
- Academic Editor
Ischemic stroke (IS) is the predominant cause of morbidity and mortality worldwide. Ferroptosis, a new type of programmed cell death, has been shown to play a crucial role in IS pathogenesis. Traditionally, research focused on neurons did not uncover specific positive results for IS. However, glial cells have recently received interest as promising targets for IS treatment, not only for their structural function but also in the iron transfer between glia and neurons, which indicates a promising glia–neuron crosstalk in mediating the IS process and ischemia/reperfusion-associated neuropathology, showing their affiliation with ferroptosis. This review addresses the major phenomena of iron metabolism and the process and regulation of ferroptosis, with a particular focus on their impact on IS pathology. The review discusses iron homeostasis, the biology of reactive oxygen species, and lipid peroxidation for modulating the process of IS-induced ferroptosis in different glial cells. We then review recent therapies that leverage ferroptosis modulation for the treatment of IS. Extensive preclinical and clinical research is necessary to fully understand the roles of glia–neuron crosstalk and ferroptosis in IS.
Stroke is a predominant cause of disability and death worldwide [1]. Of all stroke types, the most common is ischemic stroke (IS) and evidence based treatments such as intravenous thrombolysis with recombinant tissue plasminogen activator and endovascular thrombectomy are the most effective treatments, currently with a low proportion of eligible patients and a short time window [2]. Hence, more effective targets for IS treatment are required and need further investigation. Potential mechanisms of IS pathogenesis have been gradually uncovered, including neuroinflammation, metal ion imbalance, mitochondrial dysfunction, oxidative injury, and lipid peroxidation [3, 4]. Although research focused on neurons has not presented a high rate of positive results for IS, glia cells have recently received interest as promising therapeutic effectors for IS treatments. Activated glia have multiple functions because they can release neurotrophic factors and proinflammatory cytokines and phagocytose with different pathways during the onset and progression of IS [5]. Clinical studies have reported that high serum ferritin levels result in a poor IS prognosis [6, 7, 8, 9]. Notably, ferroptosis is an iron-dependent programmed cell death (PCD) and have been demonstrated to play a role in regulating iron metabolism and lipid peroxidation. Recently, researchers have proven that suppressing ferroptosis reverses ischemic injuries [10, 11, 12]. In this review, we discuss iron homeostasis, the biology of reactive oxygen species (ROS), and lipid peroxidation for modulating IS-induced ferroptosis. We also address iron metabolism, the mechanism of ferroptosis in glial cells and neurons, and their impact on IS pathologies. Lastly, we review recent therapies that focus on ferroptosis for IS treatment.
Ferroptosis is a regulated and iron-dependent form of PCD that is distinct from apoptosis, necroptosis, autophagy, and other forms of cell death [13]. With respect to its morphology, ferroptosis targets mitochondria, which present a small volume, increases membrane density, reduces or vanishes crista, and thus ruptures the outer membrane while the nuclei structure stays intact [13, 14, 15]. By contrast, margination, condensation, and fragmentation of chromatin, as well as the generation of apoptotic bodies and plasma membrane blebbing, are typically involved in apoptosis [16]. The characteristics of necroptosis are membrane rupture and the production of membrane bubbles, which are stained positive for annexin V [17]. As another form of PCD, autophagy is characterized by the de novo synthesis of cellular components such as macroproteins or organelles sequestered into a double membrane and fused with a lysosome for digestion and recycling [18]. These characteristics are not present in ferroptosis, which has unique morphological characteristics.
Iron accumulation and excessive lipid peroxidation are the primary bioenergetic features of ferroptosis (Fig. 1). It has been demonstrated that three major mechanisms may control ferroptosis: iron regulation, ROS biology, and metabolism [19].
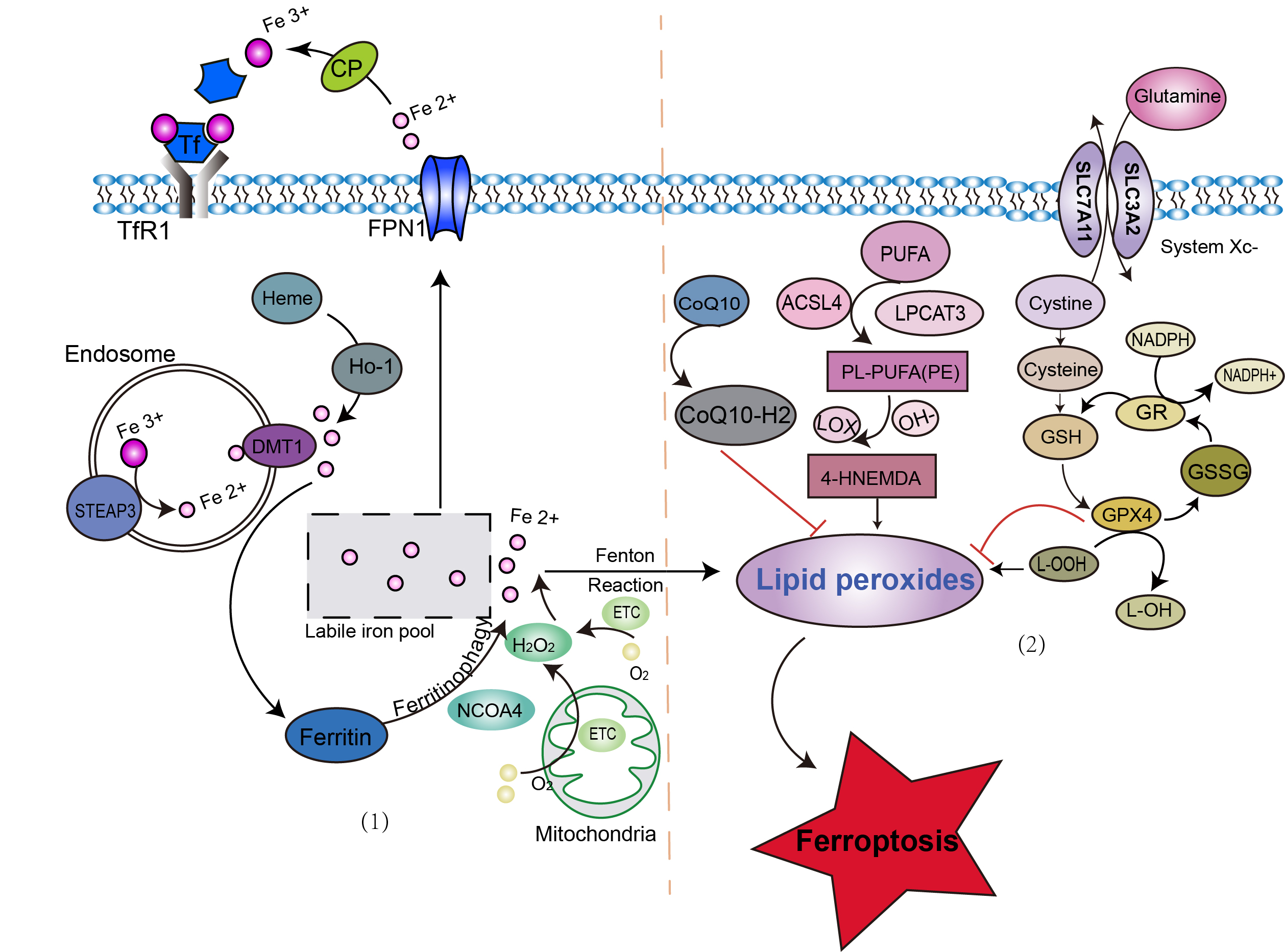 Fig. 1.
Fig. 1.Possible signaling pathway of ferroptosis. Ferroptosis occurs in two major pathways: (1) through the accumulation of ferrous ions via iron-transport molecules, such as transferrin, transferrin receptor, and ferroprotein; and (2) through the accumulation of lipid peroxides. ACSL4, acyl-CoA synthetase long-chain family member 4; CoQ10, coenzyme Q10; CP, ceruloplasmin; DMT1, divalent metal transporter 1; ETC, electron transport chain; FPN1, ferroportin 1; GPX4, glutathione peroxidase 4; GR, glutathione reductase; GSH, glutathione; GSSG, glutathione disulfide; HNE, hydroxynonenal; HO-1, heme oxygenase 1; LOX, lipoxygenase; LPCAT3, lysophosphatidylcholine acyltransferase 3; MDA, malondialdehyde; NCOA4, nuclear receptor co-activator 4; PUFA, polyunsaturated fatty acids; Se, selenium; SLC3A2, subunit solute carrier family 3 member 2; SLC7A11, solute carrier family 7 member 11; STEAP3, six-transmembrane epithelial antigen of prostate 3; Tf, transferrin; TfR1, transferrin receptor 1.
Although the exact molecular event of neuronal ferroptosis remains obscure, the contribution of glial cells to mediate iron-induced toxicity to neurons has been uncovered. It is very well known that neuroglia contributes to the maintenance of brain iron metabolism and physiological function. Moreover, glia express and distribute almost all iron transporters and various iron metabolism proteins. Accumulated studies have identified glia as essential for regulating the ferroptosis of neuronal death [20, 21].
Ferroptosis is involved in the pathological process of IS, which is a novel type of cell death after ischemia onset [22]. Table 1 (Ref. [12, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35]) presents several targets of ferroptosis genes that are associated with IS. In addition, the characteristics of ferroptosis are highly consistent with the pathological changes in patients with IS. For instance, the prognosis of IS is worse in older patients, who have cerebral iron deposition, indicating that iron accumulation may exacerbate ferroptosis of neurons after IS injury [6]. Moreover, an association between high ferritin levels and poorI S prognosis has been found [7]. Gill et al. [8] reported that according to observational studies, a higher transferrin level was associated with a decreased stroke risk. It was also shown that the level of the iron-regulatory hormone hepcidin was significantly higher in patients with IS [9]. Additionally, ferroptosis occurred in different IS animal and cellular models, such as middle cerebral artery occlusion/reperfusion (MCAO/R) rats and PC12 cells treated with oxygen glucose deprivation and reperfusion (OGD/R), including iron accumulation [23, 36], glutathione (GSH) [24, 37, 38], lipid peroxidation [25, 26, 39], oxidative stress [27, 40], and coenzyme Q10 (CoQ10) [41]. Edaravone, as the only drug among ferroptosis inhibitors, has been approved for acute IS therapy [42, 43] and demonstrated to suppress ferroptosis in the ischemic cerebral area. Other drugs were only found to be effective preclinically, such as deferoxamine [44] and selenium [11], but clinical tests are still needed. Although evidence indicates that iron homeostasis and ferroptosis play a prominent role in mediating neuronal death in IS, how these mechanisms occur at each stage of stroke remains largely unknown. Research on the biological processes targeting iron accumulation and associated cell death is urgently needed.
| Gene | Function | Model | Refs |
| PVT1 | PVT1 mediates ferroptosis via miR-214 and by regulating p53 and TfR1 levels. | Patients with AIS and OGD/R PC12 cells | [23] |
| ACSL4 | ACSL4 promotes ferroptosis via enhancing lipid peroxidation and contributes to microglia-mediated inflammation. | MCAO | [25] |
| Inhibiting ACSL4 upregulates GPX4 expression in ischemic stroke, suppresses iron accumulation, and decreases GPX activity to inhibit ferroptosis. | MCAO | [12] | |
| NCOA4 | The NCOA4 deletion abrogates ischemic stroke-induced ferritinophagy and suppresses ferroptosis both in vivo and in vitro. | MCAO and OGD/R | [28] |
| Tau | Tau promotes iron export to stabilize Fpn, which results in iron accumulation. | MCAO | [29] |
| 9a | 9a blocks ferroptosis via disrupting the NCOA4-FTH1 protein-protein interaction. | MCAO | [30] |
| PGE2 | PGE2 inhibits Fe |
Patients with cranial hypertension and MCAO. | [31] |
| LncRNA Meg3 | Treatment with Meg3-siRNA inhibits ferroptotic events through regulating GPX4 expression at ischemic onset. | OGD/R RBMVECs | [24] |
| cPKCγ | The cPKCγ deletion decreases GPX4, COX2, and ACSL4 expressions. | OGD/R primary cultured cortical neurons | [26] |
| UBIAD1 | UBIAD1 significantly suppresses ferroptosis via elevating SOD, T-AOC, the production of CoQ10, and eNOS-regulated NO generation. | MCAO and OGD/R. | [27] |
| PTRF | PTRF targets PLA2G4A to promote lipid metabolism and alter mitochondrial bioenergetics and ferroptosis in neurons. | MCAO and OGD/R HT22 and BV2 cells | [32] |
| PMU2 | PMU2 exacerbates neuroinflammation via SLC7A11-dependent inhibition of ferroptosis at ischemic stroke onset. | MCAO/R and cells with OGD/R-induced cortical neuron injury | [33] |
| TLR4 | TLR4 induces neuronal ferroptosis through promoting oxidative stress and neuroinflammation. | HIBD and OGD/R HT22 cells | [34] |
| circ-Carm1 | circ-Carm1 stimulates ferroptosis. | Patients with ACI and OGD/R HT22 cells | [35] |
PVT1, plasmacytoma variant 1; Fpn, ferroportin; GPX4, Glutathione peroxidase 4; NCOA4, nuclear receptor coactivator 4; FTH1, Porcine Ferritin heavy chain; PGE2, prostaglandin E2; RBMVEC, rat brain microvascular endothelial cells; OGD, oxygen-glucose deprivation; cPKCγ, conventional protein kinase cγ; COX2, cyclooxygenase 2; UBIAD1, UbiA prenyltransferase domain containing 1; SOD, superoxide dismutase; CoQ10, coenzyme Q10; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; PTRF, polymerase I and transcript release factor; PMU2, pumilio 2; SIRT1, sirtuin 1; TLR4, toll-like receptor 4; MCAO, middle cerebral artery occlusion and reperfusion; HIBD, hypoxic-ischemic brain damage.
Glial activation is involved in the ischemic cascade reaction of IS through various pathways (Fig. 2). Activation of microglia and astrocytes, which are typically classified into M1/A1 and M2/A2 phenotypes, is one of the earliest events after brain ischemia [45, 46, 47]. M1/A1 and M2/A2 release pro- and anti-inflammatory cytokines, respectively, to present neurotoxic and neuroprotective functions after brain ischemia [47, 48]. Oligodendrocytes are known to produce superoxide radicals, lipid peroxidation, and iron oxidation at ischemia onset [49]. Different types of glia may have effects on IS pathogenesis via various pathways, and glial activation may also affect neuronal ferroptosis via iron homeostasis, lipid metabolism, and oxidative damage.
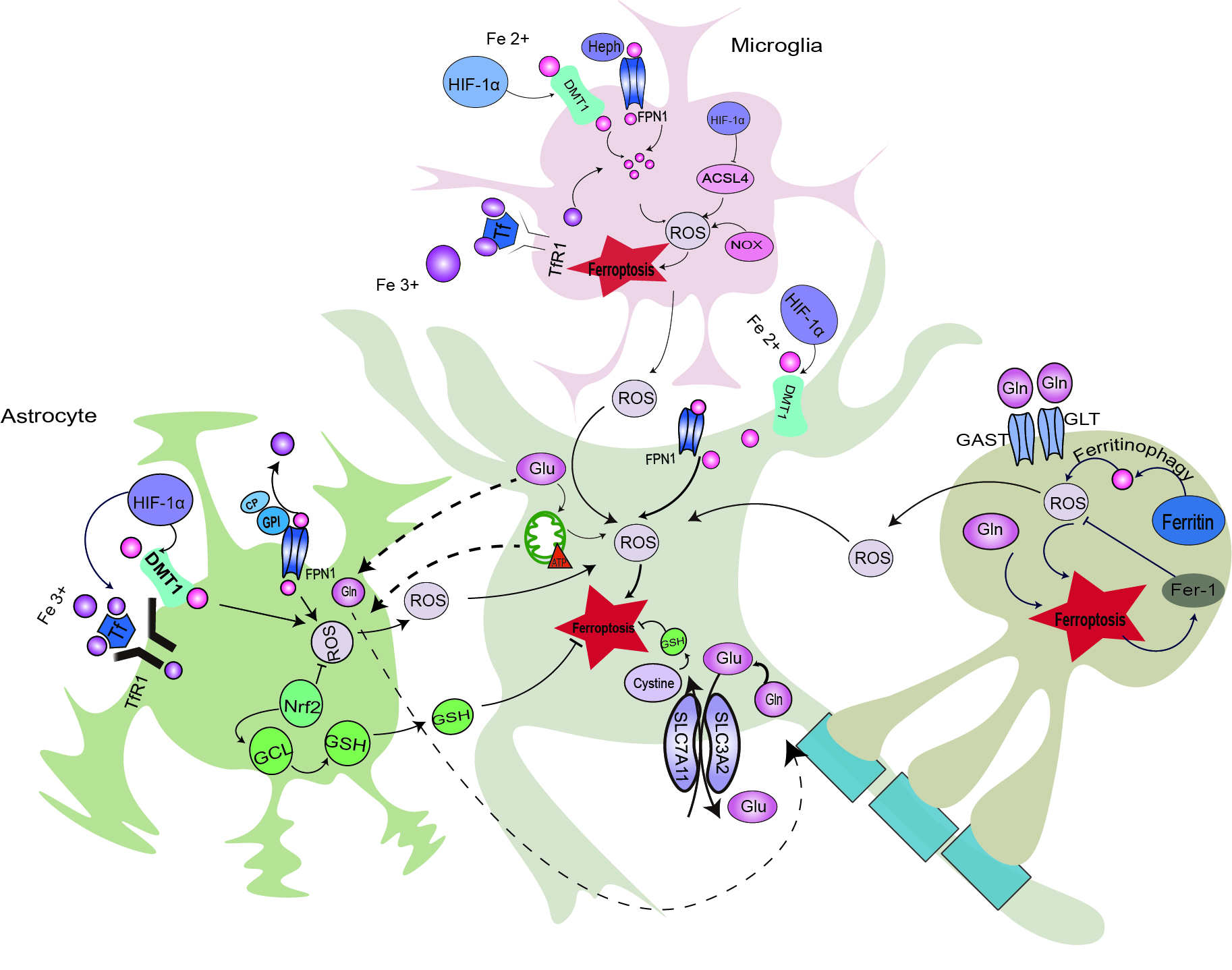 Fig. 2.
Fig. 2.Possible neuron–glia crosstalk in ischemia-induced
ferroptosis. HIF-1
It is known that iron can cross the blood-brain barrier (BBB) and be released into the brain’s interstitial fluid. Neurons and glial cells can absorb and unevenly distribute iron [50]. Studies have confirmed that almost all the iron transporters and proteins involved in iron metabolism are expressed in glia [51, 52, 53]. It has been reported that iron retention is much higher in microglia than in neurons [54]. Hence, glial cells are critical to maintaining iron homeostasis and physiological and pathological function in the brain. In addition, ferritin is directly driven by hypoxia [55], indicating that glial cells have effects on IS via regulating iron metabolism.
Microglia serve a key role in the regulation of iron homeostasis in the central nervous system (CNS) [56]. Intuitively, iron homeostasis relies on the expression of iron-uptake and iron-release proteins on the cellular membrane. The mechanism of iron uptake in microglia has been uncovered, and microglial iron accumulation occurs in cerebral ischemia. It was found that microglia are able to take up non-transferrin-bound iron (NTBI) via a transferrin-independent mechanism [57, 58]. Moreover, transferrin receptor 1 (TfR1) has been identified in microglia both in vivo and in vitro [59, 60, 61]. It was reported that the expression of TfR1 and intracellular iron are both upregulated in hypoxia-induced microglia [60]. Divalent metal transporter 1 (DMT1), a pivotal protein for both transferrin-dependent and transferrin-independent iron uptake, has been identified in microglia [56, 62, 63, 64]. Hypoxia-inducible factor-1 (HIF-1) is known as a critical regulator, which is responsible for the facilitation, adaptation, and survival of cells from normoxia to hypoxia [65, 66]. Qian et al. [67] demonstrated that DMT1 is a target gene of HIF-1 and that silencing HIF-1 down-regulates DMT1 expression and ferrous uptake. Hence, the identification of TfR1, DMT1, and iron content in hypoxia indicates that microglia might take up transferrin-Fe via NTBI and TfR1/DMT1 pathways and might be regulated in hypoxia. Ferroportin 1 (Fpn1) is currently the only known microglial iron exporter [68]. It was also verified that Hephaestin (Heph), which is a critical ferroxidase, is expressed in microglia [69]. Yang et al. [70] reported that the levels of Fpn1 and Heph proteins were downregulated in MCAO models. Another study found that ischemic onset significantly upregulated the levels of the iron-import proteins TfR1 and DMT1 and decreased the expression of Fpn1 in reactive microglia, also indicating that iron deposition occurred in activated microglia after IS [71]. Thus, Fpn1/Heph may be the pathway of iron efflux in physiological and ischemia-induced microglia. Moreover, accumulated iron in hypoxia-induced microglia can stimulate ROS and modulate inflammatory processes via the generation of pro-inflammatory and anti-inflammatory cytokines [72]. In hypoxic-ischemic conditions, an excessive gliosis process can result in tertiary cerebral injury, and astrocyte cytotoxicity would be induced by microglial signaling, while the regular microglial response promotes the regeneration process. On the other hand, studies suggest that microglia activation prevents excessive iron accumulation in response to hypoxic damage [73, 74]. It was reported that excessively retained microglial iron is a key mediator of oligodendrocyte cytotoxicity in periventricular white matter following hypoxia [74]. Taken together, activated microglia play a crucial role in physiological and cerebral ischemia via regulating iron metabolism, and this has been partially corroborated, implying that a treatment strategy targeting microglial iron metabolism and ferroptosis modulation may have potential therapeutic value in treating IS.
Astrocytes, one of the most important components of the BBB, influence the
regulation of iron homeostasis in the cerebral area [75, 76]. Studies have shown
that astrocytes are responsible for acquiring nutrients such as iron from the
circulating blood [77]. Astrocytes are also able to transport various types of
iron, such as Tf-Fe [78], NTBI [79], and heme [80], through coupling with
proteins such as ceruloplasmin (CP), which is known as the form of
glycosylphosphatidylinositol-anchored proteins in the astrocyte membrane [81, 82, 83].
In addition, astrocytes can take up iron via the TfR1/DMT1 pathway, and it is
transferred to proteins, including Fpn1 and CP [84]. Moreover, findings suggest
that iron effluxes from astrocytes via the Fpn1/CP pathway [85]. CP is highly
expressed in astrocytes and suppresses iron-induced lipid peroxidation [86].
Patients with aceruloplasminemia present serious iron accumulation in astrocytes
and iron deposition, which is also observed in the astrocytes of mice
consistently lacking CP [87, 88]. Ryan et al. [89] reported that the CP
level was significantly upregulated in permanent MCAO models. Conversely, loss of
CP dilated the lesion size and aggravated neurological function recovery with a
decline in ferroportin and a raise in DMT1 proteins, indicating the
neuroprotective effects of CP in IS via iron regulation in astrocytes [89]. In
addition, hypoxia can stimulate the expression of HIF-1
Oligodendrocytes, a myelin-forming cell type in the cerebral area, have high iron requirements for normal function. Moreover, they require two- to three-fold higher energy levels than other cell types in the CNS during myelination. Oligodendrocytes also synthesize cholesterol, which is a highly metabolically demanding process and makes them vulnerable to hypoperfusion. The white matter, which is comprised of critical axonal projections, is especially vulnerable to ischemia and has attracted attention lately. About 95% of patients with IS suffer from white matter injury, accounting for half of the stroke total mean infarct volume [91]. It has been considered that white matter injury after stroke is highly correlated with axon degeneration, oligodendrocyte damage, and nerve demyelination [92]. A recent study showed that ischemia increases intracellular iron uptake with the upregulation of oligodendrocytes and myelin markers (markers of oligodendrocyte differentiation [MBP], oligodendrocyte-specific protein [OSP], and helix-loop-helix transcription factor) during the recovery phase of IS [93]. Michalski et al. [94] reported that OSP and MBP display early expression after the ischemic onset and promote protective and regenerative reactions in IS pathogenesis. Therefore, oligodendrocytes might have effects on white matter injury, such as myelination, after IS via iron regulation.
It is known that ferroptosis is ultimately driven by oxidative stress [19]. After IS, the damage-associated molecular patterns are activated, inducing a sequence of pathological changes such as oxidative stress [95]. Oxidative stress refers to the relative excess of ROS induced by disrupting the balance between ROS production and depletion. Recent studies have shown that glial cells are involved in ROS regulation, which might further induce ferroptosis in IS [96].
Activated microglia are frequently present during oxidative stress and the inflammatory processes of IS [97]. Specifically, they are closely connected to an excessive ROS response in the early stage of IS [98, 99]. NADPH oxidase (NOX), which is known as a primary source of ROS production, has been consistently reported to participate in the pathogenesis of cerebral ischemic injury [100]. Recently, NOX was also identified as a biomarker predicting ferroptosis via GPX4-independent control of the lipid ROS accumulation system [101, 102]. During ischemic conditions, NOX-mediated ROS production is reported to occur in microglia, and targeting NOX-dependent ROS generation suppresses microglial activation and polarization and ferroptosis [103]. Peroxiredoxin-1 (Prdx1) is known as one of the typical antioxidant enzymes, and it can inhibit ROS deposition and keep the reduction-oxidation status in both normal oxidative metabolism and oxidative stress [104, 105]. In a recent study, Kim et al. [106] identified stroke-associated microglia (SAM) and showed that Prdx1 deficiency decreased the extent of SAM in IS and exacerbated cerebral injury, which indicates that SAM might have potential antioxidant effects on stroke-induced microglia. Under neuroinflammatory conditions, reduced ROS levels and fewer apoptotic neurons present in CX3CR1-deficient MCAO mice suppressed the proliferation and regulated the polarization of microglia, which inhibited the process of releasing inflammatory cytokines to improve the ischemic brain injury outcome [107]. Consistently, inhibiting the production of microglial proinflammatory cytokines and lipid peroxidation via silencing acyl-CoA synthetase long-chain family member 4 (ACSL4) in IS has proved to inhibit ferroptosis in neurons [108].
Inhibiting excessive ROS production after IS has been proposed as the most effective strategy to improve the prognosis outcome. Elastin-derived peptides are shown as the scavengers of free radicals via the induced activities of antioxidant enzymes, which are detectable in the cerebrospinal fluid of patients after IS [109, 110]. In mouse astrocytes induced by OGD, the Val-Gly-Val-Ala-Pro-Gly (VGVAPG) peptide upregulated nitric oxide release during ROS accumulation [111]. However, the effects were reversed by decreasing ROS production when knocking down the galactosidase beta 1 gene, which indicates that the VGVAPG peptide has pro-survival effects on activating astrocytes via mediating ROS regulation [111].
The functional impairment of mitochondria, resulting in increased ROS deposition
and oxidative stress in cerebral ischemia, has been shown to initiate and enhance
the vulnerability of cells to ferroptosis [19, 112]. Some important modulators,
such as triiodo-l-thyronine and inositol trisphosphate receptor type 2, which can
promote astrocyte mitochondrial adenosine triphosphate (ATP) production and the
disruption of astrocytic Ca
Oligodendrocytes and myelin damage cause the functional impairment in IS.
Glutamate, which has been recently proposed to promote ferroptosis [115], also
contributes to oligodendrocyte excitotoxicity and axonal dysfunction during IS
onset [116, 117]. In addition, oligodendrocytes are known to express purinergic
receptors, and ATP release and P2X7 receptor activation result in white matter
injury in ischemia [118, 119]. Ca
After IS, iron accumulation and ROS deposition can further contribute to lipid peroxidation via the Fenton reaction, which has been proposed as the vital lethal factor inducing ferroptosis [122]. Studies focusing on the suppression of lipid peroxidation-induced ferroptosis might provide a new target for IS therapies [29, 123, 124]. Recently, it has been reported that glial lipid metabolism is specially connected to cerebral function, implying that lipid metabolism-derived ferroptosis might be involved in glia after ischemia onset.
Evidence has suggested that fatty acids (FAs) have a close relationship with microglia [125]. Basically, FAs are present in high amounts in the cellular membranes of microglia, which can further be metabolized into bioactive derivatives to mediate the localization and function of proteins and downstream signaling pathways [126]. Microglia can also express various lipid-sensitive receptors, including toll-like receptors, triggering receptors expressed on myeloid cells 2 and receptors for FA derivatives [127]. In addition, it has been shown that FAs can be stored within lipid droplets (LDs) in microglia to regulate the neuroinflammatory response [128]. Saturated, monounsaturated, and polyunsaturated fatty acids (PUFA) are the three main families of FAs, all of which have shown strong effects on microglial cells. Microglia-activated phorbol myristate acetate treatment induces iron accumulation and leads to iron-dependent lipid peroxidation [129]. Moreover, inducible nitric oxide synthase is greatly increased to inhibit ferroptosis by decreasing arachidinate 15-lipoxygenase activity in microglia [130]. These studies indicate that microglia activation may be involved in ferroptosis via inducing lipid peroxidation.
ACSL4 is an important isozyme for the metabolism of PUFA, which results in ferroptosis-mediated cerebral damage at ischemia onset when activated [25, 131]. Cui et al. [25] reported that the ACSL4 level is inhibited in the early phase of IS through hypoxia-inducible factor 1 alpha. They demonstrated that ACSL4 aggravated neuronal death by exacerbating lipid peroxidation-derived ferroptosis and promoting inflammatory cytokine production in microglia simultaneously [25]. Hence, inhibiting microglial lipid peroxidation and inflammatory-induced ferroptosis may be therapeutic targets in treating IS.
Lipids play a fundamental role in astrocyte function, such as energy generation, membrane fluidity, and cell-to-cell signaling [132]. It has been reported that reactive astrocytes drive the death of neurons via lipoparticle secretion [133]. Acrolein, a neurotoxin that is endogenously produced by lipid peroxidation during ischemic onset, induces astrocytic inflammation through the NLR family pyrin domain-containing protein 3 inflammasome [134, 135], suggesting that astrocytes might have neurotoxic effects on neurons via lipid peroxidation during IS.
However, studies have illustrated that the crosstalk between astrocytes and neurons might protect neurons from lipid peroxidation at ischemia onset. LDs are one of the fundamental components of lipid metabolism in astrocytes, thus playing a critical role in maintaining astrocyte–neuron homeostasis [136]. Lipid particle-mediated FA transfer is involved in the deposition of astrocytic LDs after acute stroke and protects neurons from lipid peroxidation [137]. In the acute IS model, omega-3 PUFA present a high neuroprotective and anti-inflammatory potential via targeting the function of astrocytes to alleviate neuronal injury [138]. This indicates that neuron–astrocyte metabolic coupling might suppress ferroptosis via inhibiting lipid peroxidation. Therefore, the dual role of astrocytes in lipid metabolism during IS pathogenesis needs to be investigated.
Oligodendrocytes enwrap CNS axons with myelin, which has an extremely high lipid content and a peculiar lipid composition with a ratio of 2:2:1 cholesterol:phospholipid:glycolipid. As previously mentioned, oligodendrocytes and myelin injuries underlie the neurological damage in the ischemic cerebrum [139, 140]. ATP-binding cassette transporter A1 (ABCA1) has been shown to be one of the representative genes that play a key role in lipid metabolism when treated with the ferroptosis inducer [141]. Li et al. [142] reported that ABCA1 deficiency decreases myelination and oligodendrogenesis and deteriorates the prognosis after IS, indicating that oligodendrocytes might be involved in ferroptosis via lipid metabolism at ischemia onset, but further investigation is needed.
As we previously discussed, iron can be accumulated in glial cells and neurons. Some special routes have been investigated as iron transporters between cells, which are involved in regulating ferroptosis in the CNS [85]. It has been demonstrated that proteins responsible for iron metabolism can be transported between cells through extracellular vesicles, including exosomes [143, 144]. In addition, TfR has been reported to be transported via tunneling nanotubes, which exist in both glia and neurons [145]. This indicates a promising mode of crosstalk between glial cells and neurons in the IS pathological process.
Ferroptosis is mainly induced by the inactivation of the antioxidant system, including the cystine-glutamate antiporter (system xc-)-GSH-GPX4-dependent antioxidant defense, which results in lipid hydroperoxide deposition [146]. It is known that astrocytes can provide maintenance and energetic support for neurons through mediating oxidative stress via glucose transformation into lactate and GSH production. Mounting evidence has shown that hypoxia promotes ferroptotic cell death, which is highly connected to excessive ROS deposition by mitochondrial complex III, and enhances lipid peroxidation directly via damaging mitochondrial membranes, resulting in the elevation of hydrogen peroxide levels to initiate and aggravate Fenton reactions and/or acting through the suppression of cellular antioxidant capacity. It is known that neurons receive a continuous supply of glucose and oxygen via communication with astrocytes, indicating that astrocyte–neuron crosstalk might be indispensable for neuronal defense against oxidative stress-induced ferroptosis after ischemic onset. It is known that neurons can release damaged mitochondria and transfer them to astrocytes for disposal and recycling [147]. Notably, Hayakawa et al. [148] reported that astrocytes can produce functional extracellular mitochondria to support neuronal viability under IS conditions, indicating a new potential mechanism of neuroglial crosstalk that might be involved in ferroptosis after stroke.
In 2017, Tuo et al. [29] first reported that ferroptosis is a form of neuronal death occurring in IS. Interestingly, three hallmarks of ferroptosis—iron accumulation, ROS deposition, and lipid peroxidation—have already been studied as targets for IS treatments for decades. However, only one drug, edaravone, which acts as a ferroptosis inhibitor, has been applied for acute IS therapy [42, 43]. Others have been found to be effective in animal and cell models of IS. Catalysis by 12/15-lipoxygenase (12/15-LOX) of the stereospecific peroxidation of esterified PUFA to promote lipid peroxides and inhibitors of 12/15-LOX have been demonstrated to significantly relieve cerebral injury after IS [149, 150, 151]. Iron chelators, such as ciclopirox, 2,2-bipyridyl, and deferoxamine (DFO), have been extensively investigated for IS treatment [152, 153, 154, 155]. DFO, one of the best-known chelators, is reported to relieve brain damage and facilitate the neuronal recovery process in MCAO models [155]. In addition, iron regulatory proteins, including transferrin and CP, have been found to alleviate functional damage in IS models. Moreover, ferrostatin-1 and liproxstatin-1 can both significantly improve neurological deficits after IS onset in vivo [29]. Selenium-containing compounds have been found to decline infarct volume in the MCAO model [11]. However, with these being preclinical studies, large-scale clinical studies are warranted to examine the use of these ferroptosis inhibitors for IS treatment.
Traditional Chinese therapies have been widely applied for clinical treatments of IS; however, a specific mechanism has yet to be uncovered. Recently, researchers have shown that some traditional Chinese herbs can attenuate IS injuries by inhibiting ferroptosis. Carvacrol can suppress ferroptosis via upregulating GPX4 expression and mitigating lipid peroxidation after IS occurrence [156]. Naotaifang was reported to improve ischemia-induced neural injuries in rats via suppressing ferroptosis through the downregulation of TfR1/DMT1 and the upregulation of SCL7A11/GPX4 pathways [157]. Guan et al. [158] reported that galangin attenuates IR injury by activating SLC7A11 and GPX4 in gerbils to inhibit ferroptosis. Moreover, Carthamin yellow, which is extracted from safflower, has been reported to protect rats against IS via the mitigation of inflammation and ferroptosis [41].
Traditionally, IS research has focused on neurons; however, this has not resulted in a high rate of positive results for IS. As functional and structural supporting cells, neuroglia have received interest and offer promising therapeutic effects for IS treatments. On the other hand, glial cells play an important role in IS-associated neuropathologies, such as inflammation, excitotoxicity, necrotic tissue removal, oxidative stress maintenance, and neurogenesis, all of which have been associated with ferroptosis [5]. Here, we reviewed the mechanisms of iron homeostasis, ROS biology, and lipid peroxidation in different glial cells and the potential effects of neuron–glia crosstalk on IS-induced ferroptosis. However, the investigation of the role of ferroptosis in glial cells after IS and the underlying molecular pathways is still in the early stages, and several unresolved questions need to be answered. First, it is known that IS can induce inflammation, during which glial activation plays an important part. Many studies have shown that ferroptosis can be accompanied by neuroinflammation; however, the mechanistic link between ferroptosis and neuroinflammation in IS is still unclear. Another issue is that the causal relationship between iron deposition and neuronal death induced by ischemia remains uncovered. The question of whether iron deposits in glia form first and then influence the function of neurons or whether iron deposits in neurons cause glial overactivation so that more iron is transferred to the neurons, inducing neuronal death, needs to be addressed. In addition, the crosstalk between ferroptosis and other types of PCD in glial cells, such as apoptosis, necrosis, and autophagy, remains unclear. Associations between ferroptosis and other cell death pathways have been reported. For example, both necroptosis and ferroptosis contribute to IS [159]. Likewise, some modulators, such as p53, affect both apoptosis and ferroptosis [160]. Whether ferroptosis promotes other types of PCD or whether they can suppress or activate each other in neuroglia requires further investigation. All these questions should be addressed in further experimental and clinical studies to fully elucidate the roles of ferroptosis in neuroglia after IS.
12/15-LOX, 12/15-lipoxygenase; ABCA1, ATP-binding cassette transporter A1; ACI,
Acute cerebral infarction; ACSL4, acyl-CoA synthetase long-chain family member 4; ATP, adenosine triphosphate; BBB,
blood-brain-barrier; cPKC
YJP conceived the review and provided guidance. SYX and SMN wrote the manuscript and collected materials. CLZ was for picture processing. All authors contributed to the final manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This study was funded by National Natural Science Foundation of China (82174484, 81973932), the Peak academic Talent of Jiangsu Hospital of Traditional Chinese Medicine (grant number: K2021RC24), Jiangsu Graduate Practice Innovation Program (grant number: SJCX22_0755).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.