

 , An-Jing Ren 1,*
, An-Jing Ren 1,*1 Experimental Teaching Center, College of Basic Medical Sciences, Naval Medical University, 200433 Shanghai, China
2 Department of Nephrology, Changhai Hospital, Naval Medical University, 200433 Shanghai, China
†These authors contributed equally.
Abstract
Prohibitin-2 (PHB2) is a conserved protein in mitochondria that regulates various biological processes, including cell cycle, proliferation, apoptosis, transcription, signal transduction, and mitochondrial ridge morphogenesis. Recently, there has been growing interest in the biological function of PHB2. This article primarily discusses the recent advances in the role of PHB2 in diseases.
Keywords
- prohibitin-2
- mitochondria
- autophagy
- diseases
Prohibitin (PHB) is a conserved protein group mainly located in the inner
mitochondrial membrane and nuclei with diverse biological functions. PHB
comprises PHB1 (32 kDa) and PHB2 (37 kDa) and is a member of a superfamily
containing stomatin and flotillin [1]. PHB mostly exists as a polymer with a
rosette structure with a diameter of 20–25 nm [2, 3]. It is hypothesized that
PHB regulates the cell cycle, senescence, and tumor suppression while
specifically inhibiting the initiation of DNA synthesis [4]. PHB1 suppresses the
signaling of several steroid hormone receptors [5, 6, 7], and PHB2 selectively
represses estrogen receptor (ER) activity [8]. PHB2 participates in essential
cellular processes, including cell survival, apoptosis, metabolism, inflammation,
gene transcription and signal transduction [9, 10]. In Caenorhabditis
elegans and mice, loss of PHB has been found to cause embryonic lethality [11, 12]. To further elucidate the role of PHB2 in physiological and
pathophysiological processes, several tissue-specific PHB2 knockout mouse models
have been established. For example, forebrain-specific PHB2-deficient mice
exhibit tau hyperphosphorylation and neurodegeneration [13], loss of PHB2 in
renal podocytes results in glomerulosclerosis [14], hepatocyte-specific PHB2
knockout mice exhibit liver failure and impaired gluconeogenesis [15],
Recently, the investigation of the biological functions of PHB2 has advanced significantly. This review discusses the structure of PHB2, elucidates its role in autophagy, senescence, apoptosis, and cell metabolism, and explores how it relates to several diseases, including cardiovascular and cerebrovascular diseases, kidney diseases, diabetes, and cancers (Table 1, Ref. [8, 10, 14, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40]).
| Diseases | Mechanisms | References | |
| Autophagy, Senescence and Apoptosis | Autophagy | PHB2 interacts with LC3. | [17] |
| PHB2 mediates mitophagy through the PARL-PGAM5-PINK1 axis. | [18] | ||
| Senescence | PHB2 maintains the stability of newly assembled proteins. | [19] | |
| PHB2 maintains the imbalance in the synthesis of mitochondrial and nuclear-encoded mitochondrial proteins | [20] | ||
| Apoptosis | PHB2 interacts with BIF-1. | [21] | |
| PHB2 is a crucial mitochondrial regulator for homeostasis and lineage-specific differentiation of ES cells. | [10] | ||
| Cell Metabolism | PHB2 influences the stability of Complex IV and affects mitochondrial respiration. | [22] | |
| PHB2 maintains mitochondrial oxidative phosphorylation (OXPHOS) levels. | [23] | ||
| PHB2 inhibits hnRNPA1-induced PKM2 expression and aerobic glycolysis. | [24] | ||
| Mitochondrial-mediated Innate Immunity | PHB2 interacts with VP1. | [25] | |
| PHB2 mediates DENV-2 entry into insect cells. | [26] | ||
| PHB2 interacts with the mitochondrial antiviral signaling protein. | [27] | ||
| Cardiovascular and Cerebrovascular Disease | Cardiac fibrosis | MicroRNA-24-3p targets PHB2 to suppress cardiac fibroblast mitophagy. | [28] |
| Heart failure | PHB2 ablates impaired myocardial FAO by down-regulating carnitine palmitoyltransferase1b (CPT1b). | [8] | |
| Early brain injury | MitoQ activates mitophagy after SAH via the Keap1/Nrf2/PHB2 pathway, inhibiting oxidative stress-related neuronal death. | [29] | |
| Vascular diseases | PHB2 inhibits hnRNPA1-induced PKM2 expression and aerobic glycolysis to maintain the contractile phenotype of VSMC. | [24] | |
| COMP interacts with PHB2 in maintaining mitochondrial homeostasis and regulating VSMC phenotypic transition. | [30] | ||
| Kidney Diseases | PHB2 is localized to the slit septum; its absence results in the disappearance of the foot process. | [14, 31] | |
| PHB2 regulates mitochondrial dysfunction and NLRP3 inflammasome activation. | [32] | ||
| Diabetes | Deficiency of PHB in |
[16] | |
| Cancers | Hepatocellular carcinoma | PHB2 enhances the invasive ability of HCC cells. | [33] |
| Lung carcinoma | PHB2 promotes cell proliferation and migration. | [34] | |
| PHB2 promotes tumorigenesis by RACK1. | [35] | ||
| Breast cancer | BIG3 interacts with PHB2/REA. | [36] | |
| PHB2 regulates the expression of p21. | [37] | ||
| Prostate cancer | PHB2 promotes the migration of PCa cells by inhibiting the expression of AKT2. | [38] | |
| Leukemia | SLAMF receptors negatively regulate B cell receptor signaling through the recruitment of PHB2. | [39] | |
| Mitochondrial AURKA induces mitophagy by interacting with MAP1LC3 and PHB2. | [40] | ||
PHB2, Prohibitin-2; BIF-1, Bax-interacting factor-1; ES, embryonic stem; PKM2, Pyruvate Kinase M1/2; DENV-2, dengue serotype 2; FAO, fatty acid oxidation; MitoQ, mitoquinon; SAH, subarachnoid hemorrhage; VSMC, vascular smooth muscle cell; COMP, cartilage oligomeric matrix protein; HCC, Hepatocellular carcinoma; RACK, receptorforactivated Ckinase; RAE, repressor of estrogen receptor activity; SLAMF, Signaling lymphocytic activation molecule family; AURKA, Aurora kinase A.
The PHB2 gene is located on chromosome 12p13.31 and consists of 10 exons. The PHB2 mRNA contains 1515 bp and finally encodes a 299 amino acid protein [41]. PHB2 exhibits a high degree of PHB1 [8]. PHB1 and PHB2 are highly conserved in yeast, plants, worms, flies, and mammals [42, 43, 44, 45, 46]. Regarding amino acid sequence similarity, the full-length PHB2 between humans and mice, fruit fly, or yeast is at 100%, 71%, and 56%, respectively [1].
PHBs are known to localize to the inner mitochondrial membrane and form a large protein complex [2]. PHB proteins have a similar domain topology, including an N-terminal transmembrane domain, a structurally related PHB domain that may facilitate partitioning into lipid microdomains, and a C-terminal coiled-coil domain [47, 48]. PHB1 and PHB2 are heterodimers and can form higher structural oligomers [3]. Like PHB1, PHB2 contains a transmembrane domain necessary for mitochondrial localization, a central PHB domain, and an overlapping coiled-coil domain [41]. In contrast to PHB1, PHB2 is characterized by its distinctive possession of the ER-binding domain and a putative nuclear import sequence inside the ER-binding domain, facilitating selective inhibition of ER activity, as previously demonstrated (Fig. 1). Additionally, a crystal structure of human PHB2 has been successfully established [27]. The overlapping coiled-coil domain of PHB2 is assembled by a heptad repeat (HR) region. The HR region consists of seven amino acid residues that repeat along the length of the coiled-coil domain, forming a characteristic pattern of hydrophobic and hydrophilic residues. This pattern allows the HR region to interact with other HR regions within PHB2 and other proteins, forming larger protein complexes critical for mitochondrial function. PHB2’s ability to localize to the mitochondria or execute its regulatory functions may be compromised, leading to cellular dysfunction and potentially resulting in disease [27].
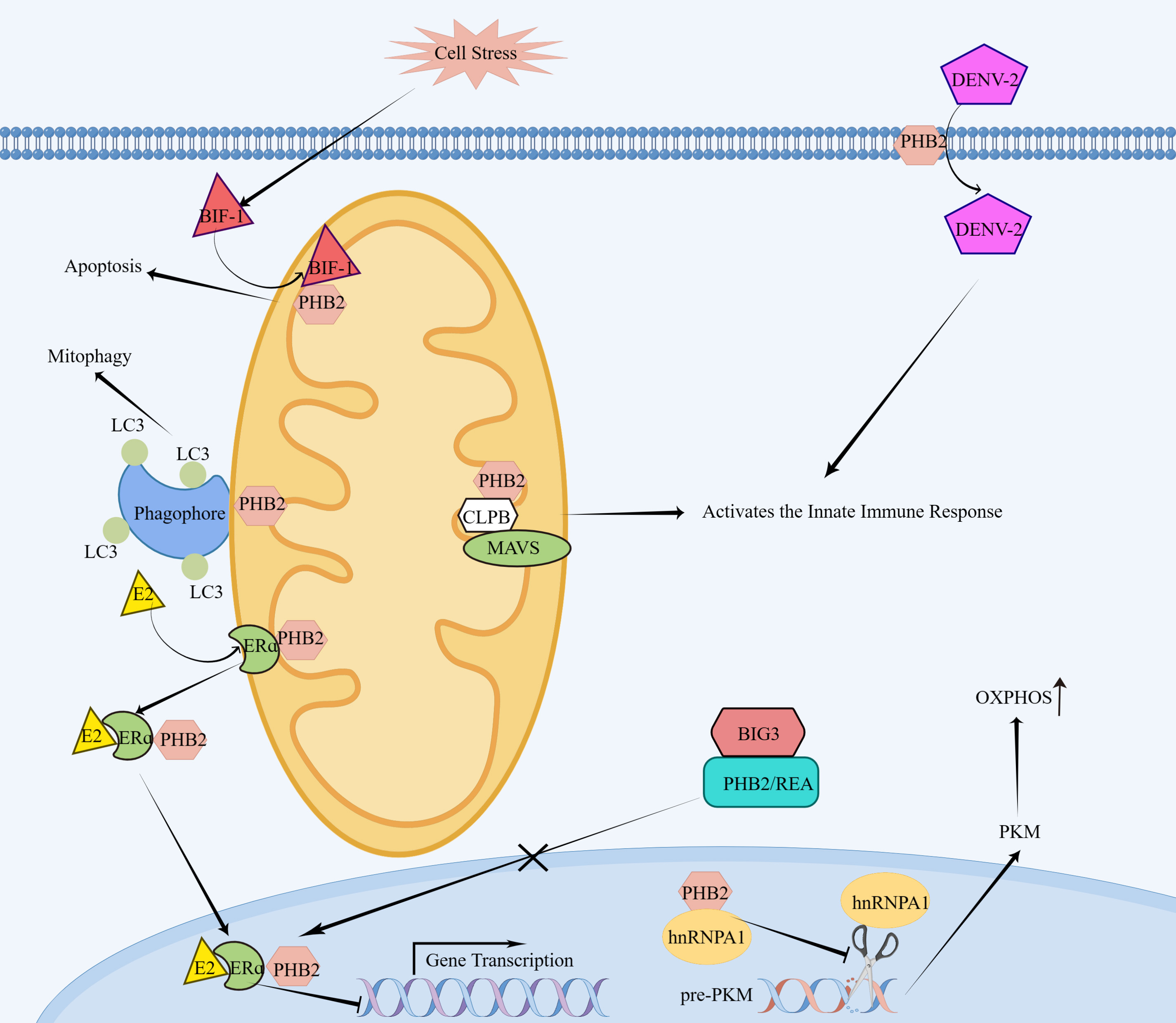 Fig. 1.
Fig. 1.Molecular mechanism of PHB2 action. On the surface of cells,
PHB2 acts as a receptor for DENV-2 and facilitates the entry of DENV-2 into
insect cells. In the mitochondria, PHB2 interacts with MAVS, which requires a
bridging protein called CLPB, and activates the innate immune response. Upon cell
stress, BIF-1 translocates to mitochondria and binds PHB2, resulting in
apoptosis. PHB2 binds the autophagosomal membrane-associated protein LC3 and
mediates mitophagy. In the absence of the brefeldin A-inhibited guanine
nucleotide-exchange protein-3 (BIG3),
Mitophagy is vital in maintaining cellular homeostasis and promoting cellular health by removing defective organelles and misfolded proteins [49, 50]. It is important for various biological processes, including development, cellular homeostasis, tumor suppression, and prevention of neurodegeneration and aging. Mitophagy has been linked to various neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [49].
Wei et al. [17] demonstrated that PHB2 is a critical receptor for mitochondrial autophagy, which binds the autophagosomal membrane-associated protein LC3 through an LC3-interaction region (LIR) domain upon mitochondrial depolarization and proteasome-dependent outer membrane rupture (Fig. 1). PHB2 is essential for Parkin-induced mitophagy in mammalian cells and the elimination of paternal mitochondria following embryonic fertilization in C.elegans [17, 51]. PHB2 facilitates PTEN-induced kinase 1 (PINK1)—Parkin RBR E3 ubiquitin protein ligase (PRKN)—mediated mitophagy by promoting PINK1 stability and increasing recruitment of PRKN to the mitochondria. In addition, PHB2-mediated mitophagy relies on the mitochondrial inner membrane proteases presenilin-associated rhomboid-like (PARL) and PGAM family member 5 (PGAM5). Thus, Yan et al. [18] proposed a pathway for PHB2-mediated mitophagy known as the PHB2-PARL-PGAM5-PINK1 axis.
Mitochondrial proteins are one of the key molecular markers of aging, and PHB is a vital mitochondrial protein. Therefore, many scholars consider PHB to be significantly associated with aging. When the function of the PHB protein decreases, the stability of newly assembled proteins in mitochondria decreases. The improper assembly of the oxidative respiratory chain leads to the failure of normal oxidative metabolism in cells. Consequently, there is an increase in free radicals, which leads to cell aging [19]. Furthermore, decreased heterogeneity of PHB1 and PHB2 has been observed in cellular senescence in human and chicken fibroblasts, potentially linked to metabolic stress response due to an imbalance in the synthesis of mitochondrial and nuclear-encoded mitochondrial proteins [20]. PHB2 overexpression promotes mitophagy and delays the senescence of cardiomyocytes, while PHB2 knockdown reverses the anti-aging effect of tetrahydroberberrubine (THBru) [52].
PHB2 is implicated in regulating apoptosis by Bax-interacting factor-1 (BIF-1). BIF-1, also identified as SH3GLB1 or endophilin B1 was initially recognized as a protein that interacts with Bax [53, 54]. BIF-1 is linked to Bax activation in regulating apoptosis, autophagy, and mitophagy [55, 56]. Cho et al. [21] found that BIF-1 translocated to mitochondria and interacted with PHB2 during cell stress, resulting in mitochondrial inner membrane cleavage, mitochondrial fragmentation and cell apoptosis (Fig. 1). The embryonic stem (ES) cells in their pluripotent state undergo significant apoptosis due to the knockdown of PHB2, whereas the overexpression of PHB2 fosters cell proliferation. PHB2 is significant in maintaining ES cell homeostasis and lineage differentiation [10] and being a key regulator for platelet mitophagy and Parkin-mediated mitophagy in urothelial cells of bovine papillomavirus-infected cattle [57, 58].
The mitochondria are the major site of energy metabolism in most cells. PHB regulates mitochondrial energy metabolism by controlling the activity of pyruvate carboxylase. PHB2 can impact the stability of Complex IV and modulate mitochondrial respiration [22]. In colorectal cancer cells, the knockdown of PHB2 significantly reduces oxidative phosphorylation (OXPHOS) levels in mitochondria [23]. PHB2 knockout in the heart impairs cardiac fatty acid oxidation (FAO) [8]. Jia et al. [24] have demonstrated that PHB2 is critical in regulating the metabolism and phenotype of vascular smooth muscle cells (VSMCs). PHB2 knockdown in VSMCs promotes neointimal formation after vascular injury. Mechanistically, PHB2 inhibits hnRNPA1 (a key modulator of pyruvate kinase M1/2)—induced pyruvate kinase M1/2 (PKM2) expression and aerobic glycolysis to maintain the contractile phenotype of VSMCs (Fig. 1) [24].
PHB2 participates in the viral infection process [25, 26]. The interaction between PHB2 and VP1, a structural protein of Enterovirus A71, is crucial for initiating autophagy and increasing the infectivity of EV-A71 [25]. PHB2 has been identified as a receptor for dengue serotype 2 (DENV-2) on the surface of cells and facilitates the entry of DENV-2 into insect cells (Fig. 1) [26]. PHB2 is involved in the retinoic acid-inducible gene (RIG-I) signaling pathway, a key component of the innate immune response to viral infections. Specifically, PHB2 interacts with the mitochondrial antiviral signaling protein (MAVS) to form a signaling complex that activates downstream signaling pathways in response to viral infection [27]. However, the interaction between PHB2 and MAVS requires a bridging protein called caseinolytic peptidase B protein homolog (CLPB), which fills the topological gap between the two complexes (Fig. 1). In addition, A-kinase-anchoring protein 1 (AKAP1) and ATPase family AAAdomain containing protein 3A (ATAD3A) have been shown to assist in this process by stabilizing the PHB2-CLPB complex. Together, these proteins combine to form a MAVS signalosome that activates the innate immune response, helping to protect cells against viral infections [27].
Cardiac fibrosis is a common pathological process observed in various cardiovascular diseases, and it may contribute to the development of heart failure. The expression of microRNA (miR)-24-3p was significantly down-regulated in mice hearts with transverse aortic constriction (TAC) and angiotensin II (AngII)-treated cardiac fibroblasts. Further studies showed that PHB2 was a direct target of miR-24-3p and knockdown of PHB2 attenuated AngII-induced fibrosis in CFS. MiR-24-3p mitigates cardiac fibrosis by inhibiting mitophagy in cardiac fibroblasts through PHB2 downregulation [28]. It has been reported that heart-specific PHB2 knockout mice experience disorders in fatty acid oxidation, mitochondrial dysfunction, heart failure, and mortality, highlighting the importance of PHB2 in maintaining standard cardiac metabolic function [8]. PHB2 was found to be a potential fatty acid oxidation (FAO) regulator in the inner mitochondrial membrane of myocardium, and ablation of PHB2 impaired myocardial FAO by down-regulating carnitine palmitoyltransferase1b (CPT1b), a rate-limiting enzyme of cardiac mitochondrial FAO [8]. Further research exploring the control of mitochondrial metabolic homeostasis by PHB2 could aid the development and optimization of treatments for heart failure.
Early brain injury (EBI) is considered to be the primary cause of high mortality and delayed neurological deficits in patients after subarachnoid hemorrhage (SAH) [59]. Oxidative stress-induced neuronal death is the primary factor leading to the weak prognosis associated with EBI [60]. Mitochondrial dysfunction primarily contributes to reactive oxygen species (ROS) production, which results in cellular damage and oxidative stress [61]. Therefore, effective treatments that reduce mitochondrial damage play a crucial role in the treatment of SAH patients. Mitoquinone (MitoQ) is an exceptional antioxidant targeting the mitochondria and effectively preventing mitochondrial dysfunction [62]. The oxidative stress complex regulatory Keap1 (the cytoskeleton-associated protein)/Nrf2 (nuclear transcription factor 2) has been identified as a significant antioxidant target in various diseases and SAH models. Given that PHB2 is a mitophagy receptor that binds to the autophagosomal membrane-associated protein LC3II [17], Zhang et al. [29] found that MitoQ activated mitophagy after SAH via the Keap1/Nrf2/PHB2 pathway, thereby inhibited oxidative stress-related neuronal death. The results of this study indicate that PHB2’s influence on mitophagy could serve as a promising therapeutic target for the treatment of SAH.
Phenotypic modulation of VSMC is an early and critical step in the pathogenesis
of numerous vascular diseases, including atherosclerosis, restenosis after
vascular injury, aneurysms, and vascular calcification. Jia et al.
[24] identified that PHB2 was expressed in the vascular media, and PHB2-knockdown
smooth muscle cells showed reduced contractile protein expression, polygonal
morphology, and enhanced proliferation and migration; meanwhile, smooth muscle
cell-specific PHB2-knockout mice showed significantly reduced aortic
contractility, suggesting that PHB2 may be able to maintain the contractile
phenotype of VSMC. The expression of smooth muscle contractile protein decreased,
the proliferation of VSMC increased, and the formation of neointima increased in
PHB2-deficient mice after balloon-induced carotid injury, indicating that PHB2
can inhibit the formation of neointima after vascular injury. Mechanistically,
PHB2 inhibits hnRNPA1-induced PKM2 expression and aerobic glycolysis to maintain
the contractile phenotype of VSMC. Cartilage oligomeric matrix protein (COMP) is
a 524 kDa pentameric noncollagenous glycoprotein expressed in the musculoskeletal
and cardiovascular systems. COMP plays a key role in maintaining VSMC homeostasis
and the contractile phenotype of VSMC through integrin
Mitochondrial dysfunction is a significant hallmark of the progression of various kidney diseases and is closely linked to cell death. PHB2 is a promising target for the treatment of kidney disease. Renal interstitial fibrosis (RIF) is the primary histopathological change in various renal disorders closely linked to renal dysfunction. PHB2 expressions in the renal interstitium of RIF rats were reduced [65]. PHB2 is necessary for maintaining the structural integrity of podocyte foot processes, and its absence can lead to the destabilization of extra-mitochondrial functions at the kidney filtration barrier [14]. PHB2 deficiency may cause glomerular disease through two different pathways. Firstly, PHB2 is localized to the slit septum, and its absence results in the disappearance of the foot process, leading to loss of filtration function and proteinuria. Secondly, PHB2 deficiency results in mitochondrial dysfunction enhances mTOR activation, and causes harmful metabolic turnover in podocytes, ultimately leading to podocyte loss [14, 31]. The combined effects of these two pathways are responsible for the development of severe glomerular disease observed in podocyte-specific PHB2-knockout mice. Membranous nephropathy (MN) is the leading cause of adult nephrotic syndrome when left treated. PHB2 is upregulated in the kidneys of MN mice treated with cationic bovine serum albumin (CBSA). The knockdown of PHB2 through short hairpin RNA (shRNA) inhibited the expression of the tumor suppressor p53 and significantly increased podocyte proliferation. The upregulation of PHB2 may be caused by the blockage of proteasome activity [66]. These findings imply that the upregulation of PHB2 is linked to CBSA-mediated podocyte toxicity and leads to the development of MN. Xu et al. [32] found that PHB2-mediated mitophagy alleviated injury in renal tubular epithelial cells by regulating mitochondrial dysfunction and NLRP3 inflammasome activation.
PHB is expressed in pancreatic
Studies have demonstrated that PHB2 is overexpressed in hepatocellular carcinoma, lung, prostate, and breast carcinoma compared to the corresponding normal tissues [33, 34, 35, 38]. Therefore, there has been a growing interest in the involvement of PHB2 in cancer.
Hepatocellular carcinoma (HCC) is a prevalent malignant tumor in humans, with an increasing incidence rate. The molecular mechanisms underlying the aggressive behavior of HCC are complex, and details about the malignant behavior of HCC remain unclear. Cheng et al. [33] found that PHB2 expression was upregulated in HCC cells and hypoxic HCC cells. Inhibition of PHB2 significantly reduced hepatoma cells’ proliferation and colony formation in both normoxic and hypoxic environments. PHB2 overexpression enhanced the adaptation of hepatoma cells to hypoxic microenvironment and resulted in increased resistance to chemotherapy-induced apoptosis. PHB2 improved the invasive ability of HCC cells, and the specifics of the malignant behavior of HCC are still unknown. These findings suggest that PHB2 is a survival factor and a potential target for drug design.
Lung cancer is a malignant tumor with the highest incidence and mortality rates. Lung cancer comprises two clinical types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). An analysis of NSCLC tissues demonstrated that PHB2 expression was notably higher than in normal lung tissues [34, 35]. In vitro, PHB2 overexpression promoted cell proliferation and migration, while PHB2 knockdown reduced parkin-mediated mitosis and inhibited cell proliferation and migration [34]. PHB2 has been discovered to promote tumorigenesis in NSCLC by interacting with and stabilizing the receptor for activated C kinase 1 (RACK1), a member of the tryptophan-aspartate repeat protein family [35].
Breast cancer is the most common cancer in women and is hormone-dependent.
Brefeldin A-inhibited guanine nucleotide-exchange protein 3 (BIG3) plays an
important role in activating the estrogen/ER signaling in breast cancer cells by
interacting with PHB2/repressor of estrogen receptor activity (PHB2/REA) [36].
ER
Several small molecules targeting PHB1/2 have been synthesized to develop a new class of drugs that can be used as anticancer agents in the future [76]. A promising example is fluorazoline, which binds directly to PHB1/2 and exerts cytotoxic effects on cancer cells [77]. Takagi et al. [78] found that fluorazoline inhibited the proliferation of MCF7 breast cancer cells and induced p21 expression. Interestingly, the knockdown of PHB2 led to significant upregulation of p21, whereas overexpression of PHB2 inhibited this change in MCF7 cells [37]. Furthermore, the growth and cell cycle progression in fluorazoline-treated MCF7 cells can be restored through the overexpression of PHB2. Fluorazoline acts by blocking the interaction between PHB2 and GGCT protein, which reduces the nuclear localization of PHB2 [78]. Fluazoline increases the expression of p21 and inhibits cell proliferation by regulating the activity of PHB2.
Prostate cancer (PCa) is a commonly diagnosed malignancy that affects the male genitourinary system. However, the factors contributing to the development of PCa have yet to be comprehensively elucidated. PCa tissue shows significant upregulation of PHB2 compared to normal tissue. Overexpression of PHB2 promoted migration of two PCa cell lines (PC3 and DU145) and decreased the protein-serine/threonine kinase (AKT2) expression, while PHB2 knockdown increased AKT2 expression and enhanced AKT2 protein stability [38]. The mechanism of PHB2 promoting the migration of PCa cells may be through the inhibition of AKT2 expression. These results have significant implications for the development of targeted drugs for the treatment of PCa.
Cytogenetically normal acute myeloid leukemia (CN-AML) is a subtype of AML that accounts for 40%–50% of all AML cases. CN-AML patients with higher PHB2 expression in AML cells showed a worse overall survival rate than those patients with relatively lower PHB2 protein expression. PHB2 protein overexpression was significantly associated with adverse prognosis in CN-AML patients [79]. The AKT/PKB kinase pathway regulates cell survival, proliferation, and differentiation. PHB2 is a novel nuclear AKT substrate during all-trans retinoic acid-induced differentiation of AML cells [80]. Signaling lymphocytic activation molecule family (SLAMF) receptors play an important role in natural killer cell biology. SLAMF receptors negatively regulate B cell receptor signaling in chronic lymphocytic leukemia by recruiting PHB2 [39]. The serine/threonine kinase AURKA has been shown to localize mitochondria and regulate mitochondrial dynamics and ATP production. AURKA overexpression is commonly observed in hematologic tumors. Bertolin et al. [40] found mitochondrial AURKA-induced mitophagy by interacting with MAP1LC3 and PHB2. Since PHB2 has been indicated to be a potential blood cancer biomarker, Yun et al. [81] designed a highly sensitive electrochemical immunosensor to detect PHB2 in blood cancer patients.
PHB2 is a pleiotropic protein expressed ubiquitously across diverse cellular locations, including mitochondria, cytoplasm, nucleus, and plasma membrane. PHB2 has a variety of biological functions, which are determined by its cellular location and cell type. The diverse functions of PHB2 include regulation of normal mitochondrial growth and development, nuclear transcription, cell proliferation and apoptosis, cell cycle, and senescence. Although recent advances have been made in understanding PHB2’s biological role, many issues remain unresolved and require further investigation. Accumulating evidence supports the importance of PHB2 in human pathology. Further studies on its biological functions and mechanisms of action will help to identify PHB2 as a novel biomarker and molecular target for therapeutic intervention in cancer, neurodegenerative diseases, cardiovascular diseases and other metabolic diseases.
AKAP1, A-kinase-anchoring protein 1; AKT, serine/threonine tyrosine kinase; ALS,
amyotrophic lateral sclerosis; AngII, angiotensin II; ATAD3A, ATPase family
AAAdomain containing protein 3A; AURKA, Aurora kinase A; BIF-1, Bax-interacting factor-1; BIG3, brefeldin
A-inhibited guanine nucleotide-exchange protein 3; CBSA, cationic bovine serum
albumin; CFS, cardiac fibrosis; CLPB, caseinolytic peptidase B protein homolog;
CN-AML, cytogenetically normal acute myeloid leukemia; COMP, cartilage oligomeric
matrix protein; CPT1b, carnitine palmitoyltransferase1b; DENV-2, dengue serotype
2; EBI, early brain injury; ER, estrogen receptor; HCC, hepatocellular carcinoma;
ERAP, ER
Conceptualization, FL, YZ, ZG and A-JR; writing-original draft preparation, FL and YZ; writing-review and editing, ZG and A-JR. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research was funded by grants from the National Natural Science Foundation of China (32071115 and 32271154).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.