risk factor for chronic diseases such as hypertension, coronary
atherosclerosis, and T2DM, Effective anti-obesity treatments are therefore
urgently needed. BAT can increase energy expenditure through thermogenesis, while
ACE2 is a promising treatment. Anti-inflammatory factors induced by obesity and
HFD-induced mice models have also been identified. Oral formulation of A1-7
treatment improves metabolism, increases GLUT4 expression, and activates the
AMPK/FOXO1/PPAR-
ACE2 protects multiple organs and contributes to physiological metabolic homeostasis through its enzyme and amino acid transport functions (Fig. 2). As the main member of the RAS, ACE2 is widely expressed in the lungs, cardiovascular system, pancreas, liver, gastrointestinal tract, adipose tissue and testis. ACE2 has a vital role in the glucose and lipid metabolism by protecting pancreatic islet functions, improving IR, promoting energy consumption, and reducing oxidative stress. It exerts its biological effects mainly by converting Ang II to A1-7, thereby antagonizing the classical RAS functions. ACE2 functions independently of the RAS, and also regulates intestinal amino acid homeostasis and the gut microbiome. However, clinical trials that investigate ACE2 in glucose and lipid metabolism have yet to be performed. ACE2 is a potentially valuable therapeutic target for metabolic diseases, particularly in T2DM and obesity. Further studies into the effects of ACE2 on metabolism and related mechanisms are warranted.
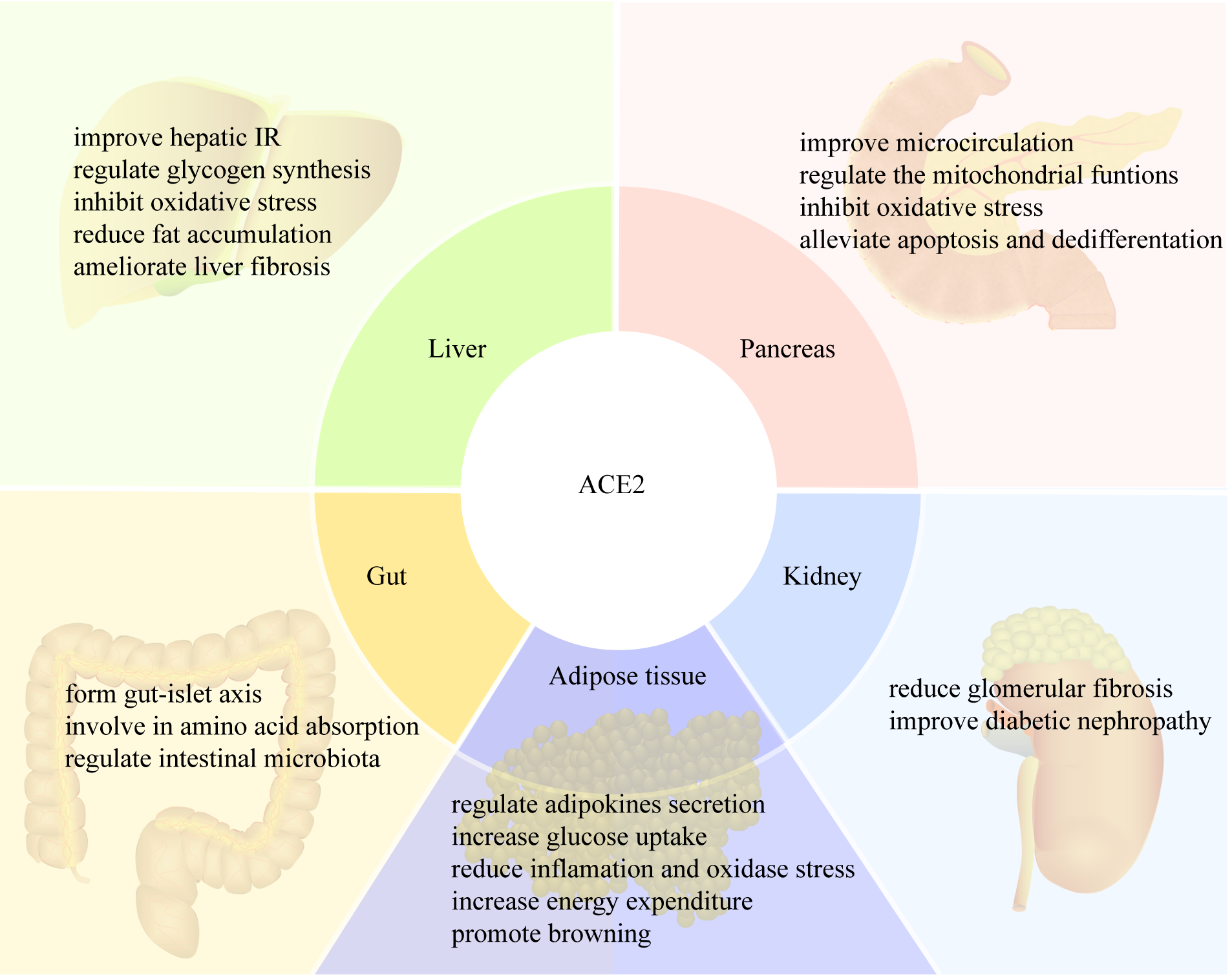 Fig. 2.
Fig. 2.ACE2 regulates metabolism in different tissues. IR, insulin resistance.
ACE2, angiotensin-converting enzyme 2; RAS, renin–angiotensin system; Ang II,
angiotensinⅡ; A1-7, angiotensin1-7; T2DM, type 2 diabetes; A1-9, angiotensin 1-9;
Ang I, angiotensin I; ACE, angiotensin-converting enzyme; NAFLD, non-alcoholic
fatty liver disease; AT1R, type 1 angiotensin receptor; IR, insulin resistance;
VEGF, vascular endothelial growth factor; eNOS, endothelial nitric oxide
synthase; PI3K, phosphatidylinositol 3-kinase; MAPK, mitogen-activated protein
kinase; JNK, c-Jun N-terminal kinase; iNOS, inducible nitric oxide synthase; ROS,
reactive oxygen species; HFD, high food diet; GABA, gamma-aminobutyric acid;
IRS-1, insulin receptors substrate-1; PEPCK, phosphoenolpyruvate carboxykinase;
G-6-P, glucose-6-phosphate; NADPH, nicotinamide adenine dinucleotide phosphate;
TLR4, Toll-like receptor 4; IL-6, interleukin-6; NF-
LY, FL and RL contributed to the study conception and design. The first draft of the manuscript was written by RL. FL and LY edited the draft and revised the manuscript for important intellectual content. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by the National Natural Science Foundation of China, No. 82170812 and No. 81974104.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.