







 , Jianping Xue 1,*
, Jianping Xue 1,*1 Anhui Provincial Engineering Laboratory for Efficient Utilization of Featured Resource Plants, College of Life Sciences, Huaibei Normal University, 235000 Huaibei, Anhui, China
Abstract
Background: High temperature and drought environments are important limiting factors for Pinellia ternata growth, whereas shading can promote growth by relieving these stresses. However, the mechanism of growth promotion by shading in P. ternata is unknown. Long non-coding RNAs (lncRNAs) play important roles in the plant’s growth and environmental response, but few analyses of lncRNAs in P. ternata have been reported. Methods: We performed lncRNAs analysis of P. ternata in response to shading using RNA-seq data from our previous studies. A total of 13,927 lncRNAs were identified, and 145 differentially expressed lncRNAs (DELs) were obtained from the comparisons of 5 days shade (D5S) vs. 5 days of natural light (D5CK), 20 days of shade (D20S) vs. 20 days of natural light (D20CK), D20S vs. D5S, and D20CK vs. D5CK. Of these, 119 DELs (82.07%) were generated from the D20S vs. D20CK comparison. Results: Gene ontology (GO) analysis indicated that the reactive oxygen (ROS) metabolism and programmed cell death (PCD) processes might regulate shade-induced growth promotion. The “signal transduction” and “environmental adaptation” in the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were used for lncRNA-mRNA regulatory network construction and showed that the lncRNAs might mediate P. ternata growth by regulating ROS accumulation and light signals. Conclusions: This study explores lncRNAs’ functions and regulatory mechanisms related to P. ternata growth and lays a foundation for further research on P. ternata.
Keywords
- Pinellia ternata
- shade
- transcriptome
- long noncoding RNAs
- growth promotion
Extensive research has shown that most regions of eukaryotic genomes (e.g., yeast) can be transcribed with a transcriptional coverage of approximately 85%. However, only 1–2% of the transcribed genome is responsible for protein coding [1]. The remaining transcripts have no protein-coding potential, with 80% being occupied by long non-coding RNAs (lncRNAs) with lengths over 200 bp [2]. Since lncRNAs were first identified in mice in 2002 [3], numerous lncRNAs have been found in various species. For instance, 6480 lncRNAs were identified in Arabidopsis using transcriptome RNA-seq [4], and Li et al. [5] identified 20,163 lncRNAs in maize by analysing transcriptome data. Moreover, with the rapid development of next-generation high-throughput sequencing technologies, lncRNAs have been extensively identified in other plant species such as rice [6], wheat [7], and Zea mays [8], with the functional study of lncRNA becoming a hot topic in plant science. Although lncRNAs cannot code proteins, they regulate gene expression at the transcription, post-transcription, and translation levels, contributing to plant growth, flowering regulation, and stress response [9, 10, 11, 12].
Pinellia ternata (Thunb.) Breit is a native medicinal herb in China, and its tuber is widely used in traditional Chinese medicine for its alkaloids, organic acid, and polysaccharose components [13, 14, 15], providing anti-tussive, anticancer, and anxiolytic effects [16, 17, 18]. P. ternata is sensitive to high temperature and light during its growth, which accelerates its senescence and causes “sprout tumble” (ST), a phenomenon that limits this plant’s cultivation [19, 20, 21]. Previous studies have pointed out that shading can effectively delay the ageing of P. ternata caused by high temperature and light intensity in summer, thereby increasing tuber production, which has been revealed at the physiological level [22, 23, 24]. Recently, Xue et al. [19] found 6384 differentially expressed genes (DEGs) as a response to shade using full-length transcriptome sequencing, providing a transcriptional-level explanation for shade-induced growth promotion in P. ternata. Chao et al. [25] revealed that demethylation occurred in the genome in response to high temperature, while Shi et al. [26] reported that a shaded environment severely increased the methylation rate, indicating that variation in DNA methylation could regulate the aging of P. ternata. The ageing of P. ternate is a complex biological process, and the mechanism of shade-induced growth promotion in P. ternata is still unknown.
This manuscript aimed to identify lncRNAs in P. ternata using full-length transcriptome sequencing and compare the responses of differentially expressed lncRNAs’ (DELs) to shade by analysing the second-generation transcriptome. This might provide a new perspective to explore the mechanism of ageing and shade-induced growth promotion in P. ternata.
P. ternata was obtained from the Experimental Farm of Huaibei Normal University. Approximately 3 weeks after sprouting, half the individuals were selected for 90% shade-treatment with sunshade net. P. ternata seedlings grown under natural light and the shaded environment were collected at 5 and 20 days of treatment for next-generation sequencing (NGS), with three biological replicates of each treatment group. These samples were mixed for single-molecule real-time (SMRT) sequencing. All transcriptome data used in this study are from our previous study [19], which was uploaded to NCBI under the accession numbers SRP215828 and PRJNA515824.
The transcriptome data generated from SMRT sequencing were examined for redundancy, and the non-redundant transcripts were used for lncRNAs identification. First, the screened transcripts were blasted to the protein database of Pfam; the ones in the database were considered protein-coding genes, while the remaining unmatched transcripts were further evaluated for protein-coding potential prediction based on the CNCI, CPC, and PLEK platforms. The threshold values of CNCI, CPC, and PLEK were all set to zero; the transcripts with values greater than zero were considered mRNAs, while those with values lower than zero were identified as lncRNAs. Based on this criterion, transcripts with coding potential were valued in every database, and the transcripts without coding potential in all three databases were finally identified as lncRNAs.
Excluding the identified lncRNAs, the remaining transcripts were chosen as
candidate target genes, and Pearson correlation was used to identify pairs of
lncRNA and target with absolute values greater than 0.9 and p-values
lower than 0.01. Target genes were further identified using the RIsearch-2.0
software (Frederiksberg C, Denmark) [26], with base number of direct interactions
between lncRNAs and mRNAs
Total RNA was isolated from P. ternata samples following the
procedure described previously [19], and cDNA was
synthesised using MonScript
To systematically identify lncRNAs in P. ternata, the full-length transcriptome was sequenced, generating 136,163 non-redundant transcripts after correction. The coding potential of the obtained transcripts was predicted with the CNCI, CPC, Pfam, and PLEK platforms. The number of non-coding genes predicted by each software was calculated, and a Wayne diagram was drawn (Fig. 1). Transcripts reported by all four prediction methods were identified as lncRNA, and 13,927 lncRNAs were identified in P. ternata (Supplementary Table 2). The remaining sequences, except lncRNAs, were analysed as target genes. The Pearson correlation and p-values between lncRNA and mRNA were calculated. The lncRNA-mRNA pairs with absolute correlation over 0.9 and p-value less than 0.01 were considered regulatory relationships, and 667,381 relationship pairs were obtained, including 87,507 target gene information (Supplementary Table 3).
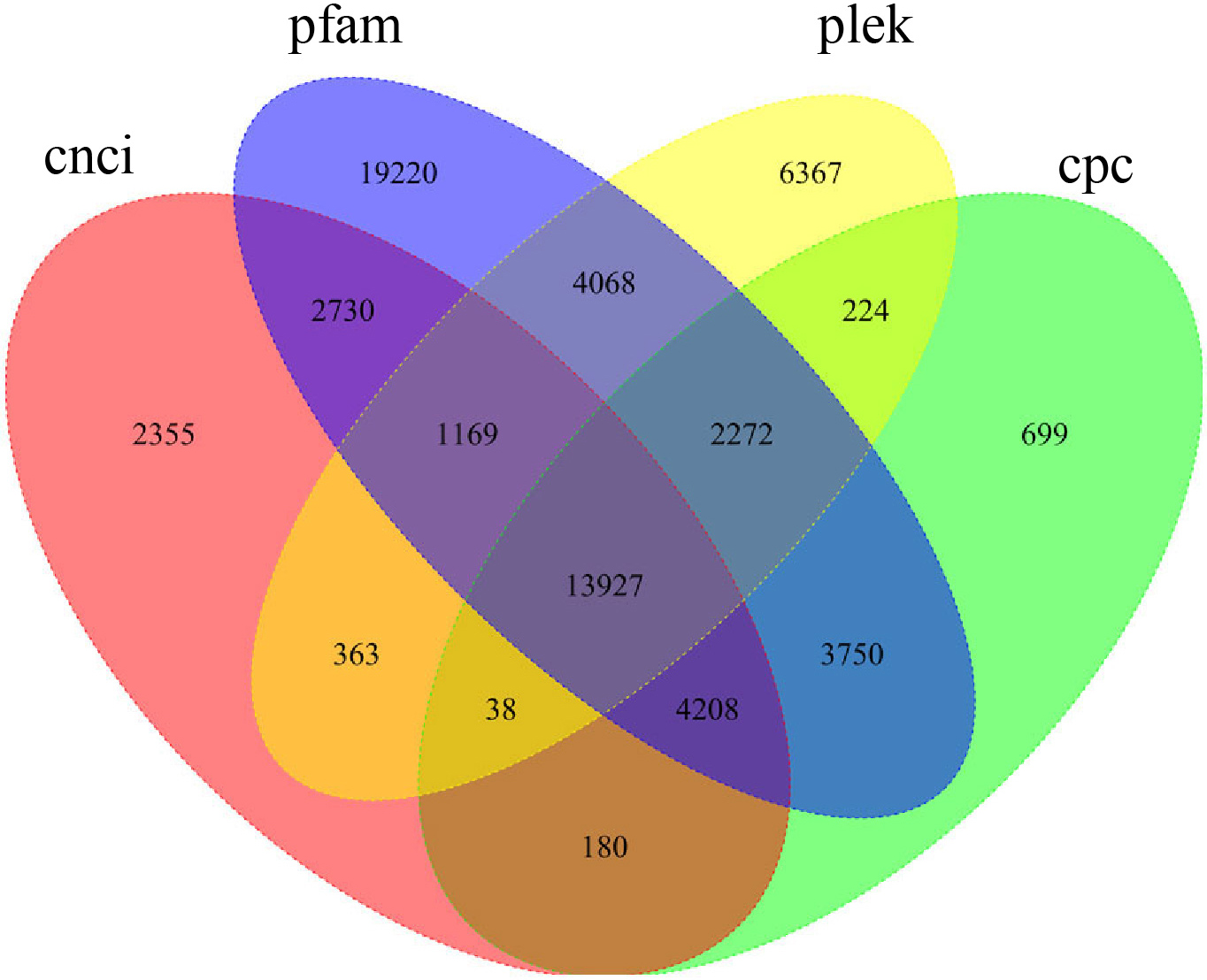 Fig. 1.
Fig. 1.Venn graph of the long non-coding RNA (lncRNA) predicted results.
DESeq2 was used for differential expression analysis of the identified lncRNAs. A total of 145 lncRNAs that had a response to shade was collected (Supplementary Table 4). The results revealed seven DELs in the comparison of D5S vs. D20S, including three upregulated and four downregulated. The D5S vs. the D5CK comparison had two downregulated lncRNAs, whereas the D5CK vs. the D20CK comparison had 17 DELs, including seven upregulated and 10 downregulated. Notably, in D20CK vs. D20S, many lncRNAs were activated or inhibited with 119 DELs, including 63 upregulated and 56 downregulated (Fig. 2A). Because of its higher number of DELs than the others, the D20S vs. D20CK comparison was selected for exploring DELs functions further (Fig. 2B).
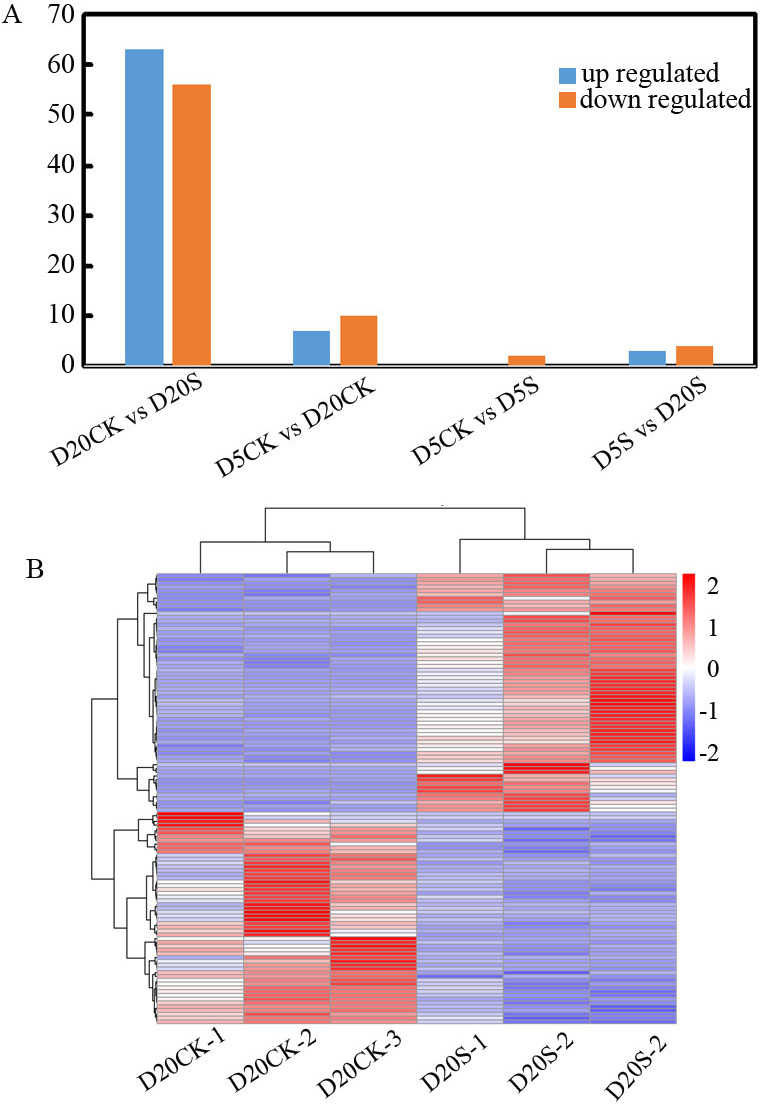 Fig. 2.
Fig. 2.Differentially expressed lncRNAs in the D5S, D5CK, D20S, and D20CK groups. (A) Hierarchical clustering graph of the 145 differentially expressed lncRNAs (DELs) based on average log10(FPKM(fragments per kilobase of exon model per million mapped fragments)+ 1) values of all lncRNAs in each cluster. (B) Statistics of upregulated- and downregulated-expression of DELs in each cluster.
To better understand DELs functions in response to shade, the target genes of
the 119 DELs generated from D20CK vs. D20S were chosen for gene
ontology (GO) functional enrichment analysis. The GO terms were classified into
three categories, namely cellular component, molecular function,
and biological process, with 51 functional
groups (Fig. 3). We focused on biological process. Biological
process comprises 24 functional groups, including “response to stimulus”,
“developmental process”, and “signalling”, which are directly linked to the
shading environment. A total of 424 GO terms were enriched with KS value
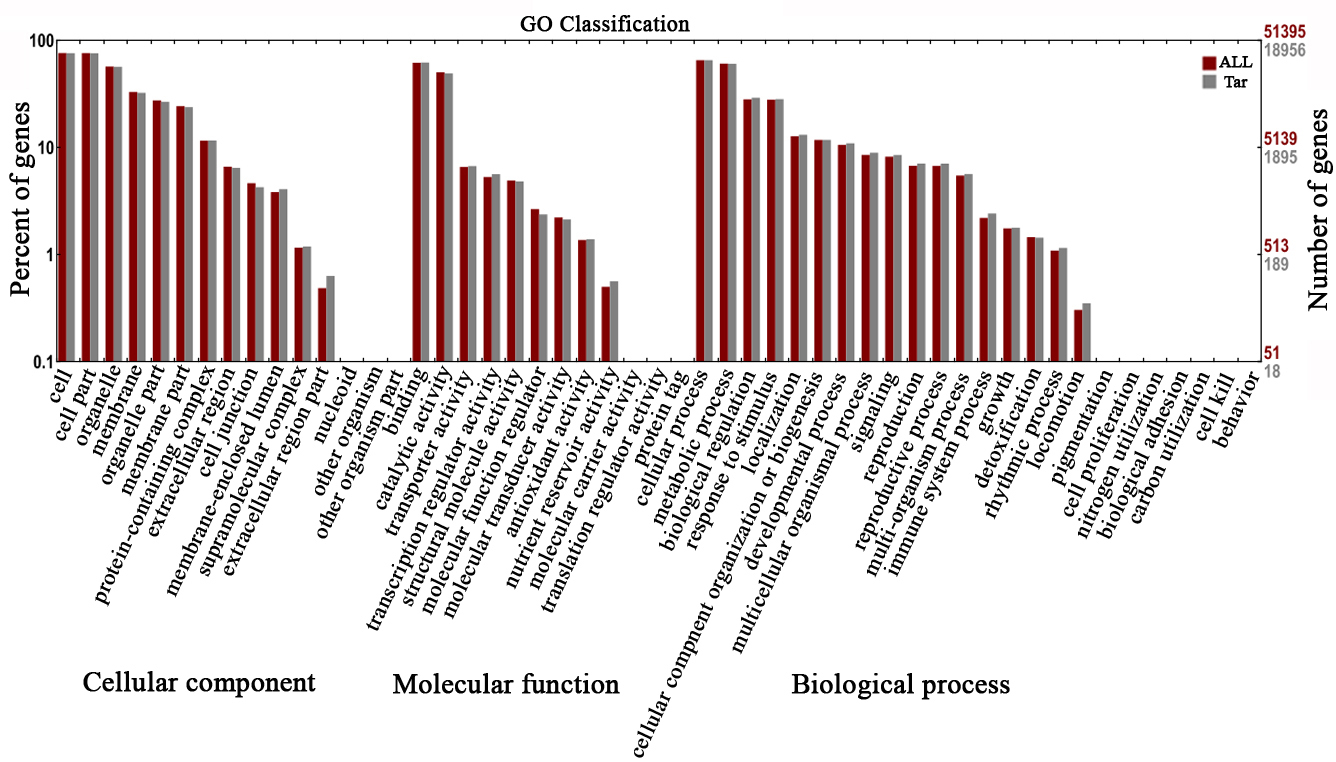 Fig. 3.
Fig. 3.Enriched gene ontology (GO) terms of lncRNA target genes generated from the D20S vs. D20CK group.
Based on the KEGG pathway enrichment analysis, 128 KEGG pathways were obtained (Supplementary Table 6) and assigned to five categories, including 18 subfamilies (Fig. 4). This study focused on environmental information processing and organismal systems and screened two subfamilies closely related to environmental response, namely “signal transduction” and “environmental adaptation”.
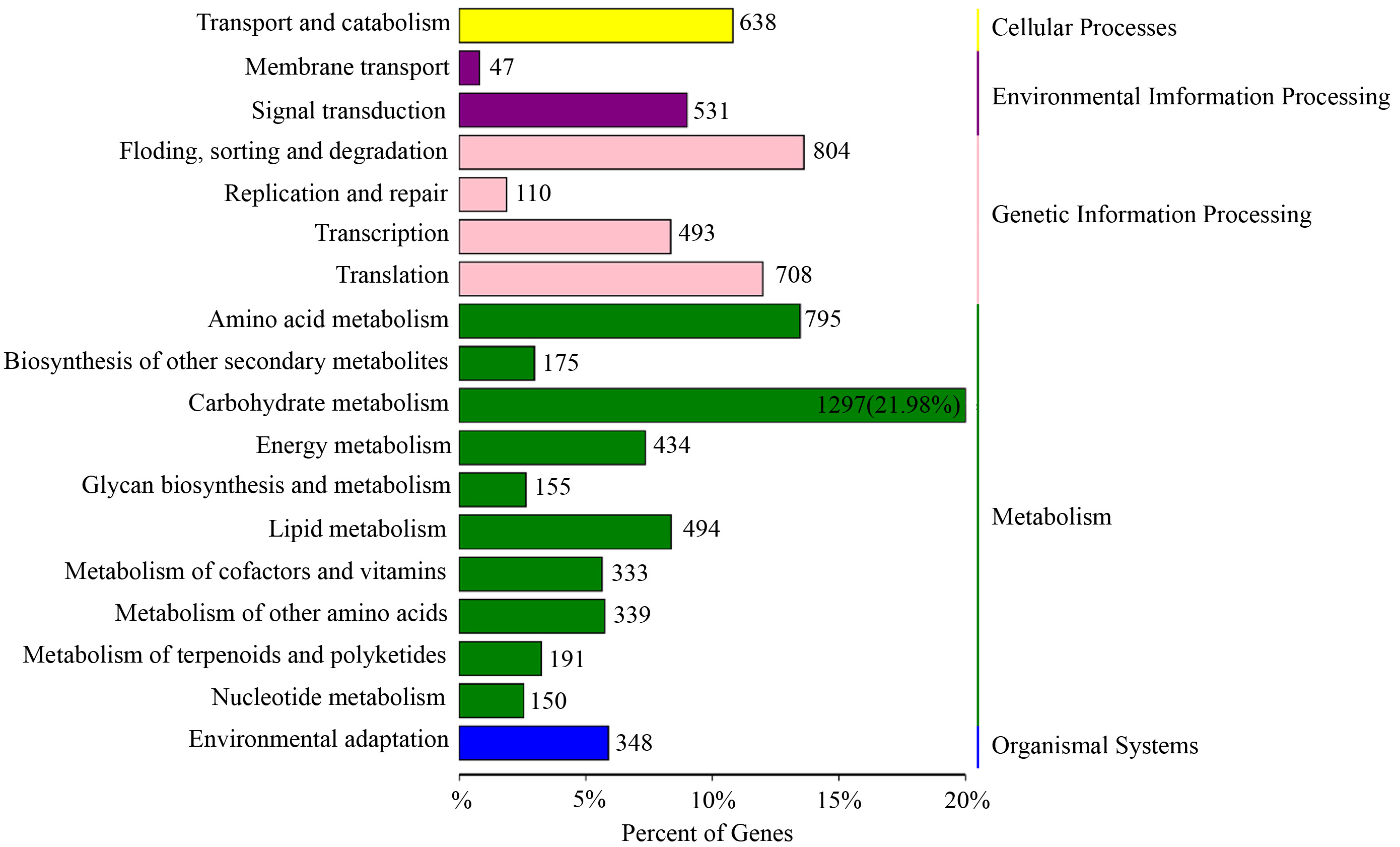 Fig. 4.
Fig. 4.Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis with the lncRNA target genes generated from the D20S vs. D20CK group.
To explore the co-expression networks of lncRNAs involved in shade response, the DELs and their target genes involved in “signal transduction” and “environmental adaptation” were selected for constructing the lncRNA-mRNA regulatory network. The networks were composed of 83 lncRNAs and 348 target mRNAs in the environmental adaptation pathway (Supplementary Table 7), and 95 lncRNAs and 531 target mRNAs in the signal transduction pathway (Supplementary Table 8). These predicted target genes were mainly annotated to hormone signals, light signals, protective enzymes, heat shock proteins, and transcription factors. For environmental adaptation pathway, the lncRNA of i0_LQ_Pts1_c4467/f1p5/265 (lncRNA265) targets HSP90 (i2_HQ_Pts1_c113746/f2p64/2456), HSP83 (i3_LQ_Pts1_c22913/f1p4/3105), and WRKY24 (i2_LQ_Pts1_c44853/f1p17/2068), while lncRNAs of i0_LQ_Pts1_c17019/f1p114/222 (lncRNA222, i0_LQ_Pts1_c11463/f1p2/226 (lncRNA226), i1_LQ_Pts1_c42191/f1p16/1785 (lncRNA1785), i1_HQ_Pts1_c6713/f2p3/1631 (lncRNA1631), and i0_HQ_Pts1_c210/f5p0/974 (lncRNA974) target genes of heat shock protein and WRKY factors (Fig. 5A). In the signal transduction pathway, lncRNA265, lncRNA974, and i1_LQ_Pts1_c64130/f1p10/1092 (lncRNA1092) regulated ethylene insensitive (i2_LQ_Pts1_c111059/f1p24/2627) and auxin response factor (i2_LQ_Pts1_c19140/f1p6/2841), jasmonic acid-amido synthetase JAR1 (i2_HQ_Pts1_c86287/f9p17/2219) and AUX/IAA family (i1_LQ_Pts1_c41312/f1p3/1976), and abscisic acid-insensitive 5-like protein (i2_HQ_Pts1_c86287/f9p17/2219) and auxin response factor (i3_LQ_Pts1_c9435/f1p3/3529), respectively. This indicates that the above lncRNAs can modify hormone signals of ethylene, auxin, jasmonic acid (JA), and abscisic acid (ABA) in response to shading. PIF4 (i2_LQ_Pts1_c2083/f1p1/2449 and i9_LQ_Pts1_c106/f1p0/9572) was regulated by both lncRNA226 and lncRNA1785, while lncRNA226 and lncRNA222 regulated CAT (i1_LQ_Pts1_c57682/f1p22/1751 and i2_LQ_Pts1_c11146/f1p40/2047). Furthermore, lncRNA265 and lncRNA1785 participated in the signal transduction pathway by WRKY (i2_LQ_Pts1_c44853/f1p17/2068) and bZIP (i2_LQ_Pts1_c57046/f1p2/2181) transcription factors (Fig. 5B).
 Fig. 5.
Fig. 5.The lncRNAs-mRNAs regulatory networks in the (A) environmental adaptation and (B) signal transduction pathways in response to shade. Triangular nodes represent lncRNAs, while circular nodes represent mRNAs.
To validate the transcriptome data, six lncRNAs and six target genes involved in the environmental adaptation and signal transduction pathways were selected for qRT-PCR analysis. Compared with the control group, four lncRNA222, lncRNA226, lncRNA265, and lncRNA1631 were upregulated in the shade, while lncRNA1092 and lncRNA1785 decreased in the shade (Fig. 6A). The target genes of PtJAR1, PtABI5, PtWRKY, PtCAT1, and PtPIF4 were upregulated, and PtHSP was downregulated (Fig. 6B). Moreover, the expression trends were consistent with RNA-seq results, indicating that the transcriptome data was reliable.
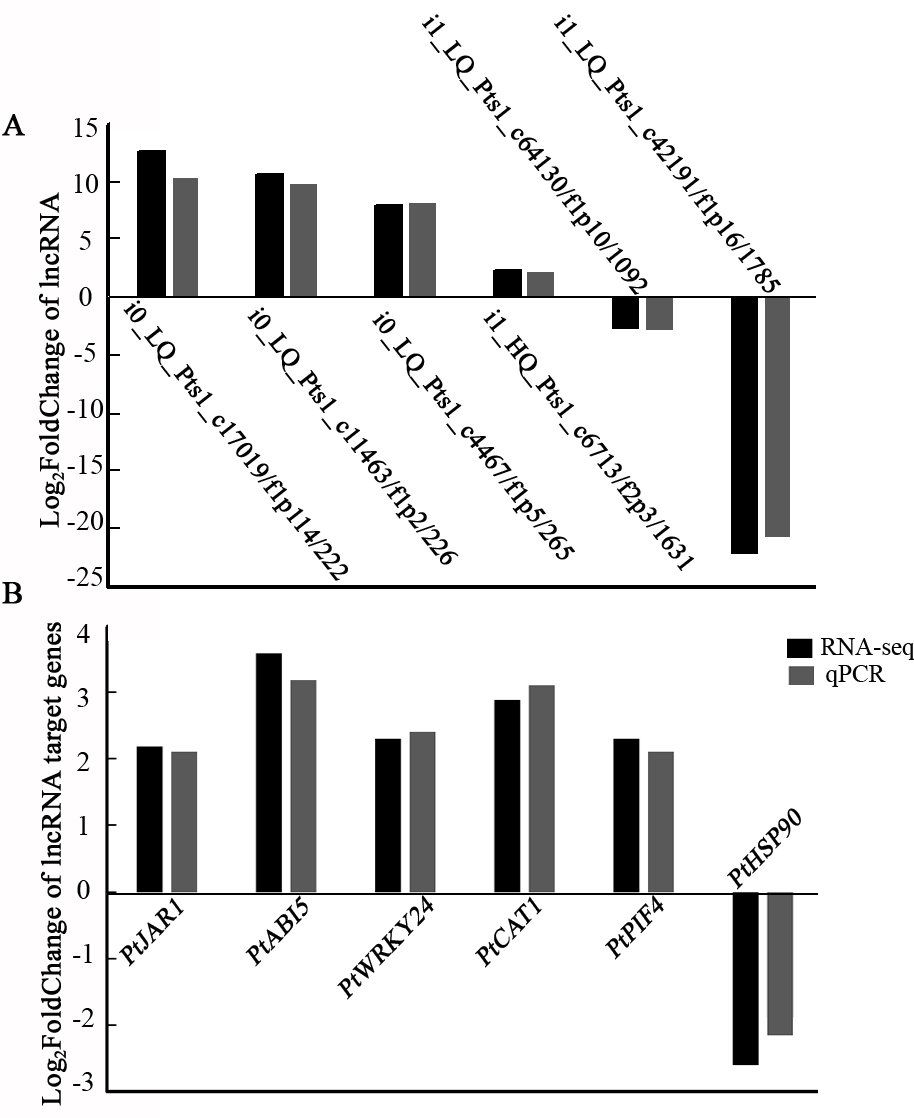 Fig. 6.
Fig. 6.Expression validation of lncRNAs and target mRNAs by quantitative RT-PCR. (A) and target mRNAs (B)by quantitative RT-PCR.
Many studies have shown that high temperature and soil water deficit cause the ST of P. ternata [28] and that shading can effectively delay the ST of P. ternata, thereby increasing its production [19, 22]. The indexes of endogenous hormones, photosynthetic parameters, and protective enzymes in response to shade have also been reported [22, 23], and Xue et al. [19] identified some DEGs responsible for ST. However, the regulation mechanism of shading on the ST of P. ternata is still unknown. Recently, lncRNAs were successively identified in multiple species, and many studies revealed their functions in regulating plant response to stress [11, 12, 29, 30, 31, 32]. In the present study, we identified and analysed the lncRNAs of P. ternata based on our previous transcriptome data [19], which enriched our understanding of shading regulation of P. ternata growth.
By comparing DELs generated in different groups, we found that the DELs in the D20S vs. D20CK comparison accounted for 82.07%, which was in accordance with the DEGs distribution reported by Xue et al. [19]. The similar high percentage of DELs and DEGs in the D20S vs. D20CK comparison hinted a possible regulation of lncRNAs on the mRNAs. The ST of P. ternata has been reported as a PCD process related to the accumulation of ROS [19, 28]. The processes of ROS metabolism and PCD were notably enriched in the GO enrichment of DELs target genes, indicating that the DELs possibly regulated PCD by modifying ROS accumulation.
A shaded environment promotes growth mainly by delaying high temperature, light, and drought stresses. Therefore, the environmental variation would induce the responsible signal response in P. ternata. The KEGG pathway analysis focused on two environment-related pathways: “signal transduction” and “environmental adaptation”. The DELs of both pathways mediated hormone signals, light signals, antioxidant enzymes, and transcription factors by regulating target genes (Fig. 7). JA and ABA are important plant hormones that can enhance plant stress responses by decreasing ROS accumulation [33, 34, 35, 36, 37]. PtJAR1 and PtABI5 acted as positive regulators in the signal transduction of JA and ABA [38, 39, 40], respectively, and their transcripts regulated by lncRNAs increased in the shaded environment, demonstrating that the lncRNAs could promote P. ternata growth by enhancing JA and ABA signals.
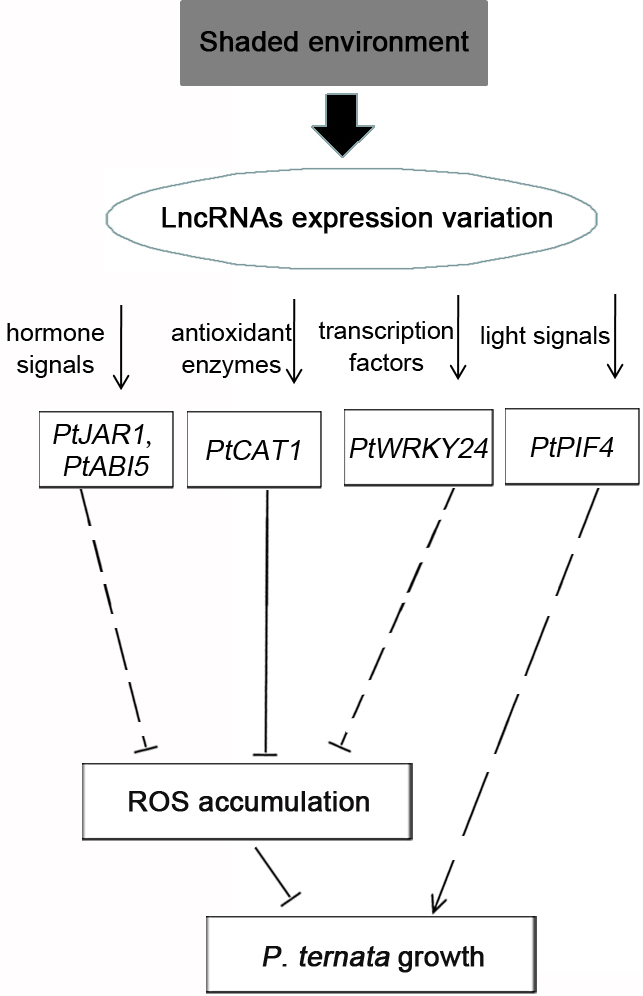 Fig. 7.
Fig. 7.The putative lncRNA interaction model for shade-induced growth promotion in P. ternata. In response to the shaded environment, the expression of lncRNAs was altered, thereby modulating hormone signals, antioxidant enzymes, transcription factors, and light signals by increasing the transcripts of PtJAR1, PtABI5, PtCAT, PtWRKY, and PtPIF4. Based on the regulation, shading can promote P. ternata growth by directly eliminating reactive oxygen (ROS) accumulation and inducing auxin accumulation.
The ST of P. ternata is directly linked with ROS accumulation induced by environmental stresses [19, 28]. The up-expression of PtCAT1 in response to shade suppressed ROS accumulation, aligning with a previous report that showed that shade-induced PtCAT activity [19], together indicated the lncRNAs could regulate ROS accumulation. WRKY transcription factors can be induced by abiotic stress and are involved in regulating plant abiotic stress response [41, 42]. In the present study, some WRKYs were upregulated in response to shade, demonstrating that they probably regulated the high temperature and drought stress response, which coincided with the finding from Wang’s study [43]. Many studies reported that WRKY transcription factors could regulate the content of various antioxidant enzymes in plants to remove excess accumulated ROS, thereby maintaining ROS balance in plants under abiotic stress conditions and improving plant tolerance to abiotic stress [41, 42]. In the present study, the differentially expressed WRKYs probably suppressed ROS accumulation indirectly by activating the antioxidant enzymes of P. ternata. The genes related to ROS accumulation were also in line with the top process of ROS metabolism in the GO analysis.
The shade directly changed light intensity, and PtPIF4 was induced for light and high temperature responses. The high expression of PtPIF4 in the shade would contribute to auxin accumulation [44, 45], thereby promoting growth of P. ternata, in accordance with our previous research findings [19]. To summarise, lncRNAs could respond to shade and mediate ROS accumulation by inducing JA and ABA signals and increasing transcripts of PtCAT1 and PtWRKY24 to delay the ST of P. ternata. lncRNAs act as important upstream-regulators in the shade-induced growth promotion of P. ternata.
lncRNAs of P. ternata were identified, and their regulation in the shade-induced growth promotion of P. ternata was analysed based on a transcriptome analysis. The GO analysis showed that the DELs of the D20S vs. D20CK groups mainly participated in regulating ROS and PCD processes in response to shade. To explore the functions of lncRNAs, we constructed putative lncRNA-mRNA regulatory networks involved in the shade response and, thus, regulation of P. ternata growth. This study lays a foundation for exploring the functions and regulatory mechanisms of lncRNAs in P. ternata growth.
The raw sequence data reported in this paper can be accessed in the National Center for Biotechnology Information under the accession number SRP215828 and PRJNA515824 that deposited by our previous studies.
Conceptualization—JY, TX and JX; methodology—WC and QY; software—JY and YZ; validation—ML and XL; formal analysis—FZ and YZ; investigation—YD; resources—JX; writing - original draft preparation—JY; writing - review and editing—TX and JX; visualization—WC, QY and ML; supervision—XL and YD; project administration—YD; funding acquisition—TX, JX, YZ and FZ. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
The authors declare the plant materials were obtained from the wild without any specifically permissive requirement. And all methods were performed in accordance with relevant guidelines and regulation.
Not applicable.
This work was supported by the National Natural Science Foundation of China (81803665, 81573518), the Project of Natural Science Research of Universities in Anhui Province, China (KJ2021A0532, KJ2021A0533). Excellent Scientific Research and Innovation Team of University in Anhui Province (2022AH010029). Open project of National and local Joint Engineering Laboratory of Anti-stress Breeding and Disaster Reduction (NELCOF20210102).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.