

1 Shenzhen Bay Laboratory, 518132 Shenzhen, Guangdong, China
Abstract
Tumor-associated macrophages (TAMs) are the most abundant infiltrating immune cells in the tumor microenvironment (TME) and play an important role in tumor progression. Clinically, the increase of TAMs infiltration is linked to poor prognosis of patients with various cancer types. Multiple studies have demonstrated that reducing or reprogramming TAMs can inhibit the occurrence or development of tumors. Therefore, TAMs have been identified as novel targets for the treatment of cancer therapy. In this review, the origin, polarization, roles, and targeting of TAMs in malignancies, are discussed.
Keywords
- tumor-associated macrophages
- tumor microenvironment
- macrophage roles
- macrophage targeting
- cancer therapy
The morbidity and mortality of malignant tumors are rising worldwide, threatening human life and health and becoming a leading cause of death [1]. Growing evidence suggests that the tumor microenvironment (TME) has a major role in determining tumor progress, in addition to the aggressive biological behavior of tumor cells [2]. The TME, which is made up of blood vessels, immune cells, fibroblasts, inflammatory cells generated from bone marrow, different signaling chemicals and extracellular matrix, is a complex microenvironment that surrounds the tumor site [3]. Immunosuppressive TME promotes the proliferation and metastasis of tumor cells [4]. Macrophages are the most abundant immune cells in the TME and play a central regulatory role. Macrophages infiltrating TME are defined as tumor-associated macrophages (TAMs), accounting for approximately 30–50% of total immune cell counts [5]. TAMs have been proven to be related to the occurrence, development, angiogenesis, and metastasis of tumors [6], suggesting that TAMs could be a potential therapeutic target and prognostic biomarker for tumors.
Current antitumor strategies targeting TAMs include inhibiting the recruitment of macrophages, promoting TAMs depletion, regulating its polarization, and enhancing TAM phagocytosis. Targeting TAMs has become one of the main anti-tumor therapeutic strategies. In this review, we attempt to discuss the origins, polarization, roles, and reprogramming of TAMs, as well as the therapeutic implications of targeting TAMs in malignancies.
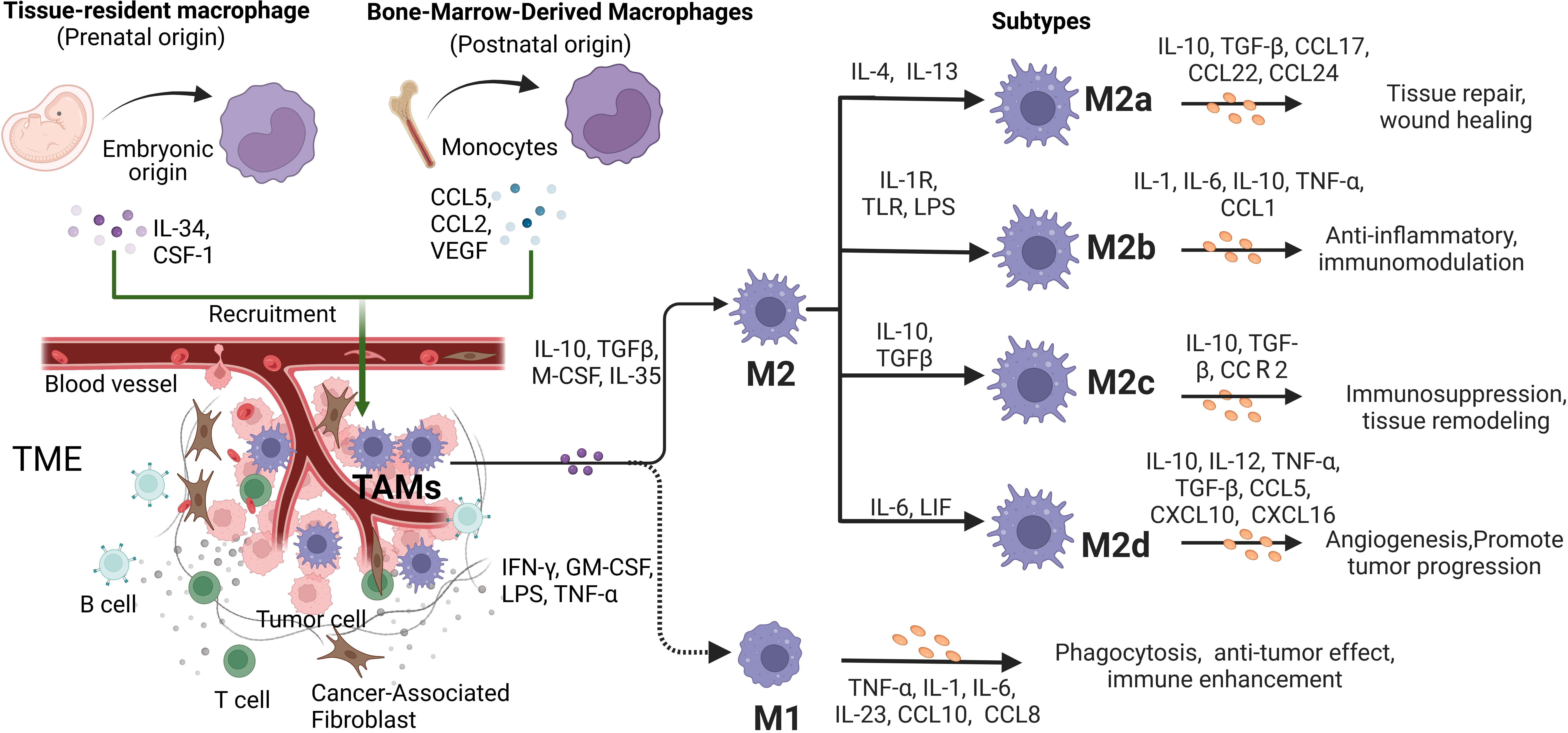
There has been controversy over the exact origins of TAMs. However, with the development of modern lineage-tracing techniques, our understanding of the origins of TAMs has been improved significantly. It is currently believed that TAMs are mainly composed of two parts, namely, tissue-resident macrophages (TRMs) and bone marrow-derived macrophages (BMDMs) (Fig. 1). TRMs originate from the embryonic precursors of yolk sac or fetal liver and persist in specific organs, such as Langerhans cells in the skin, Kupffer cells in the liver, alveolar macrophages, and microglia in the brain [7]. In the early stage of tumorigenesis, resident macrophages from surrounding tissues constitute the initial TAMs inside the tumor, forming a microenvironment conducive to tumor growth. As the tumor grows, various chemokines secreted by stromal and tumor cells in the inflammatory state of the TME induce the recruitment of a large number of BMDMs to form the typical TAMs. Although various types of macrophages coexist in tumors, BMDMs make up the majority of TAMs and its role in different types and stages of tumors needs further study [8].
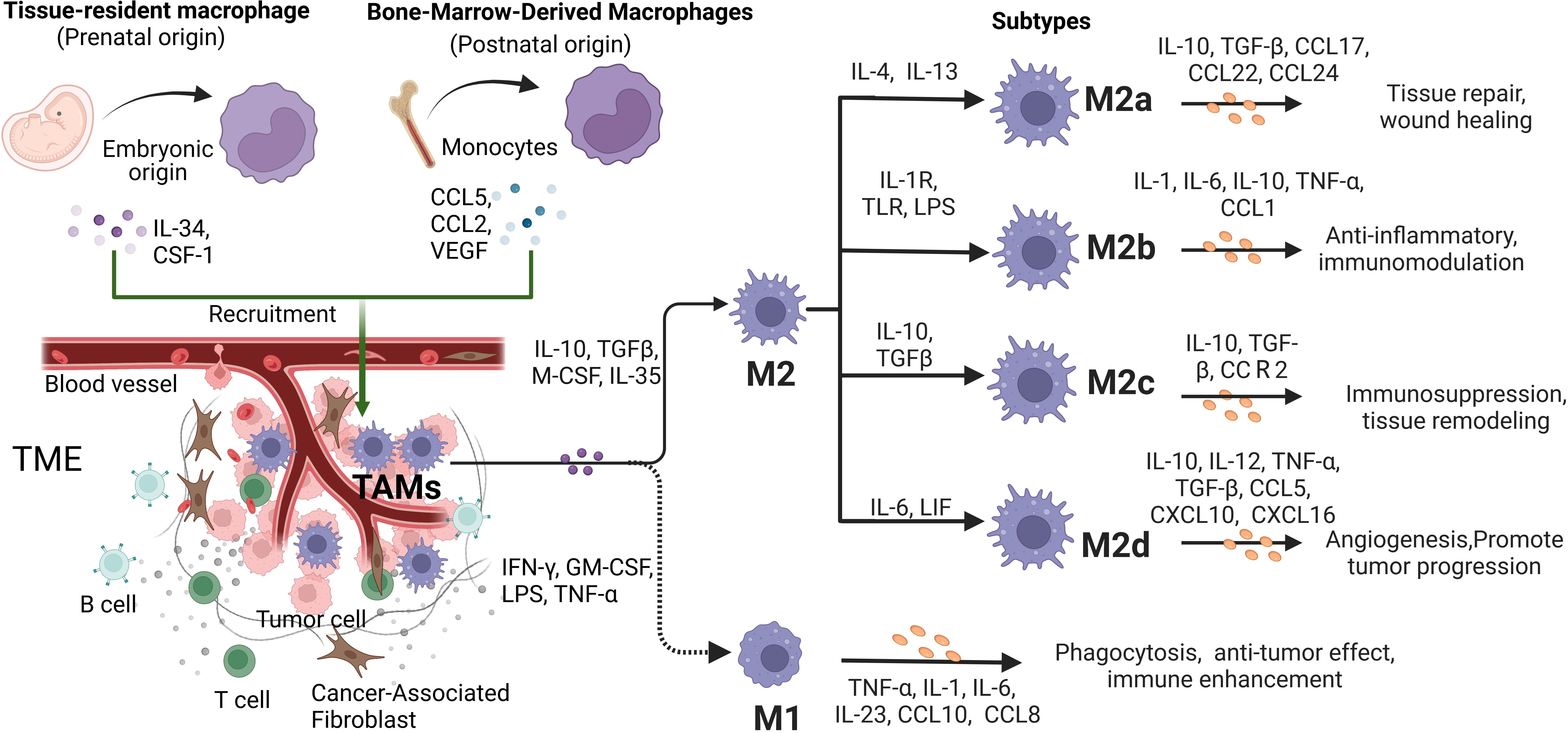 Fig. 1.
Fig. 1.The origins, differentiation and function characteristics of Tumor-associated macrophages (TAMs). TAMs can be developed from embryonic-derived tissue-specific resident macrophages and bone marrow-derived macrophages (BMDMs). As the ratio of cytokine types and concentrations in tumor microenvironment (TME) changes, the activation phenotype of TAMs changes dynamically to play different functional roles. In conventional binary model, macrophages polarize into two subtypes, M1-type and M2-type. In response to stimulation by different factors, M2-type TAMs further polarize to four phenotypes, including M2a, M2b, M2c and M2d. They secrete various factors that have significant impacts on the tumor progression.
TAMs are usually divided into the classically activated type 1 (M1-type)
macrophages, and the alternatively activated type 2 (M2-type) macrophages
according to their secreted products and functions. M1-type TAMs are primarily
responsible for tumor killing and inhibition, while M2-type TAMs are involved in
tumor incidence, development and metastasis [9]. Moreover, M1-type TAMs can be
induced and activated by interferon-
M2-type TAMs can be further divided into types M2a, M2b, M2c, and M2d. Among them, M2a-type TAMs are related to type II inflammation, that is, the Th2 reaction accompanied by the release of IL-4 and IL-13 and an allergic reaction. M2b-type TAMs are involved in Th2 activation and immunomodulation through immune complexes and toll-like receptor ligands. M2c-type TAMs induce immunosuppressive tissue repair through IL-10. M2d-type TAMs are activated by IL-6, which promotes tumor growth through angiogenesis [11] (Fig. 1).
M1-type TAMs exhibit a pro-inflammatory and anti-tumor phenotype, which is
characterized by the expression and production of high levels of pro-inflammatory
cytokines. M2-type TAMs take parts in promoting tumor progression and suppressing
immunity by expressing mannose receptors, CD206, CD163, and arginase, while also
producing IL-10 and TGF-
TAMs have remarkable heterogeneity and plasticity, and they change dynamically
under the stimulation of different signals in a specific microenvironment. TAMs
can be polarized from M1-type to M2-type [15]. At the initial stage of tumor
progression, M1-type is the dominant phenotype. Tumor cells or CD4
Human and mouse TAMs can be identified by cell surface markers, including CD11b,
CD86, CD11c and chemokine (C-C motif) receptor (CCR) 5. As for human M1-type TAMs,
CD14, CD80 and CD68 are used as markers, while M2-type TAMs express CD11b, CD206,
IL-4Ra and CD14. In mice, CD11b, CD86, F4/80, CCR5 and major histocompatibility
complex class II (MHC-II) are used to isolate M1-type TAMs, while M2-type TAMs
can be distinguished by CD11b, CD206, F4/80, IL-4R
TAMs are the largest number of antigen-presenting cells in the TME. M1-type TAMs enhance antigen presentation by increasing MHC-II and costimulatory molecules CD80 and CD86 on the cell surface, while M2-type TAMs decrease T-cell antigen presentation. M1 TAMs phagocytize tumor antigen, and the most characteristic antigen peptides are retained after lysosomal decomposition. The peptides are then combined with MHC-II and presented to T lymphocytes in the form of antigen peptide-MCH-II molecular complex on the surface of M1-type TAMs. When the complex is recognized by T cell receptor (TCR), T lymphocytes are activated [17]. Exhaustion of T cells produced a large number of myeloid-related factors, which recruited monocytes and induced them to differentiate into antigen-presenting TAMs. TAMs continued to stimulate TCR through antigen presentation, thus aggravating the exhaustion of T cells [18].
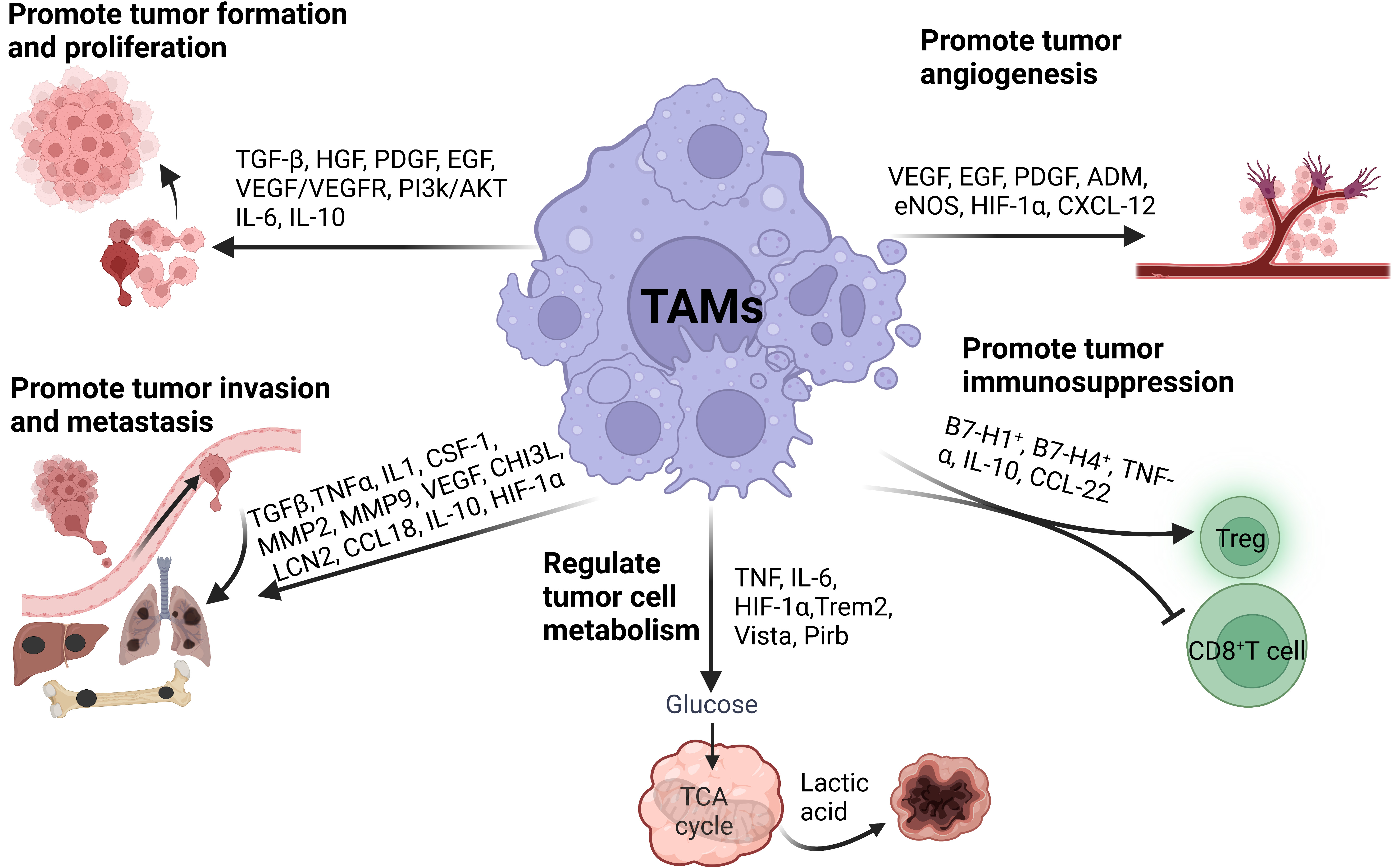
After infiltrating into tumor tissue, macrophages develop a new phenotype, aiding in tumor occurrence and development. TAMs promote tumor cell proliferation, invasion and migration, angiogenesis and immunosuppression and regulate tumor cell metabolism through complex autocrine and paracrine pathways (Fig. 2).
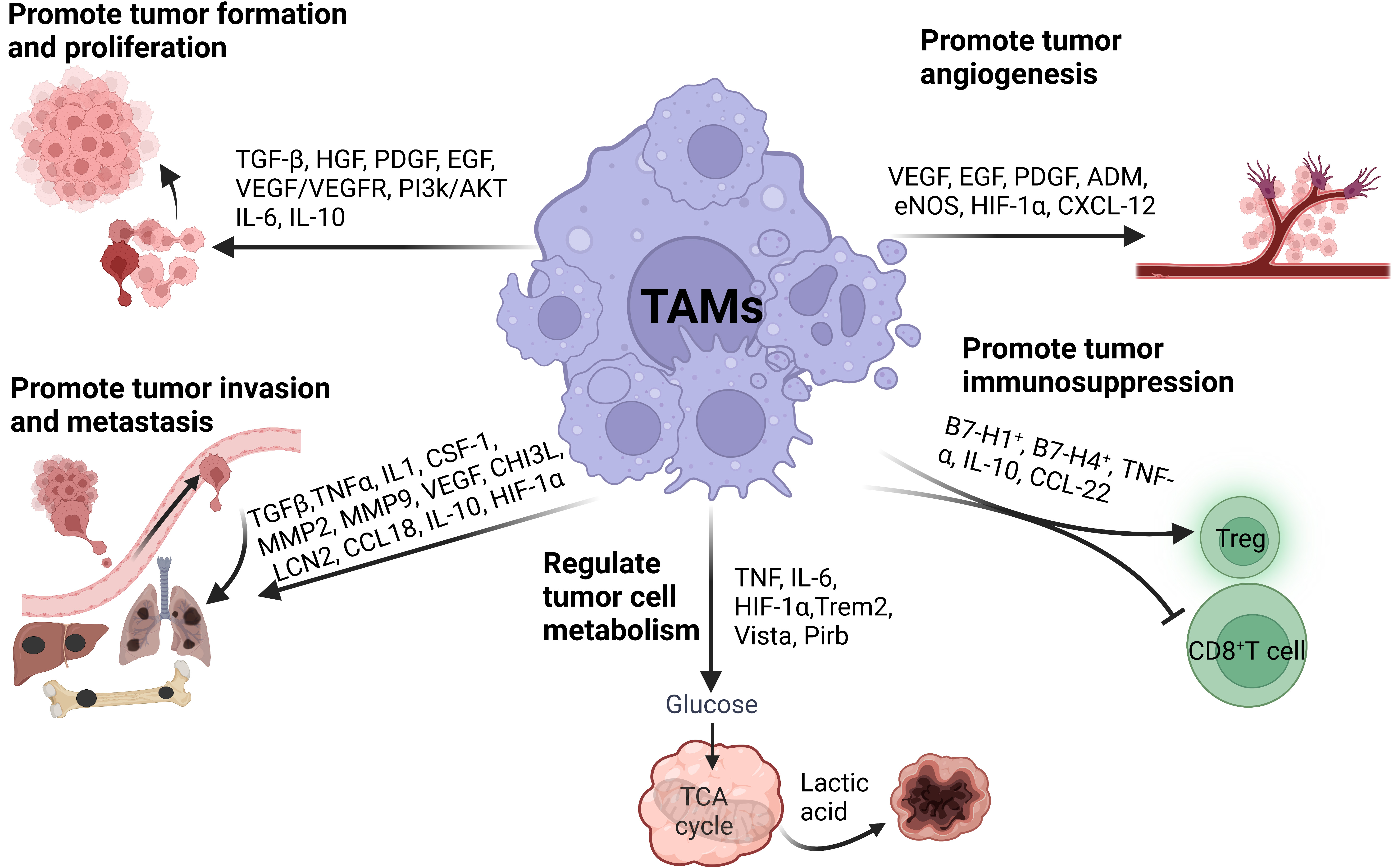 Fig. 2.
Fig. 2.The roles of TAMs in tumor progression. TAMs facilitate tumor progression by promoting tumor cell proliferation, invasion, migration, angiogenesis, immunosuppression, and metabolism.
A large number of animal experiments and clinical studies have proved that all stages of tumorigenesis are affected by tumor-related inflammation. TAMs release a large number of reactive oxygen species and reactive nitrogen mediators, which lead to DNA damage and genome instability and increase the frequency of gene mutation. Accumulatively, the DNA repair function becomes abnormal, thus forming cells with the potential to develop into tumors. If the damage continues, it will further expand the cloning of cells with tumor potential, increase genomic instability and cell atypia, and finally form tumor cells, thus starting tumor occurrence. It was reported that the pro-inflammatory effect of M1-type TAMs increased the genomic instability of malignant tumor cells and became the driving force of tumorigenesis [19].
TAMs are closely related to tumor growth. TAMs stimulate tumor cell
proliferation by interacting with them and secreting cytokines such as epidermal
growth factor (EGF), platelet-derived growth factor (PDGF), TGF-
Tumor cells usually leave the primary site and migrate to other sites. The
ability of tumor cells to spread distantly is dependent on the TME. As the main
component, TAMs play an important role in tumor metastasis. TAMs promote the
invasion and migration of tumor cells by secreting TNF-
TAMs induce epithelial-mesenchymal transition (EMT) in the TME. Breast cancer cells activated macrophages to transform into TAMs through GM-CSF, and TAMs secreted CCL18 to induce EMT and promote tumor metastasis [27]. Experiments in mice in vitro and in vivo showed that TAMs induced EMT in colon cancers by regulating STAT3/miR-506-3p/FoxQ1 axis, and then promoted the production of CCL2 to recruit more TAMs, which significantly increased the rate of tumor invasion and metastasis [28]. Ovarian cancer cells induced macrophages to polarize into M2-type TAMs by secreting macrophage colony-stimulating factor (M-CSF). M2 TAMs induced EMT by releasing CCL18, which further increased M-CSF transcription in ovarian cancer cells through ZEB1 protein. Thus, the CCL18-ZEB1-M-CSF interaction loop between ovarian cancer cells and TAMs increased tumor metastasis [29].
TRMs are also involved in the progression of different tumors and have organ
specificity. At the early stages of NSCLCs, TRMs gathered nearby tumor cells and
promoted tumor invasion and Treg cell reaction. TRMs promoted Treg cell
differentiation by up-regulating the expression of CD73 and cytotoxic
T-lymphocyte-associated protein 4 (CTLA-4) in Treg cells, and protected tumor
cells from being killed by CD8
Microglia, the resident macrophages in the central nervous system, played a similar role in promoting the progress of glioblastoma. When microglia and glioma cells were co-cultured in vitro, microglia lost phagocytosis activity and simultaneously secreted factors such as MMP-9, EGF, IL-6 and VEGF to enhance the invasion of glioblastoma [32].
In one mouse pancreatic cancer model, TRMs were amplified in the tumor tissue. Depletion of pancreatic resident macrophages led to a significant reduction in the tumor load and showed a transcription profile of cancer fibrosis [33].
The foundation of tumor angiogenesis is endothelial cells in the TME, where new blood vessels serve as the primary route for tumor invasion and metastasis as well as supplying nutrients and oxygen for tumor growth. The density of TAMs and tumor blood vascularity are highly correlated in malignancies. TAMs promote the growth of tumor microvessels and lymphatic vessels by secreting VEGF and EGF to accelerate the proliferation of tumor cells (Fig. 2). IL-17 expressed by TAMs promoted angiogenesis in laryngeal squamous cell carcinoma [34]. In the co-culture system of TAMs and melanoma cells, TAMs promoted angiogenesis and melanoma growth by secreting adrenomedullin (ADM), which stimulated the endothelial nitric oxide synthase (eNOS) signal pathway [35]. Under hypoxic conditions, TAMs expressed the transcription factor HIF-1, which induced the transcription of VEGF, PDGF, and EGF and promoted angiogenesis. Furthermore, TAMs secreted MAPK-activated protein kinase-2 in a mice colon cancer model to increase the expression of chemokine (C-X-C motif) ligand 12(CXCL-12) in the tumor, thus promoting tumor progression and angiogenesis [36].
TAMs promote tumor progression through immunosuppression. TAMs are the main
immunoregulatory cells in tumors and participate in the suppression of cytotoxic
T lymphocytes (CTLs) responses in the TME (Fig. 2). Effective antitumor immunity
mainly depends on the activation of CD8
TAMs promote tumor growth by regulating tumor cell metabolism and increasing the aerobic glycolysis (Fig. 2). During tumor growth, the TME presents different degrees of hypoxia. Hypoxia inhibited the uptake of glucose by TAMs, leading to an increase in the content of glucose in the TME, which further increased the utilization of glucose by tumor cells, and ultimately promoted tumor angiogenesis and metastasis [41]. TAMs with tumor-inducing metabolic features, including purine metabolism, amino acid (AA) metabolism, and glycolysis, expressed high levels of pro-angiogenic genes. During the transition from lipid metabolism to purine metabolism, macrophages gradually lost the expression of genes related to antigen presentation, while gained the expression of genes related to angiogenesis and immunosuppression, including Trem2, v-domain Ig suppressor of T cell activation (Vista) and paired immunoglobulin like receptor b (Pirb) [13]. In patients with medullary thyroid carcinoma, lactic acid produced by tumor cells caused the glucose metabolism mode of TAMs to change from oxidative phosphorylation to glycolysis, which led TAMs to secrete more lactic acid, TNF, and IL-6 to further promote tumor development [42].
In TME, the metabolism of TAMs is always in a dynamic process, which plays an important role in tumor development and anti-tumor immune response. Nutrients such as glucose, lipids and amino acids are absorbed by TAMs and catabolized or converted into biosynthetic intermediates or signal metabolites to regulate TAMs function. M1-type TAMs had a high level of aerobic glycolysis activity, which produced reactive oxygen species to kill pathogens. M2-type TAMs relied on oxidative phosphorylation and produced IL-10 and VEGF to promote the growth of malignant tumor cells [43]. In another study, M2-type TAMs expressed high-level glucose transporter and had stronger glucose-uptake capacity than M1-like TAMs. Glucose uptake promoted O-GlcN protein acylation in M2-type TAMs, which further promoted tumor metastasis [44].
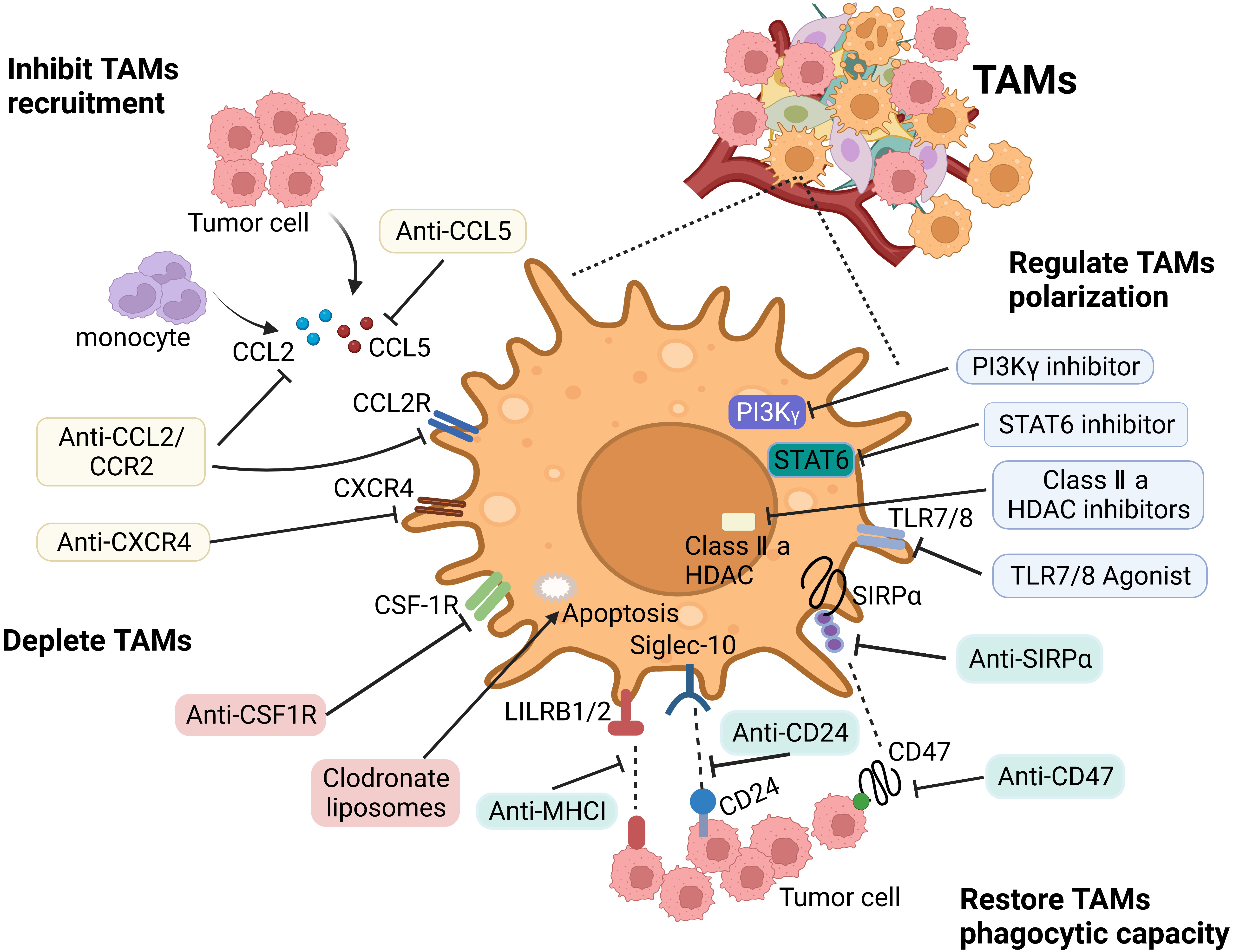
TAMs play an important role in the occurrence and development of tumors. Due to their remarkable heterogeneity and plasticity, the current treatment strategies for TAMs mainly include inhibiting their recruitment, depleting or reprogramming TAMs, and improving their phagocytic ability (Fig. 3).
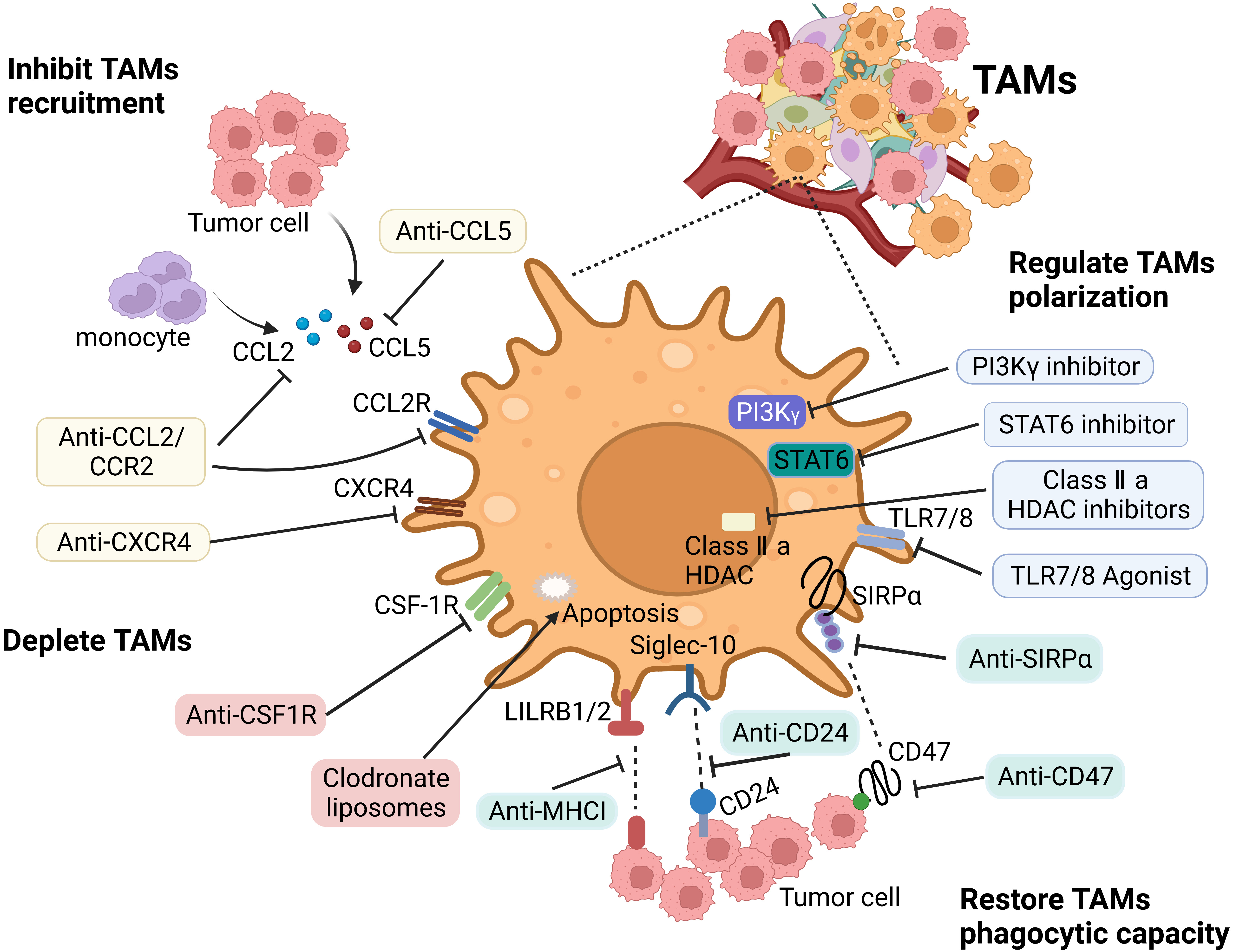 Fig. 3.
Fig. 3.Main therapeutic strategies to target TAMs. The therapeutic
strategies targeting TAMs include: (1) Inhibiting macrophages recruitment via
targeting CCL2/CCR2 axis and CXCL12/CXCR4 pathway. (2) Depleting TAMs by blocking
CSF-1/CSF-1R axis, or inducing their apoptosis by drugs, such as trabectedin and
bisphosphonates. (3) Repolarizing macrophages by anti-CD47 antibodies, CD40
agonists, PI3K
In TME, various chemokines such as CCL2, CCL5, and CSF-1 secreted by tumor cells and stromal cells recruit monocytes circulating in the blood to the tumor area to differentiate into TAMs. Blocking the relevant signaling pathways with small molecule inhibitors or specific antibodies can inhibit the recruitment of TAMs.
In the liver cancer xenotransplantation mice model, the CCR2 antagonist RDC018
blocked the CCL2/CCR2 axis, and inhibited the recruitment of inflammatory
monocytes and the infiltration and M2-type polarization of TAMs. The blockade
resulted in the reversal of the immunosuppressive status of the TME and the
activation of an antitumor CD8
TAMs depletion can reduce the density of TAMs in the tumor tissue and the
immunosuppressive TME. CSF-1 is necessary for the maturation, differentiation,
and survival of the mononuclear phagocytes, which express the CSF-1 receptor
(CSF-1R) exclusively. TAMs were massively apoptotic when the CSF-1/CSF-1R axis
was blocked [50]. Given its importance in macrophages, many clinical drugs
targeting CSF-1R have been developed, such as BLZ945, PLX3397, PLX7486 and
PLX7486. In a mouse model of PDAC, CSF-1 neutralizing antibodies not only reduced
the number of T cells but also reprogramed the remaining T cells to enhance
antigen presentation and anti-tumor responses. The combination of CSF-1
neutralizing antibodies and CTLA-4 antagonists resulted in tumor regression [51].
The monoclonal antibody (RG-7155), which inhibits CSF-1R activation, induced
macrophage apoptosis in vitro, and significantly reduced TAMs density
and increased the ratio of CD8
Another strategy to promote the depletion of TAMs is the application of
bisphosphonates, including zoledronic acid and clodronate. Both in vitro
and in vivo studies have shown that bisphosphonate treatment induced
apoptosis in macrophages. In mouse PDAC model, clodronate liposomes significantly
reduced the density of TAMs and enhanced the anti-tumor effect of CD8
Given the plasticity of macrophages, reprogramming TAMs to an antitumor
phenotype is a highly desirable cancer treatment strategy. In addition to
inducing TAMs to differentiate into M1-type, M2-type TAMs also have the potential
to be repolarized into M1-type. Recent research has shown that the cyclodextrin
nanoparticles (CDNP-R848) loaded with R848, an agonist of Toll-like receptors
TLR7 and TLR8, delivered drugs to TAMs efficiently in vivo. Application
of CDNP-R848 in multiple mouse tumor models polarized TAMs towards the
M1-phenotype and inhibited tumor growth. Furthermore, the combination of
CDNP-R848 and anti-PD-1 antibodies had a synergistic effect and led to tumor
shrinkage [55]. In a xenograft mice model of breast cancer, the nanomedicine of
STAT6 inhibitor AS1517499 and inhibitor of kappa B kinase (IKK) siRNA effectively
induced the polarization of TAMs from M2-type to M1-type, reshaped the TME and
significantly inhibited tumor growth [56]. Inhibition of PI3k-
Checkpoint molecules, including PD-1, PD-L1, Tim-3 and Tim-4 are expressed in
TAMs, which inhibit TAMs phagocytosis and promote tumor immune escape. Recent
studies showed that PD-L1 antibody therapy increased the proliferation, survival
and size of TAMs from mouse bone marrow. Moreover, PD-L1 antibody therapy
increased the phosphorylation level of Akt and mTOR, thus inducing TAMs
activation by up-regulating the expression of MHC-II and CD86. TAMs produced more
TNF
Another study showed that almost all PD-1
Tim-3 knocked-down macrophages and hepatocellular carcinoma H22 cells were
injected subcutaneously into mice. Compared with control macrophages, Tim-3
knocked-down macrophages significantly inhibited H22 tumor growth. Mechanism
study found that the inhibition of Tim-3 significantly reduced the
phosphorylation level of STAT-6 in macrophages, and impaired the polarization of
macrophages to M2-type, thus hindering tumor growth [63]. Tim-4
Increasing the phagocytic ability of TAMs is another important anti-tumor
strategy, which can directly kill tumor cells. At present, three main pathways
inhibit the phagocytosis of TAMs: the signal regulatory protein
The high expression of MHC-I in tumor cells inhibited the phagocytosis of TAMs.
MHC-I is coupled with two members of the leukocyte immunoglobulin-like receptor
(LILR) family, LILRB1 and LILRB2, and contains ITIMs to help inhibit
intracellular signal transduction. Blockade of the MHC-1/LILRB1 pathway in the
tumor that exhibited drug resistance after blocking the SIRP/CD47 axis enhanced
the phagocytosis of MHC-I
CD24 is a highly glycosylated surface protein that interacts with Siglec-10. Tumor cells highly express CD24, while TAMs highly express Siglec-10. After binding to CD24, the ITIM of Siglec-10 recruited and activated the SH2-domain-containing tyrosine phosphatase SHP-1 or SHP-2, thereby inhibiting the phagocytosis of TAMs [69]. Knocking out the CD24/Siglec-10 gene or blocking the CD24/Siglec-10 axis with monoclonal antibodies increased the ability of TAMs to phagocytize tumor cells and reduced tumor growth [70]. Recent studies showed that both mouse and human TAMs expressed PD-1, which was related to the decrease of phagocytosis. The expression level of PD-1 in M2-type TAMs was significantly higher than that in M1-type TAMs. In vivo blocking of PD-1/PD-L1 signaling increased macrophage phagocytosis, reduced tumor growth, and prolonged mouse survival time in a macrophage-dependent manner [63] (Fig. 3).
The interaction between tumor cells and TAMs is very complicated. TAMs in different activation states in the TME have different functions. They are a two-edged sword in that they can act as M1-type TAMs, recognize tumor antigens, and phagocytize or kill tumor cells, while they can also be domesticated by TME into M2-type to stimulate tumor development. Which aspect plays the dominant role depends on the activation state of macrophages, the stage of the tumor, and the influences of the TME. Targeting M2-type TAMs is a promising strategy for cancer therapy. Given many targets of TAMs still have not been discovered, or suitable targeted drugs have not yet been developed, it will be necessary to further study the molecular mechanism of the interaction between tumor cells and TAMs to find more effective new targets and drugs.
M-CSF, Macrophage colony-stimulating factor; TGF-
QG, YL and RW designed the study. YL wrote the manuscript. RW provided help with the writing. QG reviewed and revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.