
- Academic Editor
Advances in gene sequencing technology and decreasing costs have resulted in a proliferation of genomic data as an integral component of big data. The availability of vast amounts of genomic data and more sophisticated genomic analysis techniques has facilitated the transition of genomics from the laboratory to clinical settings. More comprehensive and precise DNA sequencing empowers patients to address health issues at the molecular level, facilitating early diagnosis, timely intervention, and personalized healthcare management strategies. Further exploration of disease mechanisms through identification of associated genes may facilitate the discovery of therapeutic targets. The prediction of an individual’s disease risk allows for improved stratification and personalized prevention measures. Given the vast amount of genomic data, artificial intelligence, as a burgeoning technology for data analysis, is poised to make a significant impact in genomics.
In recent years, genomic data has exploded due to the advancement of high-throughput sequencing technologies, a reduction in sequencing costs, and the proliferation of consumer-oriented sequencing platforms. There are already estimates that genomics will generate more data than social media applications and astronomy within a few years [1]. Genomic data becomes an integral part of Big Data. Gene interactions, gene-environment interactions, and non-coding regions of the genome are studied by genomics. In contrast, genetics focuses on individual traits [2]. A limitless opportunity has been created by genomic big data for the life science field through its efficient use. As big data has taken hold, genomics has witnessed unprecedented progress, including the establishment of large-scale global biobanks that contain genetic and phenotypic information [3, 4, 5, 6, 7, 8], as well as a variety of advanced computational and statistical approaches to predict disease risks and map disease genetics [9]. Consequently, genomic data are more accessible and the role of genes in disease and health is better understood. For example, there are more than 7000 single-gene diseases with known molecular etiologies and shapes (https://www.omim.org/statistics/geneMap). Transforming genomics into medicine involves applying a large amount of genomics data to solve healthcare problems.
Translational medicine epitomizes a form of medical inquiry that transmogrifies the products of fundamental research into tangible modalities encompassing ailment abatement, ailment discernment, therapeutics, and prognosis assessment for living patients. Its quintessential trait resides in interdisciplinarity, exhaustive foundational exploration concerning clinical quandaries, and the swift dissemination of research outcomes into practicality. The nucleus of translational medicine resides in the scrutiny of biomarkers. To this end, it encompasses the evolution and utilization of diverse “omics” methodologies and molecular biology repositories to scrutinize a spectrum of biomarkers for estimating ailment susceptibility, ailment diagnosis, and categorization, evaluating responses to therapeutics, prophesying the trajectory of ailment, and the evolution of innovative remedial methodologies and novel pharmacotherapeutics. The translational and clinical sciences, introduced by Zerhouni in 2005, are a significant part of biomedicine and are a promising new interdisciplinary approach for translating superior scientific innovations into health benefits [10]. Within the precincts of translational medicine, the harnessing of voluminous data is deemed a potent cornerstone, bequeathing invaluable insights to fortify the realms of medical inquiry and therapeutic paradigms.
Within the confines of this scrutiny, we have delved into the realm of clinical governance, a sphere profoundly impacted by germane technologies and methodologies within the milieu of genomics and vast datasets. Synthesizing the amalgamation of genomic erudition with pragmatic clinical implementation, alongside the adversities encountered in the analogous realm of genomic application, could serve as a beacon guiding the further transmutation of genomic information. It points out the direction for additional transformation of genomics data (Fig. 1).
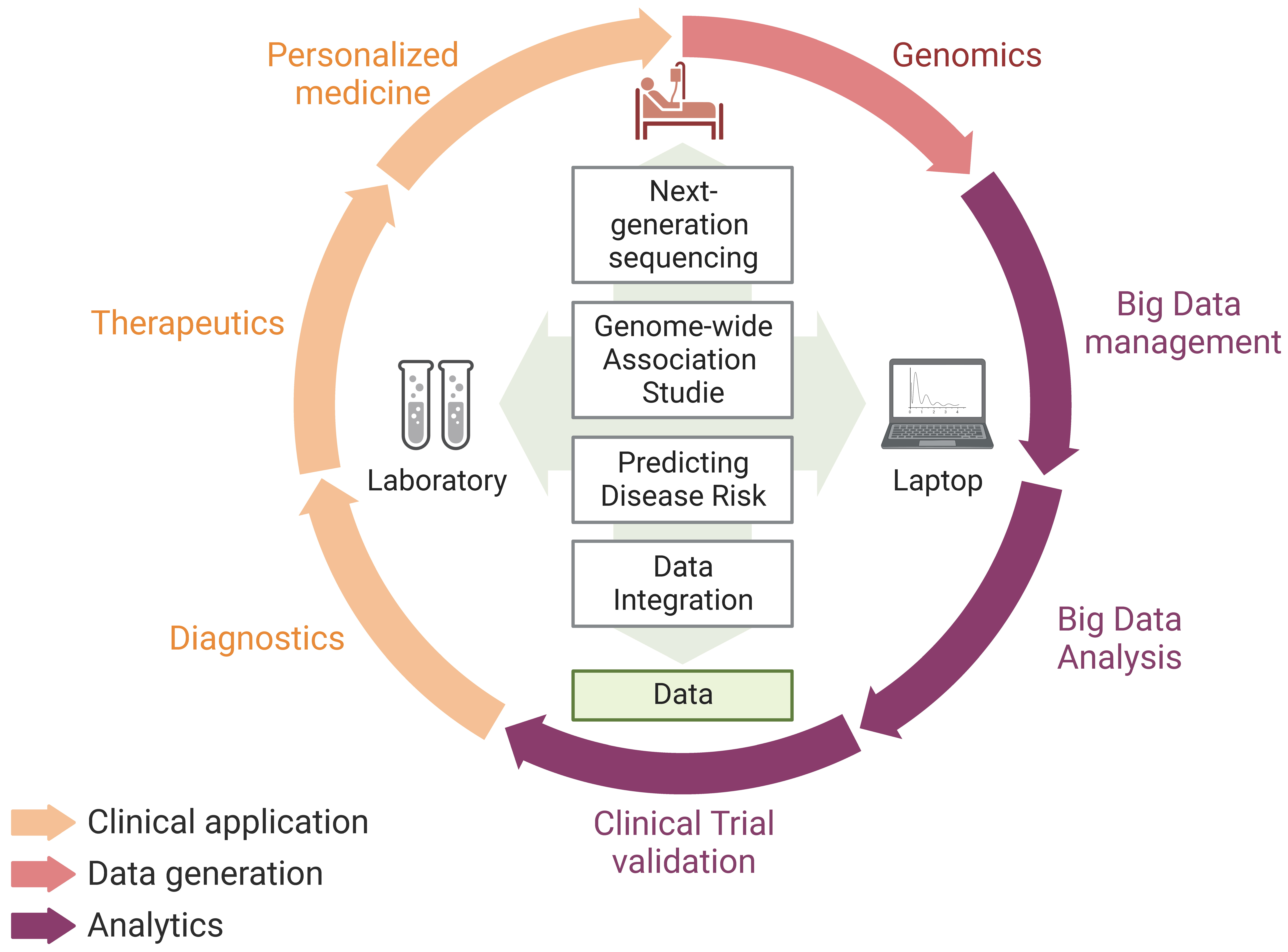 Fig. 1.
Fig. 1.Application of Genomic Data in Translational Medicine including data generation, analytics and clinical application.
The continuous improvement of genome sequencing technology allows patients to choose different next-generation sequencing (NGS) technologies such as targeted sequencing, whole-exome sequencing, and whole-genome sequencing to obtain their genome information according to their actual needs. This has the potential to change the health outcomes of patients to a great extent for only a few hundred dollars. The major advantage of NGS is its ability to simplify, accelerate, and expand the range and number of sequences that can be assessed in comparison to conventional methods of sequencing. The use of genome sequencing technology is crucial to translational medicine in genomics, as it provides high-resolution genetic information highly relevant to diseases, as well as improving disease screening, molecular diagnosis, treatment, and management [11, 12, 13, 14].
Precision prevention is a prevention strategy for primary (disease prevention) and secondary (early detection) prevention of disease by combining non-genetic and genetic characteristics of individuals. Many cancers, including hereditary breast cancer and Lynch syndrome, can be detected and risk assessed using genetic testing. It is recommended that carriers of BRCA1 or BRCA2 germline variants undergo prophylactic surgery to mitigate their risk of developing breast cancer, as well as testing relatives at risk and implementing preventive interventions for those who carry variants [15]. An individual who carries cancer susceptibility genes is typically identified based on his family history [16], however, the family-history model has been recognized to have some limitations [17]. Between 50% and 60% of patients miss the opportunity for genetic testing because they do not meet the criteria as assessed by the family history model [18, 19, 20]. Hence, in light of the increasing ease of access to NGS technologies, it has been proposed to incorporate them into a population-based screening approach [21]. With population-based genetic testing, it is possible to significantly improve health outcomes for cancer susceptibility carriers while enabling accurate cancer prevention through risk stratification. Studies have shown that population-based genetic testing reduces cancer incidence without increasing mental stress is cost-effective, and is capable of identifying more mutation carriers [19, 21, 22]. NGS also plays an irreplaceable role in the screening and diagnosis of additional diseases. For example, its use in sudden cardiac death (SCD), which kills millions of people every year [23]. It is estimated that up to 70 percent of SCD cases in people under 50 years of age are potentially due to genetic causes [24]. As the majority of SCD occurs in the general population rather than in those diagnosed with heart disease [25], a high priority should be given to optimising risk stratification and prevention of SCD in the general population as an essential clinical and public health goal. A study has shown that genetic screening using a targeted NGS panel can identify molecular genetic causes in a significant proportion of patients with suspected inherited heart disease and can be applied to cascade screening of family members of genotype-positive pre-diagnosed individuals [26]. It also guides the choice of preventive measures, such as treatment with antiarrhythmic drugs or implantable cardio verterde fibrillators (ICDs) [26]. With additional genetic and phenotypic correlation studies, NGS is expected to be a rapid and cost-effective molecular diagnostic tool for sudden cardiac death [27] that can reduce the probability of sudden cardiac death in patients through population-based screening.
Similarly, NGS has been proposed for newborn screening. Virtual rapid whole-genome sequencing for newborn screening performed in UK Biobank identified 15 disease cases missed by conventional screening. The study concluded that if whole-genome sequencing had been conducted on day 5 of life, symptoms in seven critically ill children could have been completely prevented [28]. Although neonatal genome sequencing is beneficial in detecting many diseases at an early stage, its use as a routine application in NGS requires further research on the long-term impact of preventive genomic screening on families as well as children, the long-term management of genetic sequencing results due to updated knowledge, and the unique ethical challenges that may arise [29]. It is possible to apply genomics to clinical practice as a means of precision prevention through population-based genetic testing, enabling more precise identification of diseased potential groups and earlier clinical intervention, leading to a health strategy that maximizes benefits for more patients.
Information from the molecular level of disease can more accurately characterize the disease, improve the efficiency of disease diagnosis, refine disease typing, and provide information for treatment decisions. There has always been a strong focus on genomic research in cancer. Origins of tissues, types of cells, and morphologies are traditionally used to classify tumors. Cancer diagnostics and prognostics have become increasingly reliant on mutations in the context of cancer’s genomic basis. In the molecular classification of endometrial carcinoma by The Cancer Genome Atlas (TCGA), POLE gene mutation indicates POLE hypermutation, which is associated with a more favorable prognosis. Conversely, the high copy type with TP53 mutations is linked to the poorest prognosis [30]. As well, breast cancers frequently have somatic mutations in TP53 and PIK3C genes. It should be noted that the frequency of these gene mutations varies depending on the subtype of breast cancer. With NGS, mutational variants can be identified, thus making subtype classification easier [31].
It is challenging to collect samples of tissue from patients suffering from cancer during the early stages of their disease so novel non-invasive biomarkers are being explored. It is possible to use plasma cell-free DNA (cfDNA) genotyping instead of tissue genotyping in circumstances where tissue specimens are insufficient or inaccessible [32, 33]. Plasma cfDNA, which is released from cells, contains circulating tumor DNA [34]. NGS technology has been used to detect a variety of mutations associated with cancer in cfDNA from individuals with various types of cancer, including copy number changes, single nucleotide mutations, DNA fragmentation patterns, and methylation changes [35, 36, 37]. Leighl et al. [38] compared tissue genotyping with cfDNA genotyping by NGS in untreated metastatic non-small-cell lung cancer and found highly consistent results and more efficient cfDNA genotyping. Additionally, cfDNA genotyping can enable targeted treatment matching and monitoring of resistance to therapy to provide personalized treatment [39, 40]. In a short time and for a low cost, NGS can quickly identify a wide variety of biomarkers from small biological samples due to its high sensitivity and specificity. As a result, NGS became the technique of choice for analyzing cfDNA, known as liquid biopsy, a new trend in oncology [41].
The implementation of next-generation sequencing in prenatal diagnosis represents a significant achievement in the application of genomics within clinical settings. Conducting noninvasive prenatal screenings (NIPSs) to detect fetal chromosomal defects through the analysis of maternal cfDNA has changed the way chromosomal and other genetic disorders are diagnosed and treated during pregnancy [42, 43]. Furthermore, whole-genome sequencing (WGS) has been shown to assist in determining the cause of rare diseases as well as performing accurate molecular diagnoses. In comparison to traditional diagnostic methods, WGS is more efficient in diagnosing, reducing diagnostic odyssey costs, changing medical management, and ultimately benefiting patients and their families [44, 45, 46]. There are already established guidelines recommending the utilization of clinical whole genome sequencing as either a primary or secondary diagnostic tool for rare disease sufferers [47, 48].
Personalized therapeutic decisions are increasingly being guided by NGS. Genomic analysis of cancer is crucial to selecting drugs that target somatic gene mutations that drive cancer development or progression. According to research, NGS can help identify at least one actionable target for targeted therapy [49, 50, 51]. Further, targeted therapy appears to be associated with higher progression-free survival and response rates [52]. If patients do not benefit from molecularly targeted agents, immunotherapy may also be considered. Patients for whom immunotherapy may be effective can be identified based on genomic information about the tumor obtained through NGS. Recently, a clinical trial demonstrated that patients with HER2-negative progressive gastric cancer achieved significantly improved overall survival with tislelizumab, an immune checkpoint inhibitor, compared to the chemotherapy group [53]. The utilization of immunotherapy has expanded the treatment strategies available to cancer patients, and it is increasingly being employed as a first-line therapy for many types of cancer [54].
With the advent of various biotechnologies, such as variant analysis methods, and gene editing, gene therapy has entered a new era. Genetic analysis through NGS facilitates the identification of target mutations that drive disease progression, furnishes highly precise DNA sequence data, and constitutes a crucial tool for enhancing gene therapy. As a result, many novel gene therapy products have been approved for clinical use after successful laboratory testing [55]. Gene therapy is now used not only for single genetic disorders, and cancer [56] but also for common complex diseases such as osteoarthritis [57] and diabetic neuropathy [58]. Gene therapy uses normally functioning genes to repair or replace defective genes to treat genetic diseases that are generally untreatable by drugs and are less likely to face drug resistance problems [59]. However, the field is still in an immature stage, with challenges that include ineffective delivery systems [60], and the inability to ensure continuous and stable gene expression and host immune responses. To facilitate the comprehensive market penetration of gene therapy, it is necessary to deepen basic research, overcome technical difficulties, precise safety supervision to reduce the harm caused by possible side effects [61], and appropriate pricing to increase the accessibility of treatment.
In summary, genome sequencing technology which is the key to facilitating faster translation of genomic knowledge for clinical practice provides genomic information that provides unprecedented insight into the biology and pathogenesis of many diseases [62, 63, 64] and has been used in the fields of cancer detection and treatment, assisted reproduction, prenatal and perinatal screening, inpatient management of critically ill infants, management of Mendelian diseases, rare diseases, and other clinical aspects [65, 66, 67, 68]. The current NGS still suffers from technical defects such as short long-read and the inability to completely analyze complex repetitive sequences [69]. In addition to the detection and analysis capabilities of sequencing technology itself, there are still problems other than technology that hinder its application in clinical practice. While NGS has become more affordable, its economic viability isn’t always guaranteed in all clinical and research settings [70]. The accumulation of genetic data has produced an increasing proportion of variants of uncertain significance (VUS), which overshadows clinicians’ decision-making. There is a need to classify the VUS found and review them regularly to ensure a beneficial impact on patient health guidance.
The clinical translation of genetic mechanisms for monogenic diseases has been accelerated by the dramatic impact of rare variants identified through linkage analysis. This is exemplified by ongoing clinical trials or FDA approval of gene therapies for rare diseases such as hemophilia, cystic fibrosis, and spinal muscular atrophy sclerosis, which hold significant promise for affected individuals and their families [71]. However, research into common complex diseases remains a formidable challenge. Genetic variation, mutations associated with disease, and genotype-phenotype associations can be studied with a large amount of genomic data [72, 73, 74, 75]. Genetic research on complex diseases has ushered in a new era in recent years following the discovery of two single nucleotide polymorphisms (SNPs) by the genome-wide association study (GWAS) associated with age-related macular degeneration [76]. Up to now, GWAS has successfully identified risk genes for a large number of diseases and traits, including Parkinson’s disease [77], obesity [78], autoimmune diseases [79], height [80], and others.
Large sample sizes and genomic data from large biobanks have been an important basis for supporting discoveries in GWAS. Among these biobanks, the UK Biobank plays a leading role, containing deep genetic and phenotypic data of 500,000 individuals [74]. Expanding the sample size of GWAS can facilitate the identification of additional risk loci and generate a more comprehensive list for discovering novel therapeutic targets. A genome-wide association analysis with a large sample size of 100,285 subjects revealed novel loci shared by lung function and obesity. However, the complexity of the linkage disequilibrium pattern and deficiencies in the imputation data impeded the identification of true causal variants [81]. Therefore, while we continue to explore novel associations, we must prioritize causal studies of established associations. These studies will undoubtedly provide in-depth insights into the biological mechanisms of disease and enhance clinical translation. In addition, it should be noted that susceptibility loci identified by GWAS do not necessarily correspond to causative genes. Functional genomics studies are required to accurately map these loci to specific variants and genes. The majority of association signals are located in regions of the genome that are not coding for proteins, which poses a challenge for deciphering the functional role of target genes. Moreover, due to the potent capacity of GWAS to detect subtle effects, the identified genes may not necessarily belong to the core pathways that regulate the phenotype [82]. Therefore, identifying causal relationships between variant genes and phenotypes is extremely complex and challenging. A growing number of functional datasets and genomics resources (e.g., ENCODE [83] and the GTEx [84], Epigenome RoadMap [85], FANTOM5 [86]) make it possible to combine GWAS findings with functional genomics data to advance variant functional annotations. What’s more, improved bioinformatics approaches such as computer annotation of gene regulatory regions [87, 88, 89], enrichment of causal variants in epigenome annotation [90, 91], colocalization of GWAS and expression quantitative trait locus (eQTL) signals [92, 93], gene expression prediction [94, 95], and fine mapping of causal variants [96, 97] have provided ideas for downstream analysis of GWAS. Several approaches are currently being studied to establish the connection between regulatory elements and their target genes, such as the 3C-based identification of chromatin loops and the CRISPR/Cas9 system [98, 99]. These innovative approaches with integration with GWAS will further increase the functional understanding of disease-related genes and facilitate biological discoveries translation. As an illustration, the investigators devised a method to decipher the molecular mechanism of disease-linked variants detected by GWAS. In this study, the Activity-by-Contact model was utilized to generate enhancer-gene profiles in cells and tissues, resulting in the identification of 5026 GWAS signals associated with 2249 genes. For inflammatory bowel disease (IBD), this map has revealed the mechanism of genomic regulation by demonstrating that enhancers harboring IBD risk variants alter PPIF expression, thereby altering immune cell mitochondrial function [100].
GWAS is gaining attention in the pharmaceutical industry. Researchers have discovered that there is a likelihood of at least twice as much in receiving approval for drugs supported by GWAS, especially in cases where causal genes have been identified [101, 102]. The prolonged duration and exorbitant cost of novel drug development and clinical trials have prompted researchers to shift their attention towards exploring alternative indications for existing drugs. GWAS has been used successfully to discover drug repurposing opportunities [103, 104]. Although GWAS do not typically provide direct information regarding causative genes or disease mechanisms, the identification of causal variants and biological pathways associated with diseases can be achieved through a combination of GWAS findings, subsequent bioinformatics approaches, and functional experiments. This strategy enables the matching of new drug-disease relationships using a catalog of known drug targets and the disease gene associations identified by GWAS. The high efficiency of drug repurposing satisfies the urgent demand for Corona Virus Disease 2019 (COVID-19) treatment. The TYK2 gene, which has been identified by GWAS as associated with host-driven inflammatory lung injury, is causally linked to critical illness COVID-19 and represents a promising therapeutic target for this disease [105]. TYK2 belongs to the Janus kinase (JAK) family, and baricitinib is a JAK inhibitor that specifically inhibits TYK2 expression [106]. Baricitinib was initially approved for the treatment of rheumatoid arthritis in 2018 [107]. After two phase III clinical trials that were randomized, double-blind, and placebo-controlled confirmed the significant reduction in mortality of patients hospitalized with COVID-19 by baricitinib, it has now been approved by the Food and Drug Administration (FDA) for treating severe cases of COVID-19 [108, 109, 110]. Furthermore, GWAS aids in the prediction of adverse drug reactions and enables the screening of safer drug targets, thereby mitigating foreseeable safety events and enhancing the efficiency of drug development [111].
GWAS is anticipated to offer valuable guidance for precise therapy by identifying genetic variants that are associated with drug response. The observation that patients with the same cancer type and receiving the same chemotherapeutic agent often exhibit varying degrees of response suggests that drug regimens based solely on disease phenotype are not only inefficient in terms of drug utilization but may also exacerbate patient outcomes due to delayed treatment. Accurately identifying patients who are sensitive to a specific chemotherapeutic agent is crucial for improving clinical outcomes. A genome-wide association study (GWAS) has identified two variants in ADCY1 that may affect the responsiveness of non-small cell lung cancer patients to platinum-based chemotherapy. In vitro cellular experiments have confirmed that high expression of ADCY1 is associated with increased sensitivity to cisplatin [112]. More evidence is required to elucidate the mechanisms underlying the modulation of chemotherapeutic drug sensitivity by the two SNPs and to validate the clinical utility of genotype-guided chemotherapeutic drug selection. Additionally, a GWAS study identified a CYP2C19 gene variant associated with poor clopidogrel efficacy [113]. Moreover, clinical trials designed based on this research finding have demonstrated the benefits of antiplatelet therapy regimens formulated according to the CYP2C19 genotype for patients undergoing percutaneous coronary intervention [114]. The Clinical Pharmacogenetics Implementation Consortium (CPIC) has developed a series of dosing guidelines that incorporate genetic variants associated with drug response in an evidence-based and rigorous manner, aimed at assisting clinicians in comprehending the significance of genetic test results and optimizing drug therapy [115]. These guidelines include the clopidogrel regimen based on the CYP2C19 genotype, as previously mentioned. Therefore, GWAS provides a novel approach to achieve precise drug selection in an agnostic manner and allows the integration of research findings into clinical practice.
The genetic variants identified through GWAS may be valuable in discerning individuals who have an elevated risk of specific diseases. According to a study, the LOXL1 gene contains two nonsynonymous SNPs that contribute 99% of the population-attributable risk for exfoliative glaucoma [116]. It is generally the case, however, that individual common variants exhibit low effect sizes and collectively they account for a moderate portion of heritability [117]. The persistent issue of “missing heritability” hinders the clinical applicability of GWAS in predicting diseases. Understanding the origins of deletion heritability is crucial for investigating genetic architecture and phenotypes of complex diseases. This can be attributed to several factors: (1) rare variants serve as the primary source of deletion heritability [118]; (2) neglecting the contribution of small genetic loci that are not captured by GWAS to the phenotype, and (3) intricate gene-environment interactions [119]. When considering associations beyond those of genome-wide significance, polygenic risk scores (PRS), derived from GWAS are anticipated to address the limitations of GWAS in predicting phenotypes through genetic effects.
PRS can improve disease risk prediction, guide treatment decisions, and even refine prognosis when we are not limited to variants of genome-wide significance. PRS quantifies the contribution of the genome to complex disease risk assessment by combining the cumulative effects of genome variants [120]. For coronary artery disease (CAD), PRS has the potential to identify a significantly larger number of individuals at equivalent or higher risk than rare single mutation carriers [121], suggesting that it may offer substantial benefits for high-risk individuals through the promotion of healthy lifestyles and treatment with statins [122, 123]. Damask et al. [124] found that high PRS in CAD was correlated to an elevated risk of recurrent major adverse cardiovascular events (MACE) following acute coronary syndrome. The reduction of MACE risk by alirocumab treatment was more pronounced in patients with higher PRS than those with lower PRS. However, there are challenges to incorporating PRS into medical decision-making in the real world. Complex diseases arise from the interplay of genetic and environmental factors. Therefore, to maximize predictive value, risk models should incorporate not only genetic but also environmental and other relevant factors that contribute to the phenotype. Modifiable environmental factors are precisely one breakthrough point in reducing disease susceptibility. For instance, environmental factors like activity levels, economic status, and dietary composition can modify the epistatic effects of pathogenic mutations on obesity as well as the methylation patterns of obesity genes [125]. Obesity, being a contributor to cardiovascular disease and diabetes, can be prevented or treated through lifestyle management interventions. Several scholars have suggested an analytical pipeline incorporating evaluations of SNP post-translational effects based on tissues and cell types, along with clinical phenotypes, into PRS calculations. This offers a practical method for enhancing the predictive accuracy of PRS [126]. Considering the current limited predictive diagnostic capacity of the PRS, it is not recommended to use it as a standalone diagnostic tool [127]. However, incorporating the PRS into an overall individual assessment strategy can provide a more comprehensive evaluation of disease risk. A successful example of considering multiple factors is the Canrisk tool for risk prediction of breast and ovarian cancer. This tool incorporates PRS, rare pathogenic variants in susceptibility genes, lifestyle factors, and clinical phenotypes to enhance the accuracy of disease prediction models [128]. In addition, whole genome sequencing (WGS) captures a greater amount of total genetic variation as well as rare variants compared to SNP genotyping [129]. Therefore, future larger scale GWAS based on the genomic data generated by WGS will significantly enhance the predictive power of PRS. Moreover, it is imperative to acknowledge that PRS tends to lose predictive accuracy when used across populations as a result of multiple factors, such as allele frequency differences and changes in polymorphism effect sizes [130]. Most PRS are currently calculated using information on individuals of European ancestry, with limited transferability across populations, which is likely to exacerbate health inequities in the genomic era [131, 132]. In a study, researchers integrated genomic data from multiple biobanks to investigate the PRS of lifespan biomarkers across diverse ethnic populations. The impact of body mass index (BMI) on lifespan reduction in the Japanese population differs from that observed in the European population, indicating a need for further exploration into this racial disparity regarding obesity-related health burdens. Therefore, enhancing the diversity of study populations is crucial for advancing our comprehension of human genetic variation [133, 134] and promoting healthcare equity.
Identifying the right therapeutic targets requires an understanding of multilevel pathogenic mechanisms, including gene regulation in noncoding regions [135], DNA methylation status [136], and RNA splicing [137]. Therefore, to achieve better clinical transformation of genomics, it is not enough to be limited to genomics itself, but also to combine other omics knowledge, such as transcriptomics, epigenomics, etc., and accurate disease subtypes are also individualized medically necessary. The development of electronic medical records, the popularization of wearable devices, and the explosive growth of high-throughput sequencing have generated an increasing amount of medical big data. Instead of being overwhelmed by piles of data, we should explore the mysteries behind a large amount of heterogeneous data to serve the cause of human health. Artificial intelligence (AI) and machine learning (ML) present the possibility to achieve this goal.
AI refers to computer output generated by imitating human behavior. ML is a subset of artificial intelligence. The essence of it lies in utilizing algorithms to learn from vast amounts of data, from which a model is constructed, and subsequently verifying and refining the model. A probability distribution can be used to estimate the most probable successful decision, therefore facilitating the identification and prediction of new data [138]. The strengths of machine learning lie in its versatility, extensibility, automatability, and capability to deal with complex dimensional datasets. ML models are primarily categorized into three types: supervised learning, unsupervised learning, and reinforcement learning. The process of supervised learning involves utilizing labeled data as the training target to facilitate algorithmic training and model fitting, ultimately enabling the model to accurately predict outcomes for new datasets [139]. For example, ML may predict the postoperative survival of cancer patients based on the patient’s genetic and phenotypic characteristics. On the contrary, unsupervised learning is used to process unlabeled data, by identifying patterns and regularities in data, clustering unlabeled data, discovering new associations, and data dimensionality reduction [140]. Reinforcement learning learns from trial and error and aims to reduce prediction errors, similar to conditioning mechanisms in psychology [141]. Notably, deep learning is gaining increasing popularity, using artificial neural networks to automatically extract data features, surpassing traditional machine learning applied to natural language processing [142] and computer vision [143].
Variants associated with diseases are typically located in non-coding genes, and it has been challenging to develop methods that can account for the impact of mutations in these genes. Machine learning provides novel insights for predicting the impact of noncoding mutations on disease. In a study, researchers utilized a deep learning model to analyze genome-wide data from 1790 families with autism spectrum disorder (ASD) and identified the contribution of novel noncoding variants on ASD pathogenesis by comparing anticipated transcriptional and post-transcriptional regulatory consequences of these variants in probands versus unaffected matched siblings [144]. In addition, Jaganathan et al. [145] successfully trained a deep learning model to identify non-coding mutations in rare genetic diseases by predicting the splicing of pre-mRNAs from genomic sequences. This shows that machine learning may help increase our understanding of the mechanisms of disease occurrence.
It has been shown that one-third of the genes in the human genome are co-regulated by microRNAs (miRNAs) [146]. MiRNAs are loaded into the Argonaute (AGO) family of proteins to generate miRNA-induced silencing complexes (miRISCs), which are base-paired with target mRNAs and regulate genes through mRNA cleavage or translational repression expression [146]. MiRNAs contribute to the pathogenesis of complex human diseases through post-transcriptional regulation of gene expression, thus serving as potential biomarkers for disease treatment [147]. The first step in clarifying miRNA pathogenesis is to accurately identify miRNA associations with specific diseases through experimental methods; however, this can be time-consuming and expensive given the numerous miRNA disease combinations. Computational modeling addresses this problem by identifying the most likely relevant miRNA-disease associations (MDA) for validation in biological experiments, thereby accelerating new MDA discovery. To fully utilize large-scale multi-source heterogeneous datasets and improve the accuracy of MDA prediction, researchers have started to focus on the potential of machine learning-based methods [148], including various classifiers (e.g., decision trees, support vector machines, plain Bayes, and neural networks) and matrix decomposition techniques, which simplify high-dimensional matrices into a few low-dimensional matrices. Computer-based miRNA analysis tools can predict the association of miRNAs with cellular functions and diseases and predict miRNA target genes and binding sites at the molecular level. For example, experimentally validated reports of miRNA-target interactions can be obtained using MiRTarBase [149]; miRDB, constructed by machine learning algorithms based on support vector machines, is an online resource for miRNA target prediction and functional annotation [150]. Numerous studies have previously described the available tools [151, 152]. In recent years, deep learning methods have emerged as promising new approaches to improve miRNA target group prediction. One study reported deep learning-based miRAW, which predicts miRNA targets by analyzing the entire miRNA transcripts and outperforms existing prediction methods in comparisons using independent datasets [153]. Machine learning-based research on the pathogenic mechanism of miRNA has been applied not only to cancer but also shown good prediction performance in other common diseases, such as coronary heart disease and dementia [154]. The investigators analyzed plasma extracellular vesicles (EVs) miRNA sequencing data from SCD patients and normal patients using bioinformatics online tools and statistical methods. The investigators used TargetScan and miRanda to identify EVs-miRNA targets, and functional enrichment analysis using Metascape to screen plasma EVsmiR-208b-3p and miR-143-3p as promising biomarkers for predicting SCD in patients with acute coronary syndrome (ACS) [155]. In a recently published article, the concept of theranomiRNAs, miRNAs for diagnostic and therapeutic purposes, was mentioned for the first time [156], which reaffirms the role of miRNAs in the diagnosis and treatment of diseases, and is a new direction in the translation of basic research into clinical practice, which is expected to play an influential role in precision medicine in the future.
63.4% of the untranslated region (UTR) variants in the ClinVar database are classified as “variants of uncertain significance” (VUS) [157]. In this context, in addition to functional annotation via databases, it is essential to assess the pathogenicity of genetic variants in non-coding regions. Constructing a pathogenicity prediction framework using non-coding variants in the human genome could provide a more comprehensive understanding of disease biology and reveal opportunities to develop new therapeutic targets. Deep learning has become a powerful tool for the functional study of non-coding mutations because genomics research is inherently characterized by features such as sequence local dependence and long-range correlation, and its large-scale and deep data characteristics fit well with the logic of the work of convolutional neural networks algorithms (CNNs). Currently, tools based on the CNN framework for prioritizing mutations in non-coding regions include DeepBind, DeepSEA, Basset, DanQ, Basenji, etc [158]. In 2020, the DeepFun model integrates data from ENCODE and Roadmap on top of existing CNN models to present a dense human-to-human epigenome mapping [159]. Successive upgrades of the model have contributed to increased accuracy in predicting the pathogenicity of non-coding region variants. The recently proposed “Junk” Annotation genome-wide Residual Variation Intolerance Score (JARVIS), which captures previously unavailable human pedigree constraint information, outperforms other human pedigree-specific scores [160]. JARVIS introduces a genome-wide residual variant intolerance score (gwRVIS) and incorporates primary genome sequence information and additional functional genome annotations to prioritize regions in the non-coding genome that may be more likely to be associated with clinically relevant effects when mutated.
Through the integration of genetic data with medical imaging, which is the largest source of data in the healthcare system, we can gain a more profound understanding of how genes influence organ morphology or function. Hallgrímsson et al. [161] used machine learning models to identify 3D facial images and thereby automatically diagnose the syndrome. The approach yielded a balanced accuracy rate of 73% when applied to a sample size of 7057 subjects. ML is capable of assisting in diagnosing diseases by utilizing multi-omics data. Khanna et al. [162] have developed a multivariate multimodal model that utilizes genomic data, imaging data, and biomarkers to predict the time of Alzheimer’s disease diagnosis. In this model, a Bayesian network model (an algorithm of machine learning) aims to reveal the interactions between genetic variants, biological pathways, and imaging-related features across biological scales.
Clinical transformation of genomics translates discoveries of chromosome/chromatin, DNA and RNA sequences, structure, and function into clinical applications for predicting, diagnosing, monitoring human disease-specific phenomena, while characterizing severity, duration, stage, and response to therapy. Progress in basic research discovery and translation has remained closely linked to clinical observations for most of its history, but there has been a serious mismatch between the phenomenal advances in basic biomedical sciences and the slow onset of translational medicine in the past 40 years. As a result, biomedical research in the third decade of the 21st century has been plagued by a disconnect between basic biomedical science and clinical practice. Although encouraging advances have been made, for example, in 2004, studies demonstrated the efficacy of gefitinib, an EGFR kinase inhibitor, in non-small cell lung cancer with specific gene mutations [163], and in 2011, ivacaftor, a cystic fibrosis transmembrane conductance regulator enhancer, was shown to be useful in the treatment of cystic fibrosis [164]. In complex polygenic disease disorders, the association of low density lipoprotein (LDL) cholesterol and triglyceride levels with CAD has been found to reflect a causal relationship through genomic data analysis methods, becoming a clinical marker and preventive therapeutic target for CAD [165]. Notably, the ability to effectively treat diseases as we now understand them is still limited: Of the approximately 8000 diseases that affect humans, fewer than 600 have any regulatory-approved treatments, most of which merely relieve symptoms. Effectively translating ideas from the laboratory into interventions for clinical practice currently takes more than 20 years and has a success rate of less than 1 percent [166]. Therefore, the use of data analysis methods in the big data era and the vast amount of genomics data from the level of the molecular mechanisms of disease will hopefully facilitate the efficient translation of genomics data.
As a method of medical practice that is data-driven, precision medicine is a field that takes into account relevant medical, genetic, behavioral, and environmental information about individuals to accurately predict disease risk in healthy populations while providing targeted treatment options for patients. Genomics big data is an important tool to achieve precision medicine. Variants in the human genome such as SNPs, insertions and deletions, structural variants, and copy number variants play an important role in disease onset, progression, and performance status. Therefore, the clinical translation of genomics big data reflects the goal of precision medicine, which usually relies on genomics analysis tools to mine the information behind the data that is closely related to disease diagnosis and treatment. Genomics technology and science are closely linked to the extent to which genomic information is used in medicine. Advances in high-throughput sequencing have generated vast amounts of genomic data for scientists to study gene structure and function variations. It can also serve as a medium for genomic insights to penetrate the clinic and become an essential tool for personalized therapy. A genome-wide association study uses extensive genomic data to investigate genotype-phenotype associations, and the findings may provide powerful support for drug discovery. Polygenic risk scores, developed based on GWAS, offer new ideas for disease diagnosis and personalized therapy. In the face of increasing big data, there is a need for high-performance data analysis methods to integrate a great deal of multidimensional data, and to discover patterns across different types of data to guide the improvement of high medical standards. Artificial intelligence and machine learning methods are helping data scientists to overcome this challenge.
Advances in genomic science technology, and research have enabled the translation of genomics from basic to clinical settings. The interplay of sequencing technologies, functional genomics, and other emerging technologies (e.g., artificial intelligence) has provided new perspectives and new understandings for the exploration of disease pathogenesis. Currently, some genomics data have been successfully applied in various aspects of clinical screening, diagnosis, and treatment to provide decision support for personalized healthcare issues. Therefore, there is a growing expectation to integrate genomics into mainstream medicine. Nevertheless, we need to clearly understand the challenges facing broader clinical integration of genomics. Genomic data of individuals hide private information related to life and health, which becomes a limiting factor for data sharing. A global medical data-sharing mechanism should be established under the premise of fully safeguarding data privacy and security, developing data science technology with data protection capability, and promoting the maximum utilization of medical data value. Genomics research results should be validated by clinical trials to confirm their true clinical utility. For example, attention should be paid to whether technologies with disease-predictive capabilities actually promote healthier changes in patient behavior or just increase excessive anxiety. Finally, improving the genomics literacy of health industry practitioners and promoting the mass dissemination and popularization of genomics knowledge is crucial to the industrial implementation of genomics data in the medical field. Overall, medical practice should always be patient-centred, and genomic-level data information can help researchers to be more focused, rigorous, and scientific in their experimental design. In the process of clinical implementation, researchers should pay attention to the feasibility (especially the cost and invasive operation that patients are concerned about), effectiveness, and universality (to minimise the inequality of medical treatment among races) of the research results. The collaboration of experts across diverse industries is imperative in fostering the harmonious advancement of fundamental biological research and translational medicine practice, thereby making significant contributions to the pursuit of human well-being.
YZ and JY contributed to the formal analysis, methodology, investigation, and writing the original draft. XX participated in conceptualization, and writing review & editing. FJ and CW designed the work, reviewed it critically, contributed to supervision and funding acquisition. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was funded by the National Natural Science Foundation of China (NO.82172539) and Jiangsu Province Hospital (the First Affiliated Hospital with Nanjing Medical University) Clinical Capacity Enhancement Project (JSPH-MC-2022-19).
The authors declare no conflict of interest. Given his/her role as Guest Editor, Feng Jiang had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Graham Pawelec.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.