



 , Youfang Chen 1,2,*
, Youfang Chen 1,2,*1 Department of Thoracic Oncology, Sun Yat-Sen University Cancer Center, 510060 Gunagzhou, Guangdong, China
2 Sun Yat-Sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, 510060 Guangzhou, Guangdong, China
†These authors contributed equally.
Abstract
The occurrence and development of esophageal cancer involve multiple genetic abnormalities that contribute to the malignant transformation of esophageal epithelial cells, followed by invasion and metastasis, leading to a poor outcome. Esophageal squamous cell carcinoma (ESCC) is the predominant histological subtype of esophageal malignancy in East Asia, with approximately half of newly diagnosed ESCC cases occurring in China. The TP53 tumor suppressor gene mutation is one of the most common mutations in ESCC. TP53 mutations are observed even in the early phases of esophageal carcinogenesis. Normal functions of the p53 network are lost in cells of ESCC patients who harbor the mutant TP53 gene, inducing tumor development, radiation resistance, chemotherapy resistance, and immune suppression, promoting progression and metastasis, thereby resulting in an overall poor prognosis. Although clinical trials of several pharmacological compounds targeting mutational TP53 have been explored, novel approaches are still urgently required to improve the observed dismal survival. A better understanding of the role of the mutant TP53 gene in human ESCC might lead to the discovery of innovative targeted therapies to treat this malignancy.
Keywords
- TP53 mutations
- esophageal squamous cell carcinoma
- targeted therapies
Esophageal squamous cell carcinoma (ESCC) is the primary pathological type of esophageal malignant neoplasms, accounting for approximately 84% of esophageal cancer cases; however, the 5-year overall survival rate is only ~20% [1, 2]. ESCC arises from the epithelial cells that line the esophagus and is primarily found in the upper two-thirds of the esophagus [3]. ESCC represents one of the most aggressive digestive neoplasms globally, characterized by a high mortality rate and geographical variations in incidence: East Asia has the worst epidemic incidence of ESCC and fatality, which burdens healthcare systems tremendously [2, 4]. Epidemiological studies have suggested that male sex, tobacco intake, alcohol consumption, hot drinks, and nutrient deficiency might contribute to ESCC [5, 6, 7, 8, 9]. Risk factors for ESCC also vary by region, and the risk of developing ESCC in China has been associated with factors such as family history and dietary habits (e.g., betelnut chewing and hot drinks). However, alcohol consumption and tobacco intake explain the overwhelming majority of ESCC cases in Western countries, but represent secondary factors in the Chinese population [10]. Thus, the occurrence and progression of ESCC appear to be related to various factors, and these risk factors induce gene mutations that encode abnormal p53 proteins that can be recognized by the immune system [11]. To improve the management of this fatal disease, the genetic anomalies involved in the carcinogenesis of ESCC, as well as those involved in its malignant transformation and development, need to be identified and investigated.
Currently, the mainstream management of ESCC comprises a multidisciplinary approach, in which the treatment strategy is based on tumor-node-metastasis (TNM) staging, the position of the tumor, and its performance, as well as the nutritional status of the diseased individual. Once ESCC is detected, radical resection is typically recommended as it is the most effective therapeutic approach for early staged patients [10]. The CROSS trial established neoadjuvant concurrent chemotherapy and radiotherapy followed by esophagectomy as the standard treatment strategy for locally advanced esophageal cancer (EC) [12, 13, 14]. As for ESCC, this paradigm of management has been further strengthened by the results of the recent NEOCRTEC-5010 randomized clinical trial, which indicated that neoadjuvant concurrent chemoradiotherapy (NCRT) followed by esophagectomy is a safe and effective approach for patients with locally advanced ESCC and has beneficial treatment effects, with acceptable postoperative complications [15, 16]. In recent years, the use of immunotherapy in EC treatment has evolved rapidly. The KEYNOTE-590 trial suggested that pembrolizumab plus chemotherapy improved progression-free survival and overall survival compared to placebo plus chemotherapy in patients with ESCC [17, 18]. Based on these results, immunotherapy plus chemotherapy has become the first-line treatment for advanced ESCC. Recently, the advantage of radiotherapy plus immune checkpoint inhibitor treatment was demonstrated [19], and the CHECKMATE-577 trial indicated the superior disease-free survival of patients with esophageal cancer achieved by administration of adjuvant immunotherapy (programmed cell death-1 (PD-1) inhibition) after multimodality therapy [20]. Nevertheless, despite the considerable improvement in surgical conditions and skills, perioperative care, and the application of multimodal integrated treatment, the outcome of advanced ESCC remains dissatisfying. Delayed diagnosis is the most dominant risk factor for the subsequent clinical outcome of ESCC. Therefore, immediate diagnosis and prompt intervention are vital to prevent further injury and ensuing clinical development. Therefore, the identification of new early diagnostic approaches and novel therapeutic targets to improve prognosis is necessary.
Over the last two decades since the first sequencing of the human genome, clinical genomics has undergone a rejuvenation with the development of next generation sequencing (NGS) technologies, which have enabled the detection of gene mutations in human ESCC samples, among which the TP53 tumor suppressor gene is the most commonly mutated gene identified in ESCC [21, 22, 23, 24, 25, 26]. TP53 gene mutations play an essential role in ESCC, and signaling pathways linked with p53 offer novel opportunities for tumor detection and targeted cancer treatment. The evolutionarily conserved TP53 tumor suppressor gene, generally referred to as the “Guardian of the Genome”, plays a crucial role in the complex cellular stress response network to protect the DNA integrity of the cell; TP53 can also mediate neoplasm suppression through a series of responses to internal and/or external environmental stimulations, leading to either the maintenance of cellular homeostasis or cellular death [27, 28]. The p53 pathway is activated via several stress signals, including oncogene activation, DNA damage, and replication stress [29], causing increased levels and intensified activation of the p53 transcription factors, which further control the activation of hundreds of target genes [30, 31]. The expression products of these genes are involved in a various of downstream cellular processes that recover from the damage caused by the stimuli or destroy the cells affected by irretrievable injury [28].
The title of “Guardian of the Genome” is insufficient to explain the complex biological abilities of p53, which performs miscellaneous functions in individual development and aging [32], and plays important roles in cell differentiation, cell cycle arrest, DNA repair, senescence, and apoptosis processes [33]. P53 also participates in many other pathways, such as cell metabolism, autophagy, ferroptosis, and pathways that involve the production of reactive oxygen species, by exerting its biological functions directly or via interaction with other proteins [33].
Somatic TP53 mutations in human malignant tumors were first described by Baker et al. [34] in colorectal cancer in 1989. Since then, research on TP53 mutations in human cancers has mushroomed over the intervening decades, and many studies have found that biallelic mutations in TP53 were the most frequent mutational signature of human malignancies, including ESCC [11, 24, 35]. In contrast to other neoplasm suppressive genes, TP53 is often found to undergo missense mutations, in which a single nucleotide is substituted by another [36, 37]. TP53 mutations can result in dissimilar consequences, from the loss-of-function (LOF) necessary for tumor suppression to the dominant-negative (DN) and even the gain-of-function (GOF) necessary for tumor growth and treatment resistance [35, 38]. More specifically, LOF mutations in cancers could derail the ability of wild-type TP53 to maintain genomic stability. In contrast, some GOF mutations can induce oncogenic activity that promotes carcinogenesis, and some specific mutations even enable cancer cells to acquire the capacity to maintain proliferation, become more invasive, and allow metastasis and resistance to therapy.
Understanding how the TP53 mutations affect the progression along the pathological continuum from normal esophageal epithelia and precancerous lesions, to malignant neoplasms will help us to explore new strategies to prevent or eliminate ESCC at its earliest stages of development. Here we give an overview of TP53 mutations in ESCC, discuss the significance of TP53 mutations in the malignant transformation of human esophageal epithelial cells and the development of ESCC, review how mutational TP53 could be used to help the management of ESCC in various aspects of clinical practice, and envisage how mutational TP53 could be exploited as a potential therapeutic target in ESCC treatment.
The TP53 gene was identified in 1979 as a 53 kDa protein conjugated to
simian virus (SV40) large T-antigen. The introduction of SV40 led to the
malignant transformation of cells [39, 40]. The anthropic p53 protein is encoded
by an evolutionarily conserved gene mapped on human chromosome 17p13.1 [41]. The
TP53 gene contains 11 exons, including a first non-coding exon (exon 1)
and 10 exons encoding the standard or full-length p53 protein monomer which
comprises 393 amino acids (FLp53, also termed p53
 Fig. 1.
Fig. 1.The brief structure and partial phosphorylation sites of p53 protein. S, serine; T, threonine; CK, casein kinase; ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related protein; DNA-PK, DNA-dependent protein kinase; CHK, checkpoint kinase; CAK, cyclin dependent kinase-activating kinase; CDK, cyclin dependent kinase; HIPK2, homeodomain-interacting protein kinase 2; DYRK2, dual-specificity tyrosine-phosphorylation kinase; PKC, protein kinase C; TAF1, TATA-box binding protein associated factor 1; JNK, c-Jun NH2-terminal kinase; ERK, extracellular signal-regulated kinase; CSN, COP9 signalosome associated kinase complex; PAK, protein activated kinase; LRRK2, leucine-rich repeat kinase 2; PKR, protein kinase R; FACT, facilitates chromatin transcription; *, mutational hot spots in human carcinomas.
The p53 protein is a DNA sequence-specific transcription factor, whose structure consists of five main regions: The transactivation domain (TAD, subdivided into TAD1 and TAD2) at the N-terminus, a proline-rich domain (PRD), a DNA binding domain (DBD), a tetramerization domain (TD), and a regulatory domain (basic domain, BD) at the C-terminus (Fig. 1) [44]. Upon sensing stress, such as oncogene activation, p53 binds to different messengers via its TADs and regulates downstream bioreactions, thereby mediating suppression of tumorigenesis. Each part of TAD allows the binding of p53 to a specific messenger induced by a particular stress, thus producing the corresponding biological effect [45]. The TAD region also binds the murine double minute 2 (MDM2) protein, decreasing p53 activity [46]. The PRD is indispensable for wild-type p53 stability, since disruption of the PRD results in p53 nuclear export, making it prone to MDM2-mediated degradation and ubiquitination [47]. The DBD enables p53 to take its responsibility as a transcription factor by specifically recognizing and binding target DNA sequences, termed p53 response elements (REs) [48]. The human canonical p53 protein is a homotetramer assembled by the association of four monomers via the TD domain. The TD helps p53 proteins to oligomerize as a tetramer, which is a prerequisite for activating the p53 to bind to DNA and interact with other proteins [49, 50, 51]. Moreover, oligomerization is essential for p53 to be efficiently ubiquitinated by MDM2 [52]. The C-terminus regulatory domain inhibits the combination of the DBD and p53 REs, and the aforementioned inhibitory effect will persist until the C-terminus regulatory domain is capitulated by post-translational modification, including phosphorylation and acetylation, which are usually mediated by a variety of stress signals [51, 53, 54, 55].
P53 inhibits neoplasm development by regulating the expression of a diverse range of target genes involved in various downstream cellular processes, such as cell cycle arrest, senescence, DNA repair, cell death, adhesion, and migration (Fig. 2) [30, 31, 33]. The p53 and MDM2 proteins form a central hub in which stress is input via MDM2 and responds to stimuli via p53 by informing and altering many associated pathways and related functions in the cell [28]. For instance, apoptosis can be activated through the communication of p53 with anti-apoptotic proteins localized within the mitochondria [56]. The MDM2–p53 hub has one of the highest numbers of communications with other signaling pathways in the cell, which might explain why TP53 is the most frequently altered gene in human malignant tumors [11, 24, 28, 35]. Moreover, improving our understanding of the molecular mechanisms underlying TP53 mutations in ESCC might help us develop effective methods to manage this disease in the future.
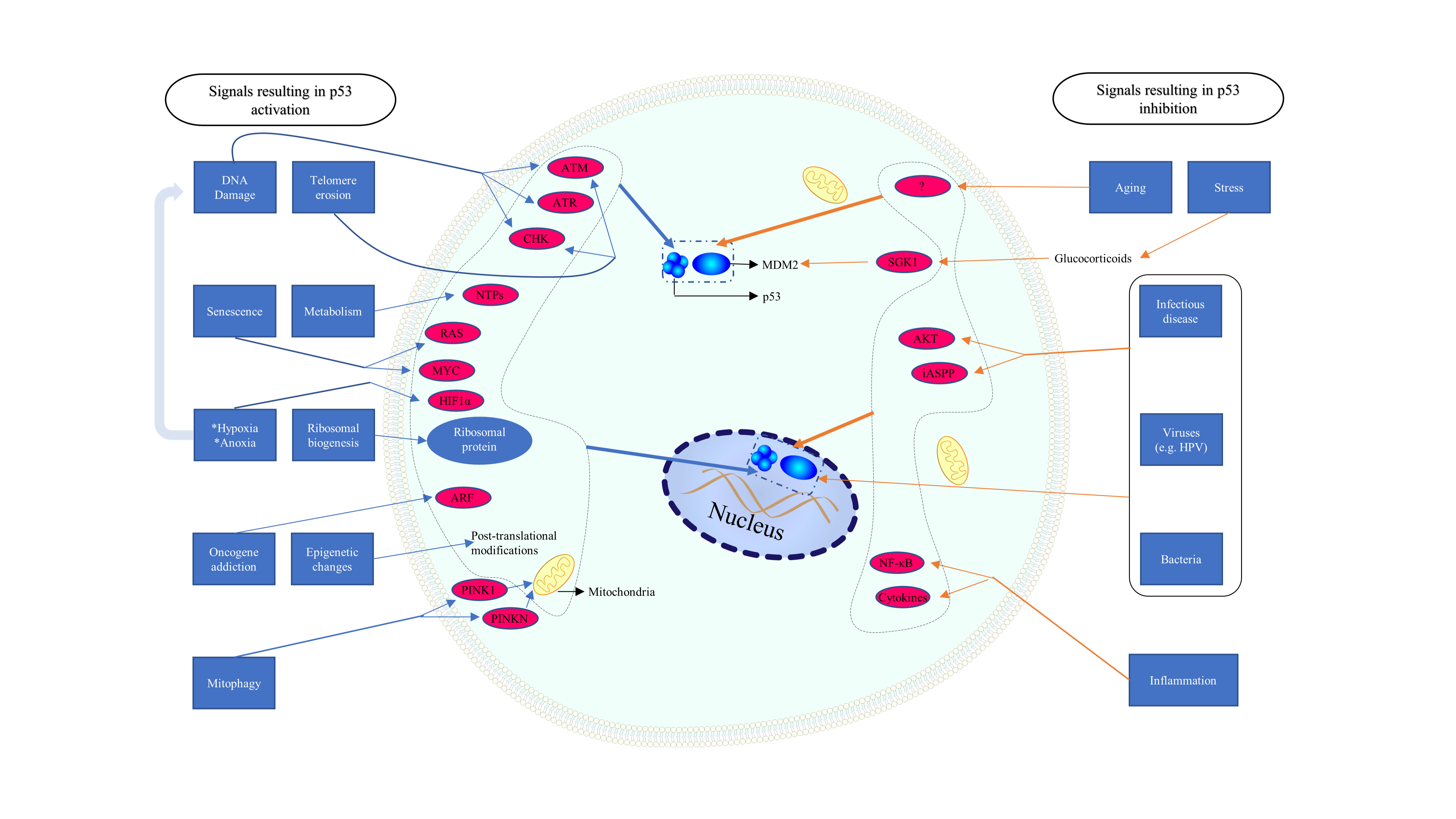 Fig. 2.
Fig. 2.Different signals inducing specific p53 reactions. ATM, ataxia telangiectasia mutated; ATR, ataxia telangiectasia and Rad3 related protein; CHK, checkpoint kinase; NTP, nucleoside triphosphate; PINK 1, PTEN induced putative kinase 1; PRKN, Parkin; ARF, alternate reading frame; HIF, hypoxia-inducible factor; SGK, serum and glucocorticoid regulated kinase; ASPP, apoptosis stimulating proteins of p53.
According to their origin, tumor-causing genetic mutations can be roughly categorized into either somatic or germline mutations. Acquired somatic mutations, together with innate germline variations, are important factors in cancer evolution.
Germline mutations in the TP53 gene have been associated with Li-Fraumeni syndrome (LFS), an inherited autosomal dominant cancer-susceptibility disorder [57, 58, 59]. About half of patients with germline TP53 mutations will develop cancer by the age of 30 years, with an 80–90% lifetime risk of cancer [60, 61]. The literature has rarely reported that a patient with LFS suffered from esophageal carcinoma, yet esophageal cancer occurs much earlier than expected in the general population [61, 62]. In 2020, a study by Katona et al. [63] analyzed the R20 International Agency for Research on Cancer (IARC) database, which contained 3043 individuals from 1243 families with germline TP53 mutations and suggested that 0.5% (15/3043) of individuals and 1.2% (15/1243) of families had a history of esophageal cancer. Remarkably, 29% of individuals with upper gastrointestinal cancers in this LFS cohort occurred before age 30. They also shared their single center cohort of 111 individuals with germline TP53 variants from 84 families, in which a history of EC was documented for 4.5% (5/111) of the individuals and 6.0% (5/84) of the families in the cohort, with no cancers reported before age 30 [63].
The mechanism of somatic mutations can be distinguished from innate germline mutations. In response to multiple intrinsic and extrinsic factors, somatic mutations start from the first division after conception and progressively accumulate as we age [64, 65, 66, 67, 68, 69, 70, 71]. The universal rule that the accumulation of somatic mutations induced by various factors is inevitable over time and applies equally in esophageal tissues, in which mutational clones colonize the majority of normal epithelium by old age [72, 73]. One of the genes most commonly altered in the aging human esophagus is TP53, which is also recurrently mutated in precancerous lesions and esophageal malignancies [22, 23, 24, 25, 68, 69, 72, 73].
Since TP53 mutations were first described in human carcinoma in 1989 [34], numerous studies have been conducted to detect P53 alterations in cancers, including ESCC. The diversity and complexity of mutational patterns and the many mutational sites make it very complicated to elucidate TP53 mutations. Studies have reported that a higher frequency of mutations occurs in exons 5–8 encoding the DBD, or more precisely, codons 175, 248, 273, 282, 245, and 249 are the top mutant sites and are referred to as hot spots, of which the most common forms are missense point mutations occurring at methylated CpG sites that encode arginine [34, 74].
Direct sequencing involving the entire gene can check unknown gene alterations and is considered the ‘gold standard’ for detecting TP53 gene mutations. Alternative methods that can reflect the presence of target mutations without sequencing are also considered valid [75]. Several studies of TP53 mutations in the human esophagus utilized immunohistochemistry, which can be easily and routinely conducted in laboratories. The wild-type p53 protein has a very short half-life, whereas in cells with p53 mutated at the DBD, the degradation period is prolonged, leading to an accumulation of p53 and positive results in p53 immunohistochemical analysis [76, 77]. Research has suggested a high correspondence between the results of genetic analysis and positive immunohistochemical staining [78]. Moreover, a series of studies reported that p53 protein expression and gene mutation occurred in multiple stages of ESCC, and p53 accumulation correlated positively with cell proliferation during esophageal carcinogenesis [79, 80, 81]. The p53 protein accumulates during ESCC malignant transformation and progression in both Asian and American populations: in 3–6% of cases with normal esophagus or esophagitis, in 10–40% of those with low and/or middle grade dysplasia, in 40–75% of cases with high grade dysplasia and/or carcinoma in situ, and in 50–90% of esophageal carcinomas [80, 81, 82, 83, 84], suggesting that P53 mutations might have occurred before the generation of the tumor and are more likely to occur in later stages of the tumor.
Evaluating TP53 mutations by detecting abnormal p53 protein accumulation is feasible but imperfect because the p53 protein can also accumulate when there is no genetic alteration [78], and about 10% of TP53 mutations are LOF mutations, some of which produce no protein [74]. High-throughput DNA next-generation sequencing (NGS) has enabled the comprehensive exploration of somatic mutations in large numbers of tumor samples. NGS-related methods applied in ESCC usually include whole-exome sequencing (WES) and/or whole-genome sequencing (WGS). Compared with WES, which only considers somatic variations in the protein-coding regions, WGS allows further evaluation of the effects of the non-coding variations on ESCC development. In the last two decades, many studies derived algorithms to detect mutational signatures from genome or exome sequences of ESCC, which demonstrated that 60–96% of patients with ESCC harbor a mutated TP53 gene (Table 1) [21, 22, 23, 24, 85, 86, 87, 88, 89, 90, 91, 92]. TP53 mutations were also identified in precursor lesions of ESCC via NGS. Chen et al. [93] reported that mutations in TP53 were the most recurrent events, being present in dysplasia samples among 95.6% of patients and in ESCCs among 97.8% of patients. For the healthy esophagus, the results were consistent with those of immunohistochemistry tests. Martincorena et al. [72] showed that 5 to 10% of the epithelium carried a TP53 mutation among the nine donors assessed, and the frequency of mutation appeared to increase with age, with the oldest donor having TP53 mutations in 20–35% of their cells. Similarly, another study found that TP53 was mutated in 30.6% of the physiologically normal esophageal epithelia [73].
| Author/year/ref. | Cases | Country | Method of detection | Frequency of mutations | Remarks |
| Agrawal (2012) [21] | 12 | USA | WES | 92% | 1. The incidence of TP53 mutations is lower in adenocarcinomas (73%). |
| 2. The most common substitutions were both C:G | |||||
| Song (2014) [24] | 158 | China | 17 WGS | 83% | 1. Head and neck squamous cell carcinoma and ESCC share some common pathogenic mechanisms. |
| 71 WES | 2. ESCC development is associated with alcohol drinking. | ||||
| 70 a-CGH | |||||
| Lin (2014) [85] | 184 | China | WES | 60% | 1. Inhibition of XPO1 with either shRNA or KPT-330 altered the expression of p53. |
| 2. Recurrent candidate druggable targets including p53. | |||||
| Gao (2014) [22] | 118 | China | WES | 93% | 1. The most common type of mutation in the exonic region was C |
| 2. No significant differences were found in either the rate or composition of mutations between smokers and non-smokers. | |||||
| Zhang (2015) [86] | 104 | China | 14 WGS | 88% | 1. A high frequency of C |
| 90 WES | 2. Genetic diversity was found between northern and southern China. | ||||
| Qin (2016) [87] | 67 | China | 10 WGS | 67% | 1. 78% of samples had genes altered in the p53 pathway. |
| 57 WES | 2. The mutant formations were nonsynonymous mutations or UTR/splicing-site mutations. | ||||
| Sawada (2016) [88] | 144 | Japan | WES | 93% | 1. A high proportion of mutations were C to T substitutions in CpG dinucleotides. |
| 2. Biallelic inactivation of TP53 was observed in most of the samples. | |||||
| Chang (2017) [23] | 94 | China | WGS | 78% | 1. The numbers of SNVs were positively associated with the mutational status of p53. |
| 2. TP53 mutations were significantly higher in drinkers. | |||||
| The Cancer Genome Atlas Research Network (2017) [89] | 90 | Worldwide | WES | 91% | 1. ESCC mutant features showed trends for geographical associations. |
| 2. ESCCs showed enrichment of C | |||||
| Guo (2018) [90] | 302 | China | WES | 61% | 1. TP53 status correlated significantly with high frequency of C |
| 2. TP53 status is associated with the activity of the “NpCpG” signature (characterized by C | |||||
| Zhang (2020) [91] | 225 | China | WES | 96% | Tumor mutational burden correlated significantly with drinking. |
| Cui (2020) [92] | 508 | China | WGS | 75% | 1. The largest dataset of genomic profiling of ESCC. |
| 2. Alcohol and tobacco were significantly interrelated. |
a-CGH, array comparative genomic hybridization analysis; UTR, untranslated region.
Technological advances bring unexpected discoveries and convenience; recent advances in clinical liquid biopsy, such as circulating cell-free DNA (cfDNA) research, enable repeatable, minimally invasive tumor gene diagnosis and monitoring. The mutational p53 allele could be found in the plasma of patients with ESCC via cfDNA detection with NGS. Haji and co-workers explored the variation of perioperative serum p53 concentration, indicating that cfDNA detection is beneficial for patients with ESCC [94]. Many reports agree that blood specimens are a reliable method for detecting targeted ESCC tumor TP53 mutations in cfDNA [95, 96, 97].
As mentioned earlier, cigarette smoking and alcohol drinking are both considered important risk factors for ESCC [5, 6, 7, 8, 9]. The International Agency for Research on Cancer (IARC) has identified more than 60 carcinogens in cigarette smoke, among which polycyclic aromatic hydrocarbons (PAHs) and N-nitrosamines are the most insidious carcinogens in humans [98, 99]. One of the major adducts of certain PAHs, such as benzo[a]pyrene, is the induction of a G-to-T transversion [100]. Indeed, about half of TP53 gene mutations in patients with ESCC are predominantly the transversion of G to T [101]. In the liver of drinkers, ethanol is oxidized by alcohol dehydrogenase to produce acetaldehyde, which acts as a carcinogen and induces gene mutations in various animals [102, 103]. A previous study proved that the mutational spectrum induced by acetaldehyde in the HPRT gene (encoding hypoxanthine-guanine phosphoribosyltransferase) in human T lymphocytes resembles that in the TP53 gene of esophageal cancers [104]. The mutant p53 was intimately related to the development of ESCC as well as muti-centric carcinogenesis of the esophagus, especially when combined with cigarette smoking and alcohol drinking [82, 105, 106]. A study demonstrated that the p53 protein was expressed in 55.1% of 89 ESCC cases examined by immunohistochemistry and further showed that ESCC in either smokers or alcohol users was 4.67–5.83 times more likely to express the p53 protein, whereas the likelihood of p53 expression in patients who use both tobacco and alcohol was more than 14.0 times higher than those who did not [107]. Another immunohistochemical study including 126 surgically treated individuals with primary ESCC has also proved the positive correlation between the expression of the p53 protein and smoking and drinking habits [108]. Moreover, the combination of alcohol and tobacco consumption was associated with a high frequency of p53 protein accumulation in ESCC [105, 108], as well as in precancerous dysplastic lesions [105]. Chang et al. [23] used WGS technology to analyze genomic alterations in ESCC and found that TP53 was mutated in 78% (73/94) of samples and the rates of TP53 mutations were significantly higher in ESCC in drinkers than that in non-drinkers, whereas the difference in smokers compared with that in non-smokers was not significant.
The prognostic value of mutational p53 in patients with esophageal cancer is still controversial; such flexibility in clinical outcomes may be correlated with the diversity among p53 mutants. Experts have different opinions about what effect mutant p53 might have on the prognosis of ESCC.
Several studies have suggested that the prognosis of ESCC did not correlate with the expression of the p53 protein, and those studies used different methods for mutational p53 examination [91, 97, 109]. There were also studies that indicated that patients who harbored mutational p53 had better outcomes. A particular study showed that patients with TP53 mutations respond better to treatment than to those without [110]. Gibson et al. [111] found patients with p53 mutations had longer overall survival after surgery than those without the mutations in a population with locally advanced ESCC. The TP53 mutation and abnormal accumulation of p53 proteins are early events in carcinogenesis rather than related to tumor progression and metastasis, which might be a reasonable explanation. On the other hand, synonymous mutations cannot induce protein changes that affect the phenotype. Moreover, some GOF mutations can confer proteins with the ability to fight carcinoma; for example, research indicated that the changes in amino acids induced by the polymorphism of the codon 72 in exon 4 of the TP53 gene could enhance the apoptosis-promoting ability of the p53 protein [112].
When GOF mutations confer p53 protein oncogenic activities that promote carcinogenesis and maintain proliferation, the cancer cell would become more invasive and allow metastasis and resistance to therapy. Those patients who harbor “bad” GOF mutations correspondingly have a worse prognosis.
The pathogenesis of ESCC is a multistep progressive process, which involves dysplasia of the normal epithelium, carcinoma in situ, and ultimately invasive ESCC. Dysplasia is characterized by nuclear atypia (enlargement, pleomorphism, hyperchromasia), loss of normal cellular polarity, and abnormal maturation but does not invade through the basement membrane. Numerous studies have demonstrated that squamous dysplasia is a precancerous lesion of ESCC [80, 113, 114]: 30% of the dysplasia cases ultimately progressed to ESCC in 2.5–6.5 years [114]; however, the mechanism of malignant transformation of esophageal dysplasia remains obscure. The recently reported mutation pattern of the TP53 gene in ESCC [93] agrees with Knudson’s two-hit hypothesis, which states that both alleles of most tumor suppressor genes need to be inactivated to promote tumorigenesis; the classical “two-hit” doctrine explains the earlier age of tumor onset in patients with LFS to an extent.
The described hot-spot mutations occur at methylated CpG sites in the TP53 gene, which encode arginine residues that contact the DNA. The normal arginine residues are indispensable for the p53 protein to recognize and bind specific target DNA sequences (response elements, REs), which is a prerequisite for the tumor-suppressive function of p53 [115]. LOF TP53 mutations might lose the ability to generate the normal p53 protein (through various mechanisms: nonsense or frameshift mutations, deletions) or reduce the ability of the mutant protein to bind the REs that mediate the transcription of tp53-regulated genes. LOF TP53 mutations thereby deprive p53 of all or most of its cellular function in response to stress and frequently also show a dominant negative effect (DNE) over wild-type p53 by forming mixed tetramers, and these factors eventually combine to disturb the normal function of the remaining wild-type p53. A study has proven that different TP53 mutants bind to REs with different efficiencies, between 0 and 75% of the wild-type [116].
Adding a cDNA with various TP53 missense mutations to cells with TP53 genes deleted (null-p53 cells) conferred a variety of new properties (phenotypes) on those null-p53 cells, which indicated that TP53 GOF mutations could provide the mutant protein with new abilities. These newly acquired abilities cause cells to lose contact inhibition, promote tumor growth and metastasis, alter metabolism, accelerate cytoskeletal alterations, facilitate wound healing, change transcriptional patterns, and/or induce treatment resistance [117]. It is easy to imagine that TP53 GOF mutations are double-edged swords for patients with cancer, with different mutations leading to various identical or opposite phenotypes, resulting in acceleration or suppression of tumor development. The altered p53 protein can also regulate cell migration, metastasis, angiogenesis, and chemoresistance via interactions with other signaling pathways, since the p53 pathway is central to integrating diverse cellular stress signals [118, 119, 120, 121].
TP53 mutations were the most common mutational signature of ESCC; further discussion is required in order to reveal the underlying mechanisms. We list two facts that provide mechanistic clues: Firstly, normal esophageal cells are proven to accumulate mutations as people age [72, 73]. Secondly, driven by various determinants, including tobacco and alcohol exposure, TP53 alterations appear at a very early stage and occur incrementally throughout the course of ESCC [80, 81, 82, 83, 84].
Some mutations in normal esophageal tissue are consistent with those in ESCC, but not all develop cancer. Most do not confer an intrinsic growth advantage and are defined as “passenger mutations” whereas a smaller number of them, known as “driver mutations” provide a growth advantage and are therefore selected during tumor evolution. Notably, even though dysplasia samples and ESCCs share TP53 gene mutations and protein accumulations, the heterogeneity between dysplasia and ESCCs in each given individual is extensive, and the evolutionary trajectories of precursor and tumors are distinct [93], which suggests that genetic mutations might precede the arrival of tumors, with the pre-existing diversified mutation background conferring the ability to evade selection pressure, such as immune surveillance on the abnormal cells, when carcinogenesis begins. Despite being phenotypically normal, cells can accrue mutations that could lead to clonal outgrowth from infancy. Clones increase their range and quantity as time passes, and the normal-appearing tissue might thus comprise a battleground for “silently” competing clones that eventually remodel almost the entire esophageal epithelium with thousands of sizable driver-mutated clones. Furthermore, risk factors, such as smoking and alcohol, substantially accelerate this remodeling process and facilitate the selection of mutations, thus resulting in different phenotypes, such as ESCC [73, 122]. Cells with specific GOF mutations correspond to particular optimal growth conditions. Thus, various internal and external environmental factors shape the observed mutational signature and genomic landscape of normal, precancerous, and malignant esophageal tissues (Fig. 3). For example, some TP53 GOF mutations confer a set of new progression-friendly functions to tumor cells, therefore are preferentially selected by cancers than loss-of-function mutations or “low-frequency mutations” [74].
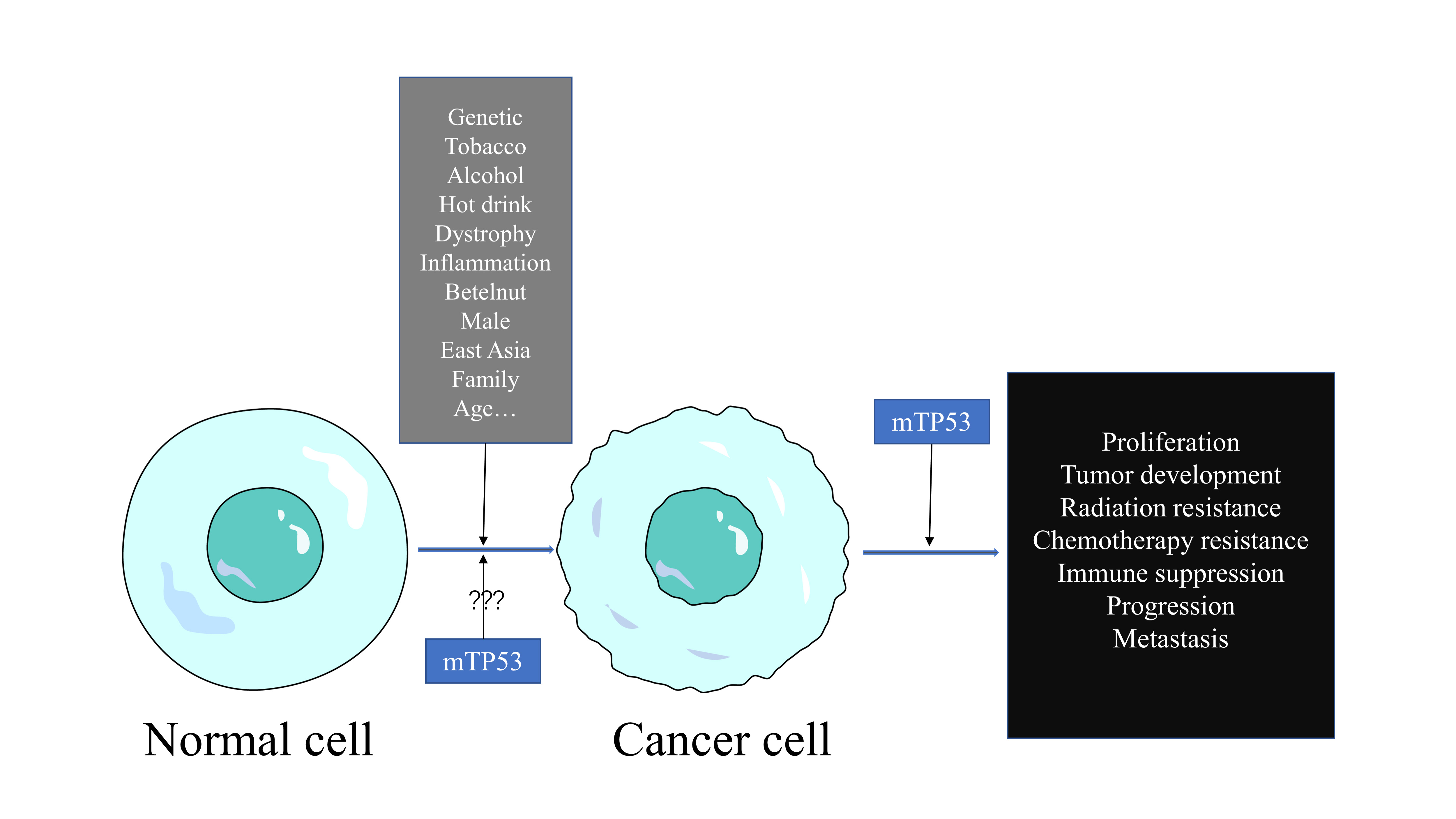 Fig. 3.
Fig. 3.Cancer cells respond and adapt to conditions using mutations optimal for success.
Traditional drugs for unresectable ESCC are characterized by limited efficacy and toxic effects [123]. Targeted therapies based on NGS of carcinoma-associated gene mutations have become the focus of research attention. Drugs targeting mutations, such as those in vascular endothelial growth factor (VEGF), epidermal growth factor receptor (EGFR), and receptor tyrosine-protein kinase erbB-2 (ERBB2), have shown promising results with minimal side effects in the management of various cancers and have been administrated recently in clinical trials in patients with EC [124, 125].
Mutational TP53 genes were identified in multiple stages of ESCC and have been shown to promote cancer growth and contribute to therapy resistance, making mutant P53 a potential target for ESCC treatment. P53 has no catalytic domain as a target for low molecular weight inhibitors. Thus, it has been considered “undruggable”; however, scientists have now illustrated several therapeutic strategies to target mutant p53 [126]. Several of these strategies have already been applied in clinical trials, in which the main approaches include restoration of wild-type-like activities to mutant p53, selective degradation of mutant p53, and inhibition of novel protein–protein interactions involved in mediating gain-of-functions of mutant p53 (Table 2) [126].
| Drug | Type | Therapeutic strategy |
| PRIMA-1 |
cysteine-targeting compounds | Restoration of wild-type like activities of mutant p53 |
| COTI-2 | Zn |
Restoration of wild-type like activities of mutant p53 |
| SAHA | Histone deacetylase (HDAC) inhibitors | Elevate methylation levels |
| Inducing mutant p53 degradation | ||
| MS-275 | HDAC inhibitors | Elevate methylation levels |
| Inducing mutant p53 degradation | ||
| Valproic acid | HDAC inhibitors | Elevate methylation levels |
| Inducing mutant p53 degradation | ||
| Sodium butyrate | HDAC inhibitors | Elevate methylation levels |
| Inducing mutant p53 degradation | ||
| BIIB021 and so on | HSP90 inhibitors | Reactivate endogenous MDM2 and CHIP |
| Inducing mutant p53 degradation | ||
| Arsenic trioxide (ATO) | / | Reactivate the mutant low/no active protein |
| Degrade the mutant protein | ||
| PC14586 | Precision targeting p53 Y220C mutation | Reactivate and stabilize the p53 Y220C mutant protein |
| Mutant p53 Reactivator | ||
| RETRA | Mutant p53 Reactivator | Release p73 from mutant p53 |
| p73 producing tumor-suppressor effects |
CHIP, carboxy-terminus of Hsp70-interacting protein.
Restoration of wild-type like activities of mutant p53 aims to restore the
normal conformation of the DBDs of p53 proteins so that the p53 proteins can
bind with REs to exercise their biological functions, which would induce the
recovery of cell homeostasis or death of the cell. Drugs such as cysteine-targeting compounds or Zn
Mitigating pro-tumorigenic functions via the degradation of mutant p53 is another option. Mitigation of function can be achieved by degrading the mutant protein or by preventing protein-protein interactions with factors involved in GOF responses. Histone deacetylase (HDAC) inhibitors (such as SAHA, MS-275, valproic acid, and sodium butyrate) elevate methylation levels in P53 mutant cell lines, suppress mutant P53 transcription in time- and dose-dependent manners, inducing mutant p53 degradation [132, 133]. The heat shock protein 90 (HSP90) recruited by mutant p53 conceals the ARF-binding site on MDM2 and inhibits its ubiquitin-protein iso-peptide ligase function, blocking the endogenous MDM2 and CHIP (carboxy-terminus of Hsp70-interacting protein) E3 ligase activity, resulting in the stabilization of both mutant p53 and MDM2 [134, 135]. HSP90 inhibitors destroy the complex, liberate mutant p53, and reactivate endogenous MDM2 and CHIP to degrade mutant p53 [134].
Arsenic trioxide (ATO) was recently shown to reactivate mutant forms of p53 possessing structural mutations, resulting in the restoration of biological function and the inhibition of tumor cell growth, both in vitro and in vivo [136]. ATO has also been reported to degrade the mutant protein [137]. Thus, ATO can neutralize the cancer promoting effects via two different mechanisms.
Recently, the United States Food and Drug Administration (FDA) granted fast track designation to PC14586 for the treatment of patients with locally advanced or metastatic solid tumors possessing a p53 Y220C mutation. Unlike the drugs discussed above, PC14586 specifically reactivates and stabilizes the p53 Y220C mutant protein, leading to the transcription of the p53 wild-type target genes as well as the induction of cell cycle arrest [138].
Inhibition of novel protein–protein interactions involved in mediating GOF mutant p53 is another therapeutic strategy. Mutant p53 can contribute to malignancy by forming complexes with a p53 family member, p73. The mutant p53 reactivator, RETRA, releases p73 from the blocking complex with mutant p53, resulting in a substantial upregulation of the level of p73, thereby producing tumor-suppressor effects similar to the functional reactivation of p53 [139].
The MDM2–p53 hub is central to integrating diverse cellular stress signals to mediate appropriate cellular outcomes. Studies have attempted to understand how p53 translates a specific stress signal into a particular cellular response. The p53 signaling pathway is indispensable to prevent malignant transformation of esophageal endothelial cells. Mutational TP53 genes are present in precancerous lesions and all stages of ESCC, playing a crucial role in the carcinogenesis, occurrence, development, progression, and migration of ESCC; however, many of these mechanisms are still unclear. More research is urgently needed to address the function of p53 mutations in ESCC.
LFS is a tumor-susceptibility disorder caused by germline TP53 mutations, and esophageal cancer surveillance by endoscopy is recommended for patients with LFS [140, 141]. The higher risk of a patient with LFS developing cancer, compared with the risk in the normal population, is 100–1000 times higher in ectodermal and mesodermal derived cell types, whereas only 2–4 times higher in endodermal cell types, like esophageal epithelial cells [142]. However, the age of onset of upper gastrointestinal cancers in patients with LFS is lower [61, 62, 63], and the optimal age of initiation of therapy and the surveillance interval by endoscopy should be determined as part of a comprehensive surveillance protocol for LFS.
Various extrinsic factors are related to esophageal carcinogenesis, including cigarette smoke and alcohol, whose carcinogenic role in ESCC acts partly through the p53 pathway [143]. The TP53 gene is one of the target genes of tobacco and alcohol in carcinogenesis, even muti-centric carcinogenesis of the esophagus [82, 105, 106]. To improve prognosis, either endoscopy with Lugol staining or narrow band imaging endoscopy is strongly recommended for smoking- and alcohol-related high-risk populations [9]. Furthermore, warning the public about the danger of these carcinogens and advising them to quit smoking and drinking are vitally important to prevent the development of ESCC. It is also important to understand the molecular mechanisms of genetic changes including, but not limited to, the TP53 gene in smoking- and alcohol-induced ESCC carcinogenesis. These mechanistic insights could be translated into practical approaches for the prevention and treatment of smoking- and alcohol-linked ESCC.
Despite efforts in recent years to develop targeted molecular therapies for ESCC, limited effective drugs have been produced. It has taken over three decades for clinical trials on targeting p53 dysfunction in cancer to begin, and the majority of ongoing trials are still at a preliminary stage; therefore, more effort is required to make significant progress. It is considerably more challenging to restore normal activity to a defective tumor suppressor protein, such as mutant p53, than to block the actions of a driver oncoprotein, and there are still many unresolved questions surrounding the role of mutant p53 in ESCC. Therefore, we encourage academic researchers and pharmaceutical companies to intensify their research into exploiting the most frequently mutated gene in cancer for therapeutic potential. Once the molecular mechanisms between p53 and ESCC have been determined, novel compounds to treat cancer will become available.
LZ and HL wrote the manuscript. WC and YA contriubuted to literature search. YC and ZW contributed to edit, surpervison and funding acquisition. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
EndNote X9 was used to arrange references.
This work was supported by grants from the National Natural Science Foundation of China (No.81871986).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.