
- Academic Editor
Inflammasomes are cytosolic multi-protein complexes that play an important role in the innate immune system, inducing cytokine maturation and pyroptosis. Trained immunity is the induction of memory in innate immune cells by epigenetic reprogramming due to repeated inflammatory stimuli that alter the inflammatory response and increase resistance to infection or disease. Although it is speculated that nucleotide-binding oligomerization domain (NOD), leucine-rich repeat (LRR), and the NLR family pyrin domain containing 3 (NLRP3) inflammasomes respond to various inflammatory stimuli and are associated with trained immunity, the exact relationship is still unclear. This paper aims to introduce data from recent research on the role of inflammasomes in trained immunity through cellular immunometabolic and epigenetic reprogramming. It also suggests a new therapeutic strategy for inflammatory diseases through the complementary regulation of inflammasomes and trained immunity.
The immune system is categorized into innate and adaptive immunity. While innate immunity is the first line of defense against pathogens and is present at birth, adaptive immunity develops over time, and has a ‘memory’ [1]. The immunological memory cells represented by T and B lymphocytes, provide a rapid and effective response against subsequent encounters with the same antigen, thereby offering a long-term defense against re-infection [1]. Interestingly, this classification of the immune system based on memory has been challenged by the discovery of ‘memory-like’ phenomena in innate immunity [2]. The most prominent example is the Bacillus Calmette-Guérin (BCG) vaccine against tuberculosis, which has shown non-specific protective effects in infants by reducing susceptibility to other respiratory infections, a response proposed to be mediated by the long-term boosting of innate immunity [3]. Also, lymphocytes and immunological memory associated with them have been believed to be present only in vertebrates, and invertebrates were believed to have no immune memory due to the absence of T and B cells. However, current evidence is to the contrary [2]. For example, although the immune response of copepods (small crustaceans) is dependent only on innate immunity, it is this immunity that has been shown to interfere with re-infection by parasitic tapeworms, indicating that even lower animals have immune memory [4]. Another such example is seen in honeybees. When exposed to bacteria, bees increase their hemocyte count and the expression of antimicrobial peptides [5, 6]. These changes are maintained long-term, allowing bees to defend themselves better against other pathogens [5, 6]. Overall, memory associated with the innate immune system is observed in invertebrates, as well as in vertebrates, including humans, indicating a clear evolutionary conservation that could be used for therapeutic benefits [7]. In general, innate immune cells respond to external stimuli and then return to an inactive state when the stimulus disappears. However, under certain circumstances, the innate immune cells undergo epigenetic and metabolic changes that result in a rapid and robust response to similar stimuli [8]. This modified innate immune response against similar stimuli is called ‘trained immunity’, a concept first introduced by Netea et al. in 2011 [2]. It is defined as ‘the immunological memory of innate immune cells to past insults’ [2] and is achieved through the epigenetic and metabolic modifications induced by proinflammatory cytokines and microbial cellular components [2].
Inflammasomes are multi-protein complexes present in the cytoplasm of innate
immune cells and some epithelial cells that initiate and amplify inflammatory
responses [9]. They are assembled by recognizing intracellular danger signals
such as pathogen-, danger-, and lifestyle-associated molecular patterns (PAMPs,
DAMPs, and LAMPs) via the induction of cytosolic homeostatic changes (e.g.,
K
The epigenetic and metabolic changes which lead to the trained immunity of the
innate immune system are primarily driven by the proinflammatory cytokines (i.e.,
IL-1
Trained immunity refers to the increase in the non-specific immune response
through metabolic changes and the epigenetic reprogramming of immune cells [8, 12]. An example of trained immunity is the non-specific protective effects of
vaccines as mentioned above (BCG vaccines administered to infants) which provide
increased protection against secondary infections through acquired immunity and
individual responsiveness [13]. The stimuli that induce trained immunity are
diverse and include infections such as bacterial (e.g., Mycobacterium
bovis in the BCG vaccine), fungal (e.g., Candida albicans), parasitic
(e.g., Nippostrongylus brasiliensis), viral (e.g., cytomegalovirus), as
well as inflammatory signals (e.g., interferon [IFN]-
Innate immune cells are reprogrammed through epigenetic mechanisms, and the
epigenetic changes train subsequent immune responses [12]. Monocytes/macrophages
rich in PRRs are rapidly activated by danger signals (PAMPs and DAMPs) and
initiate a transcriptional cascade regulated at the chromosomal level [16]. The
changes in transcription patterns such as nuclear factor (NF)-
A comparative study of BCG-vaccinated individuals who have the ability to limit
the growth of Mycobacterium tuberculosis (Mtb) and those who do not,
suggested that the differences in response to the pathogen are probably
associated with the ability of macrophages to suppress Mtb growth more
significantly in some individuals [17]. This difference was found to be
associated with increased IL-1
The epigenetic markers involved in regulating trained immunity are histone 3 with monomethylation at 4th lysine residue (H3K4me1), histone 3 with trimethylation at 4th lysine residue (H3K4me3), histone 3 with acetylation at 27th lysine residue of the N-terminus of histone H3 (H3K27ac), histone 3 with trimethylation at 9th lysine residue (H3K9me3), and histone 3 with trimethylation at 27th lysine residue (H3K27me3) [7]. H3K4me1 is an epigenetic modification that is associated with enhancers. H3K4me3 is involved in regulation of gene expression [7]. H3K27ac is defined as an active enhancer marker due to its association with higher transcriptional activation [7]. The preceding three markers increase during trained immunity [7]. H3K9me3 is associated with heterochromatin [7]. H3K27me3 is associated with the formation of heterochromatic regions and the downregulation of neighboring genes [7]. H3K9me3 and H3K27me3 decrease during the training period [7]. These markers are important indicators of trained immunity because they induce changes in the transcription levels upon re-stimulation.
The role of non-coding RNA in trained immunity is not yet fully understood, but
some studies suggest that these RNA may be involved in regulating gene expression
related to immune response [13]. For example, long non-coding RNA-cyclooxygenase
2 (Cox2) is co-expressed with the Cox-2 gene in lipopolysaccharide
(LPS)-stimulated mouse macrophages and regulates the expression of inflammatory
genes by interacting with the NF-
Metabolic rewiring within cells induces trained immunity [20]. Several
sub-signaling pathways involved in trained immunity have been studied, and some
metabolites from these pathways, namely, peptides such as muramyl dipeptide (MDP)
and BCG through the NOD2 receptor, oxidized low-density lipoprotein (oxLDL)
through direct action on the protein kinase B (PKB or Akt) [21], and a western
diet through NLRP3 leading to the Akt-mammalian target of rapamycin
(mTOR)-hypoxia-inducible factor-1
An increase in Akt phosphorylation leads to an increase in aerobic glycolysis in
macrophages [22]. Specific metabolites generated from this process, such as
acetyl coenzyme A (acetyl-CoA) and fumarate, epigenetically restructure histones
[23, 24]. These results indicate that the Akt-mTOR-HIF-1
In transcriptomic and epigenetic studies of
Citrate is generated during glycolysis and derived from other metabolites such
as glutamine and transformed into
Overall, the reprogramming of cellular metabolic pathways is considered an
important mediator of trained immunity regulation. Among these, changes in the
Akt-mTOR-HIF-1
Trained immunity was first described based on the characteristics of myeloid
cells such as monocytes and macrophages [2]. However, since monocytes and
macrophages have relatively short lifespans [39], the reason for the presence of
an effective defense against pathogens due to immune memory even after at least
one year of BCG vaccination in infants who have not yet developed adaptive
immunity could not be explained [3]. It is speculated that this long-term trained
immunity is due to epigenetic changes in the precursors of the myeloid cells. In
2018, two research groups showed that trained immunity occurs in hematopoietic
stem and progenitor cells (HSPCs) [40, 41]. Kaufmann et al. [41]
demonstrated that the intravenous administration of BCG trains HSPCs to produce
functionally reprogrammed macrophages that provide subsequent non-specific
protection. Mitroulis et al. [40] showed the expansion of HSPCs
utilizing glucose metabolism and cholesterol biosynthetic pathways induced by
The induction of trained immunity is achieved through immunometabolic and epigenetic changes in immune cells [48]. Immunometabolic changes that induce trained immunity also affect the activation of inflammasomes [49]. Although research on the effects of trained immunity on inflammasomes is limited, the impact of immunometabolic and epigenetic modifications on the priming and activation steps of inflammasome activation, as well as the reciprocal effect of these two steps on immunometabolism and epigenetics are discussed.
The priming step induces the upregulation of inflammasome components. The lack
of glucose transporter (GLUT) 1 suppresses the expression of NLRP3 and the
proform of IL-1
There is evidence to suggest that immunometabolism regulates the activation
step. The inhibition of GLUT1-dependent glycolysis suppresses NLRP3 inflammasome
activation [50]. In addition, hexokinase (HK), through its interaction with the
mitochondrial outer membrane, activates the NLRP3 inflammasomes [49], while the
inhibitors of HK attenuate inflammasome activation [51]. In mouse macrophages
stimulated by Mtb, the decreased expression of phosphofructokinase, muscle type
(PFK-m) inhibits NLRP3 inflammasome activation [52]. Pyruvate kinase (PK; PKM2,
an abundant type in macrophages) induces the assembly of NLRP3 and Absent in
melanoma 2 (AIM2) inflammasomes and stimulates the release of inflammatory
factors through the phosphorylation of protein kinase R [49]. HIF-1
Metabolites are the other regulators of inflammasome activation. Accumulation of
succinate through succinate dehydrogenase (SDH) promotes the production of mROS
by increasing mitochondrial membrane potential and succinate oxidation [49, 53].
Inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and
| Target | Mechanism | Effect on the NLRP3 inflammasome | Ref. |
| GLUT 1 | Deficiency of GLUT 1 suppresses glycolysis | Inhibition of priming step | [49, 50] |
| Inhibition of GLUT1 suppresses glycolysis | Inhibition of activation step | [50] | |
| HIF-1 |
HIF-1 |
Induction of priming step | [49] |
| Succinate | Succinate stabilized HIF-1 |
Induction of priming step | [49] |
| HK | HK disrupts the mitochondrial outer membrane | Induction of activation step | [49] |
| Inhibitors of HK | Inhibition of activation step | [51] | |
| PFK-m | Suppression glycolysis | Induction of activation step | [52] |
| PK (PKM2) | Phosphorylation of protein kinase R | Induction of activation step | [49] |
| SDH | SDH induces mitochondrial ROS | Induction of activation step | [49] |
| GAPDH | Inhibition of GAPDH suppresses NADH generation | Induction of activation step | [54] |
| Inhibition of |
Induction of activation step | [54] |
Abbreviations: NLRP3, NLR family pyrin domain containing 3; GLUT 1, glucose transporter 1; HIF-1
Components of inflammasomes are regulated at the transcriptional level by
factors such as H3K4me1, H3K4me3, and H3K27ac, which facilitate the binding of
transcription factors like NF-
| Epigenetic modification | Mechanism | Effect on NLRP3 inflammasome | Ref. |
| H3K4me1, H3K4me3, H3K27ac ↑ | Changes in chromatin structure and increased binding of NF- |
Induction of priming step | [56, 57, 58] |
| H3K27ac ↑ | Activation of NF- |
[59] | |
| H3K9me3 ↓ | Heterochromatin formation by binding with HP1 | [61] | |
| H3K27me3 ↓ | Heterochromatin formation by binding with PRC2 | [62] | |
| H3K4me1, H3K4me3, H3K27ac ↑ | NEAT1 promotor | Inhibition of priming step | [69] |
| H3K4me1 ↑ | GPR17-PRC1 increasing Polycomb repressive complex 1-mediated | Induction of activation step via ROS | [63] |
| H3K4me3 ↑ | The transcriptional regulation of genes related to ROS generation | [64] | |
| H3K4me1, H3K4me3, H3K27ac ↑ | Transcriptional activation of p53 | [67, 68] | |
| H3K9me3, H3K27me3 ↓ | Glucose-6-phosphate dehydrogenase | [65, 66] | |
| H3K4me1, H3K4me3, H3K27ac ↑ | TLR-MYD88/TRIF-IRF1 pathway | Induction of activation step via mtDNA | [56] |
Abbreviations: H3K4me1, histone 3 with methylation at the 4th lysine residue;
H3K4me3, histone 3 with trimethylation at the 4th lysine residue; H3K27ac,
histone 3 with acetylation at the 27th lysine residue; NF-
Although there may be opposing views, ROS is generally considered a common trigger that induces NLRP3 inflammasome assembly [70]. H3K4me1 was found in the promoter of the G-protein-coupled receptor (GPR) 17, which increases ROS generation through Polycomb repressive complex 1 (PRC1) [63]. Also, a demethylase (Jar1) and a methyltransferase (SETD7) of H3K4me3 regulate the transcription of genes involved in ROS generation [64]. H3K27ac was found in the promoter of a non-coding RNA called NORAD, which increases ROS generation [71]. H3K27ac was also found in the promoters of autophagy-related genes ATG5 and ATG12, which are involved in ROS generation [72]. H3K9me3 and H3K27me3 were found in the promoter of glucose-6-phosphate dehydrogenase (G6PD), a pentose phosphate pathway-related gene that suppresses ROS generation [65, 66]. In summary, in trained immunity, the increase in H3K4me1, H3K4me3, and H3K27ac and the decrease in H3K9me3 and H3K27me3 regulate the expression of genes involved in ROS generation and response, leading to an increase in ROS generation.
Some NLRP3 inflammasome activators are known to activate the inflammasomes through lysosomal destabilization [70]. H3K4me1, H3K4me3, and H3K27ac induce lysosomal destabilization through the transcriptional activation of proteins such as p53 [67, 68, 73, 74] which can induce lysosomal destabilization through various mechanisms [67, 75]. These include binding with other proteins to decrease membrane stability within the lysosome [76], or decreasing the expression of proteins involved in cholesterol transport, leading to increased cholesterol levels within the lysosome and decreased stability [76]. However, p53 also inhibits inflammasome priming by competing with the Enhancer of zeste homolog 2 (Ezh2) for binding to the NEAT1 promoter region [69].
It is known that mitochondrial damage leads to the production of oxidized mitochondrial DNA (mtDNA), which is a trigger to assemble NLRP3 inflammasomes [77]. The two adapters, myeloid differentiation primary response 88 (MYD88) and toll/interleukin-1 receptor (TIR) domain-containing adaptor protein (TRIF), in toll-like receptor (TLR) activation induce an increase in oxidized mtDNA synthesis in mitochondria, which depends on the downstream interferon regulatory factor 1 (IRF1) [77, 78]. H3K4me1, H3K4me3, and H3K27ac are enhancer regions associated with genes involved in the TLR-MYD88/TRIF-IRF1 pathway, promoting gene expression and activating the pathway [56, 79], leading to increased oxidized mtDNA production and contributing to NLRP3 activation. Thus, epigenetic changes in trained immunity regulate NLRP3 activation at both the priming and activation levels (Table 2).
Various metabolic pathways within cells are altered by immune-inducing factors through epigenetic reprogramming, thereby regulating trained immunity [12]. Thus, inflammasome activation could regulate trained immunity by influencing cellular metabolism [43].
IL-1
Inflammasomes can be involved in trained immunity through changes in
mitochondrial function. OxLDL is known to induce trained immunity through the
Akt-mTOR-HIF1
While inflammasomes indirectly induce the development of trained immunity by
inducing changes in cellular metabolism, IL-1
IL-1
| Target | Mechanism | Ref. | |
| Immunometabolism | Glycolysis | Increased expression of GLUT1 and GLUT3 | [80] |
| Upregulation of glycolysis by increasing the expression of glycolysis-related proteins | [82, 93] | ||
| Increased expression of LDH | [81] | ||
| Oxidative metabolism | Movement of the Myddosome | [87] | |
| Changes in the expression levels of mitochondrial membrane potential and fusion/fission proteins | [88] | ||
| Epigenetics | Histone modifications | H3K9ac and H3K4me3 and an increase in repressive histone modification H3K27me3 | [89] |
| DNA methylation | Promotion promoter activation, which increases pro-inflammatory gene expression | [92] | |
| HSPCs expansion | Myelopoiesis | Increased expression of myeloid lineage-specific genes | [40] |
Abbreviations: GLUT 1, glucose transporter 1; GLUT3, glucose transporter 3; LDH, lactate dehydrogenase; H3K9ac, histone 3 with acetylation at the 27th lysine residue; H3K4me3, histone 3 with trimethylation at the 4th lysine residue; HSPCs, hematopoietic stem and progenitor cells.
It has been suggested that trained immunity helps defend against various bacterial, fungal, and viral infections [12]. In the same context, trained immunity has been proposed to potentially assist in the treatment of coronavirus disease-19 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [12]. However, attempts to find a correlation between the BCG vaccination status and COVID-19 severity have yielded inconclusive results [12, 94]. Conversely, the presence of pre-existing inflammatory conditions and excessive inflammatory response induced by SARS-CoV-2 infection have been identified as poor prognostic factors for COVID-19 progression [95]. Various metabolic pathways within cells are altered by immune-inducing factors through epigenetic reprogramming, thereby regulating trained immunity [94]. This highlights the ambivalence of trained immunity, which might be either beneficial or harmful to our health depending on how it is regulated.
Among inflammasomes, the NLRP3 inflammasome has been well studied due to its
involvement in both age-related and metabolic diseases, such as type 2 diabetes,
obesity, atherosclerosis, and Alzheimer’s disease [96, 97]. Further,
inflammasomes have been reported to promote host defense responses and aid in
pathogen clearance in infectious diseases [9, 11, 70]. Thus, NLRP3 inflammasomes
respond to various pathogens such as bacteria, viruses, and fungi by secreting
IL-1
As mentioned above, trained immunity promotes the host’s defense response and aids in pathogen clearance in infectious diseases [13]. Additionally, well-regulated inflammasome activation in early immune response enhances the development of acquired/adaptive immunity, greatly increasing the host’s defense capability against infectious diseases [98]. However, excessive inflammasome activation has detrimental effects on the host [99], and the impact on the host defense capabilities varies depending on the type of pathogen and inflammasome sensor protein [98, 100]. Therefore, utilizing trained immunity through inflammasome regulation requires a meticulous approach. Further research on enhancing the defense against infectious diseases through the regulation of inflammasome activation and trained immunity is necessary.
Due to changes in modern lifestyle [10], the prevalence of inflammatory and autoimmune diseases has been steadily increasing, and consequently, the demand for treatment of these diseases is also on the rise [101]. Inflammasomes and trained immunity could be good targets for inflammatory and autoimmune diseases because they regulate the host’s response in inflammatory diseases and affect the development and progression of diseases such as atherosclerosis, rheumatoid arthritis, and neurodegenerative diseases (i.e., Alzheimer’s and Parkinson’s diseases) [70, 102, 103]. Therefore, research on the interaction between trained immunity and inflammasome regulation could provide new insights into the development of effective treatments for inflammatory and autoimmune diseases.
This review provides a comprehensive overview of the current research trends and knowledge on the role of NLRP3 inflammasomes in trained immunity (Fig. 1). However, many unresolved issues remain, and more research is needed to elucidate the relationship between trained immunity and inflammasomes. Specifically, the role of the potassium ion efflux as a major activation pathway for NLRP3 inflammasomes in trained immunity, the impact of inflammasomes other than NLRP3 on trained immunity, how trained immunity is induced and regulated in humans, how the danger signal (i.e., LAMPs) resulting from altered lifestyles affect inflammasomes and trained immunity, and how the interaction between trained immunity and inflammasomes can be clinically utilized are still questions that remain unanswered.
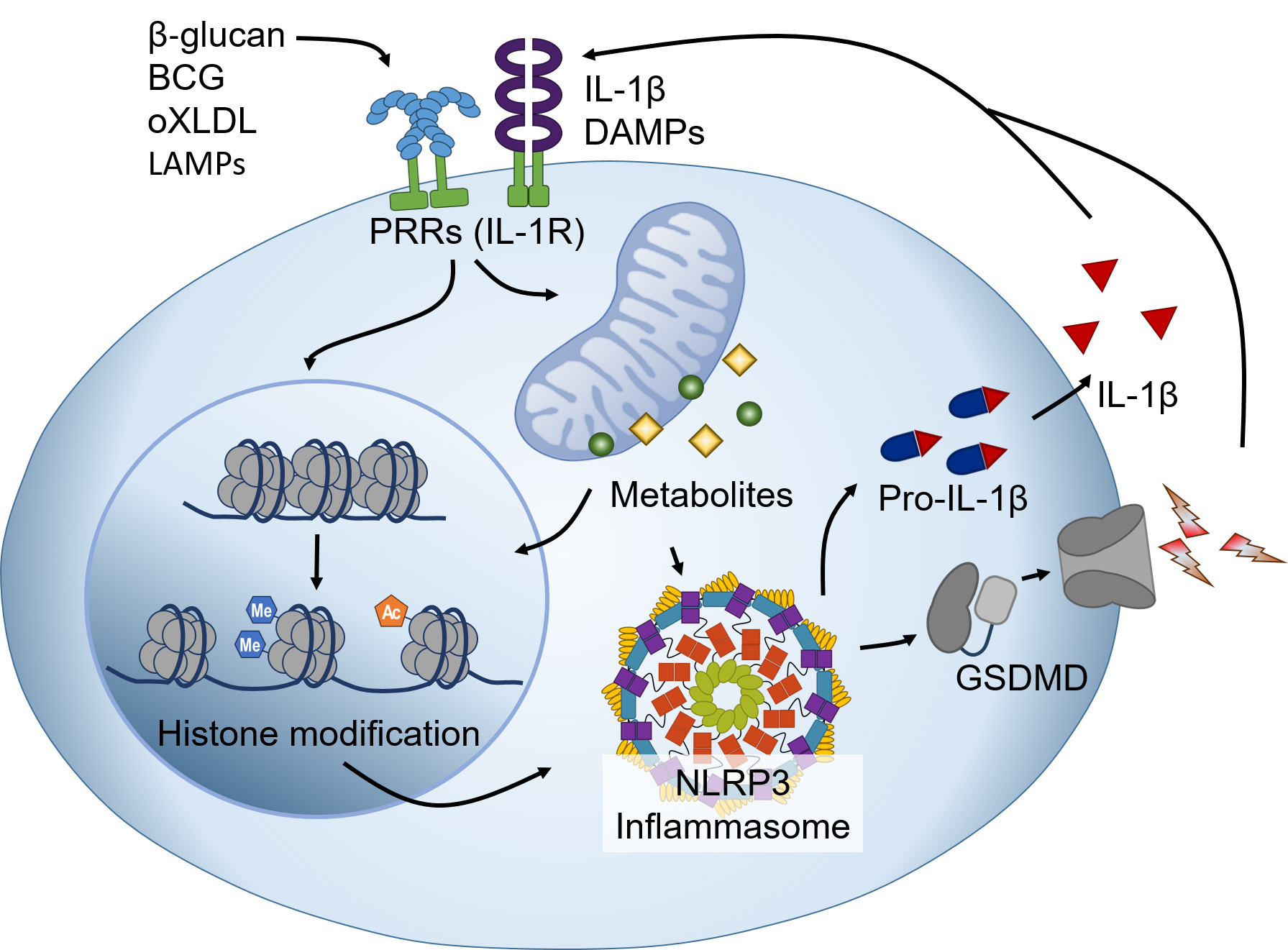 Fig. 1.
Fig. 1.Interactions of trained immunity and the NLRP3 inflammasome.
The stimuli that trigger trained immunity (e.g.,
Studies on these aspects will help to deepen our understanding of the function and regulation of trained immunity and inflammasomes and to develop new strategies for the prevention and treatment of inflammatory and infectious diseases. Trained immunity and inflammasomes are important components of the innate immune system at the intersection of immunology and metabolism and are worth exploring further.
GL: Conceptualization, Writing - Original Draft. HA: Conceptualization, Review & Editing. EL: Conceptualization, Review & Editing. GSL: Conceptualization, Writing - Original Draft, Review & Editing, Supervision. All authors have read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (No. RS-2023-00208354 and RS-2023-00244078).
The authors declare no conflict of interest. The Guest Editor, Geun-Shik Lee has not been involved in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Amedeo Amedei.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.