1. Introduction
The controlled inflammatory response is a useful process, carefully regulated by
a complex molecular cascade, which leads to the removal of harmful stimuli and
the recovery of normal physiology. The magnitude of the inflammatory response is
critical because failure to regulate acute pro-inflammatory stimulation leads to
chronic inflammation, autoimmunity and excessive tissue damage
[1, 2]. Chronic inflammation progresses slowly, lasts
longer, and can cause many diseases associated with chronic and persistent
vascular inflammation, characterized by an increase in adhesion of circulating
leukocytes to endothelial cells of the vascular wall and their subsequent
transmigration [3]. These phenomena are largely
controlled by the expression of adhesion molecules which in turn are regulated by
different transcriptional and post-transcriptional mechanisms [4]. Recent
evidence suggests that epigenetic changes, defined as heritable changes in gene
expression that are independent of changes in DNA sequence, may be involved in
chronic inflammation [4].
To preliminarily explore the role of epigenetics in the context of vascular
inflammation, we conducted a literature analysis using the Clarivate Web of
Science Core Collection database. We queried the database with the following
terms: “epigenetics”, “inflammation”, and “adhesion molecules”. The search
yielded 39 papers, which were analyzed using the bibliometric mapping tool
VOSviewer (VOSviewer version 1.6.19, Leiden University, South Holland,
Netherlands) [5]. Bioinformatic analysis of the medical subject headings (MeSH)
associated with the retrieved papers resulted in a total of 442 items (Fig. 1).
Among these, 19 keywords met the threshold level of occurrence (minimum number of
occurrences of a keyword = 3).
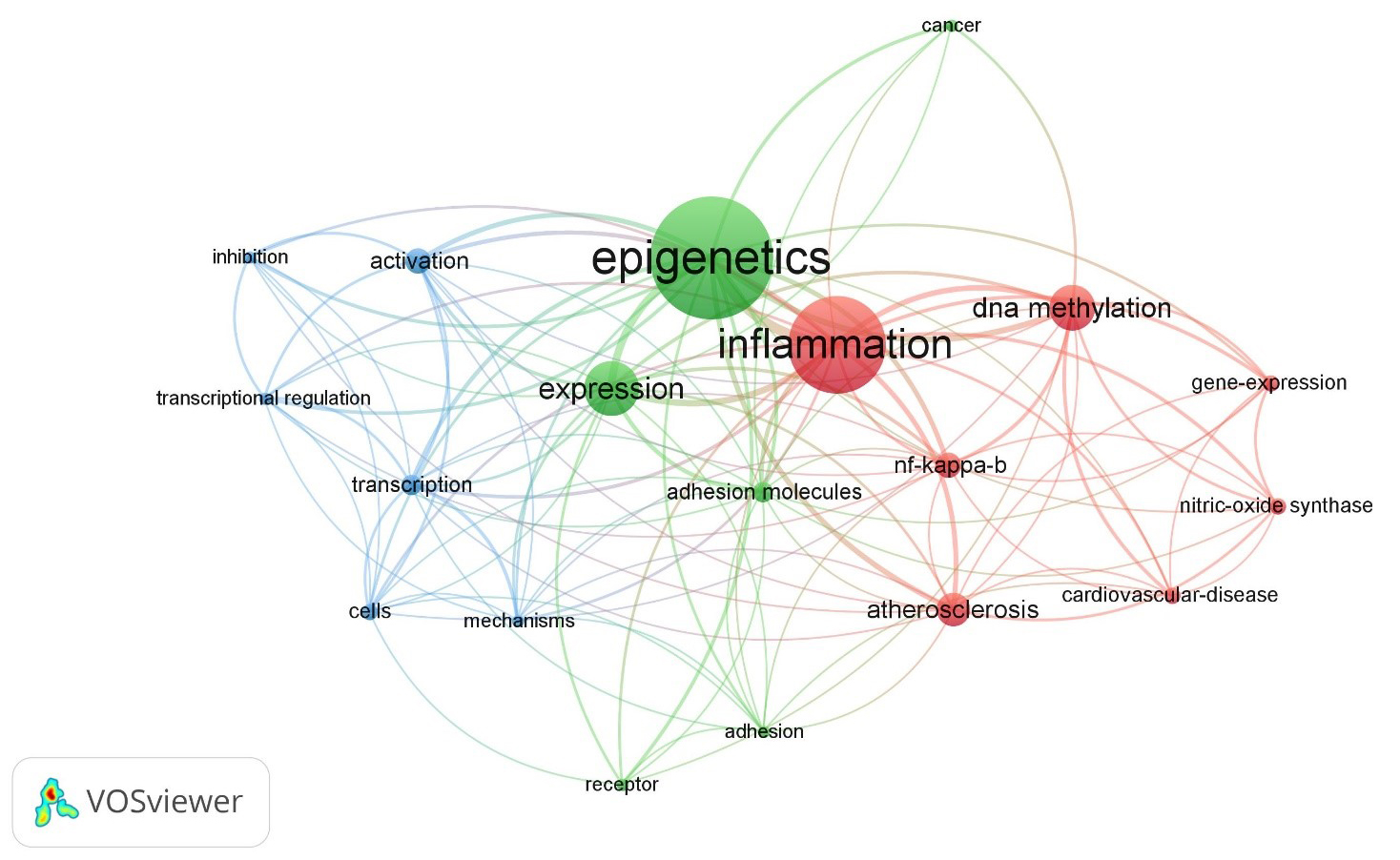 Fig. 1.
Fig. 1.
The map displays the medical subject heading (MeSH)
keywords selected from articles published on and retrieved from the Web of
Science using the search terms “epigenetics”, “inflammation”, and “adhesion
molecules” (all fields), resulting in 442 items. Applying a threshold of 3
keywords, 19 items met the criteria. The size of the bubbles indicates the
frequency of word occurrence, while the colour of the bubbles represents their
cluster affiliation. Bubbles that are closer together indicate a higher frequency
of co-occurrence between the two words. The analysis was performed using the
bibliometric mapping tool VOSviewer.
The keywords with greatest total link strength were selected and highlighted as
bubbles (Fig. 1). There are three clusters identified with three different
colours: Cluster 1, red-coloured (7 items: atherosclerosis,
cardiovascular-diseases, DNA methylation, gene expression, inflammation,
NF-B, nitric-oxide synthase); Cluster 2, green-coloured (6
items: adhesion, adhesion molecules, cancer, epigenetics, expression, receptor);
and Cluster 3, blue-coloured (6 items: activation, cells, inhibition, mechanisms,
transcription, transcriptional regulation).
The aim of this review is to present a comprehensive and current overview of the
primary types of epigenetic alterations that contribute to a dysregulated
inflammatory response, with a specific focus on the expression of endothelial
adhesion molecules.
2. Vascular Inflammation: Role of Endothelial Adhesion Molecules
In the last few years, an increasing amount of research has recognized an active
and crucial role played by the vascular wall in the inflammatory response. An
essential property of the vessel wall is the ability of the inner endothelial
lining to be activated in order to react to noxious stimuli or inflammatory
mediators [6]. In response to proinflammatory triggers, including chemokines,
cytokines, and oxidized low-density lipoproteins (ox-LDL), the endothelium is
activated, and acquires new functional properties, becoming adhesive to
leukocytes, due to the expression on its surface of cell adhesion molecules
(CAMs) that recognize counter-receptors present on different subsets of
leukocytes [4, 6]. The CAM proteins which mediate endothelium-leukocyte
interactions belong to four major families: selectins, selectin ligands,
integrins, and members of the immunoglobulin (IgG) superfamily [7].
2.1 Selectins and Selectin Ligands
The selectin family of CAMs consists of three members (E-, P- and L-selectin),
all of which mediate rolling of leukocytes along the endothelium [8]. P-selectin
is stored in granules in endothelial cells and platelets and rapidly translocates
to the cell surface in response to several inflammatory stimuli. E-selectin is
exclusively present in endothelial cells and its expression is regulated by
increased transcription after stimulation by inflammatory cytokines such as
tumour necrosis factor (TNF)- and interleukin (IL)-1 [8].
L-selectin is expressed on many subclasses of leukocytes and is rapidly shed from
the surface of the leukocyte after activation [8]. The adhesive function of all
selectins requires specialised counter-receptors. These ligands consist of
carbohydrate moieties such as the tetrasaccharide sialyl Lewis X (sLeX), whose
interaction with selectin family members is responsible for a major part of
leukocyte rolling in inflammation.
2.2 Integrins and the IgG Superfamily of CAMs
Integrins are constitutively expressed on leukocytes and many other cell types.
They are configured as dimers that contain one - and one
-subunit. Integrins are rapidly activated from a low-affinity to a
high-affinity state following cell activation and ligand binding [9]. They
mediate adhesion of cells to matrix proteins, cellular counter-receptors and many
other substrates. The interaction between integrins and IgG superfamily members
is particularly important in inflammation [8]. The IgG superfamily members are
large type I transmembrane proteins characterized by a series of repeating
extracellular IgG-like domains, a transmembrane region and a short cytoplasmic
tail. They include intercellular adhesion molecules-1, 2, and 3 (ICAM-1, 2, and
3), platelet endothelial cell adhesion molecule-1 (PECAM-1), and vascular cell
adhesion molecule-1 (VCAM-1). The expression of VCAM-1 and ICAM-1 increases after
stimulation of the endothelium by inflammatory cytokines, while PECAM-1 is
constitutively expressed on resting endothelial cells.
The main function of CAMs is to facilitate the process of leukocyte infiltration
from the bloodstream into inflamed tissue, by forming bonds with ligands on
circulating leukocytes, that allows for firm adhesion to the endothelium and
transendothelial migration [7]. This role makes CAMs crucial in acute response to
tissue damage and acute inflammation, but in chronic inflammation excessive or
prolonged infiltration of leukocytes can lead to further damage.
3. Endothelial Activation and Chronic Inflammation
The main functions of CAMs become apparent during “endothelial activation”, a
process characterized by significant functional changes in the vascular
endothelium induced by inflammatory mediators and vascular risk factors [10].
Resident immune cells, such as tissue-resident macrophages, act as “first
responders” by releasing inflammatory cytokines and chemokines which activate
endothelial cells and recruit leukocytes [10]. In non-inflammatory conditions,
the endothelium does not support the adhesion of leukocytes. However, after
activation, endothelial cells begin to express CAMs. In response to mediators
like histamine, pre-synthesized CAMs (e.g., P-selectin) are rapidly transported
to the cell surface within minutes. Other mediators, such as cytokines
(TNF-, IL-1), induce a more gradual activation of endothelial
cells, including gene expression and protein synthesis of various CAMs over a
span of hours. These CAMs include E-selectin (within 5 hours) and ICAM-1 and
VCAM-1 (within 12 hours). Furthermore, leukocytes themselves become activated and
alter the expression patterns of CAMs on their surface, such as the integrins
41 (also known as very late antigen-4 or VLA-4) and lymphocyte
function-associated antigen-1 (LFA-1), which makes them more adhesive to the
vascular endothelium. These changes collectively promote a multistep process of
trans-endothelial migration of leukocytes. In a clear sequence of events,
leukocytes initially form transient bonds with selectins on the surface of
endothelial cells, which causes them to slow down and begin rolling.
Conformational changes in integrins expressed on leukocytes then induce the
formation of high-affinity bonds by binding to other CAMs, such as VCAM-1 and
ICAM-1. In the presence of inflammatory stimuli, the activated endothelium
increases the expression of endothelium-leukocyte adhesion molecules such as
VCAM-1 and ICAM-1. The interaction of the IgG proteins ICAM-1 and VCAM-1 with
their respective integrin counter-receptors on leukocytes allows for firm
adhesion of leukocytes to the endothelium and subsequent transmigration into the
sub-endothelium. After the triggering factor for inflammation is removed, the
inflammatory process is typically resolved. However, this crucial step may be
absent or altered in several chronic immune-mediated inflammatory diseases where
CAMs play a critical role. The excessive leukocyte recruitment by CAMs can
exacerbate the inflammatory process and, in many cases, worsen tissue injury [1].
Extensive studies have confirmed the implications of CAMs in
(patho)physiological events in chronic inflammation on various levels [11].
Patients with several chronic diseases, including inflammatory bowel disease,
hypertension, diabetes, and hyperlipidaemia, among others, have been found to
have elevated plasma levels of soluble E-selectin [12]. Additionally, soluble
VCAM-1 levels have been observed to be elevated in the plasma of breast cancer
patients [13]. The evidence suggests that elevated levels of E-selectin and
ICAM-1 can serve as molecular markers for atherosclerosis and the development of
clinical coronary heart disease [14]. Lastly, it is worth noting that drugs with
anti-inflammatory effects, particularly those targeting the vasculature, can
reduce the levels of CAMs and inhibit leukocyte adhesion [15]. Similarly, certain
natural products or nutraceuticals have demonstrated the ability to reduce the
expression of CAMs on activated endothelial cells [16, 17, 18, 19, 20].
4. Nuclear Factor-B and Regulation of Vascular
Inflammation
The vascular endothelium functions as an important integrator and transducer in
response to multiple humoral and mechanical stimuli, including the inflammatory
response. As a primary inflammatory signaling factor, the transcription factor
nuclear factor-B (NF-B) is known to
significantly participate in the regulation of vascular inflammation and immune
function. NF-B is composed of homo- or hetero-dimers of RelA
(p65), RelB, c-Rel, p50/p105 (NF-B1), or p52/p100
(NF-B2), among which the heterodimer p50/p65 is the most
prominent and serves as the prototype of NF-B
[21].
These proteins carry an N-terminal Rel homology domain, which is required for
dimerization, nuclear targeting, DNA binding, and interaction with the inhibitor
of B (IB) proteins. Under physiological
conditions, the NF-B subunits are bound to
IB, which effectively sequesters NF-B in
the cytoplasm. However, when cells are stimulated by various signalling events
such as stress, bacteria, viruses, or cytokines, NF-B is
rapidly activated. It undergoes a process called translocation, where it moves
into the nucleus of the cell. Once in the nucleus, NF-B binds
to the B elements of specific genes, including those involved
in proinflammatory cytokines, chemokines, and endothelial adhesion molecules.
This binding triggers the transcription of these genes, leading to the production
of inflammatory mediators and contributing to vascular inflammation [22].
There are two distinct pathways involved in NF-B activation,
which are activated in response to different stimuli and involve distinct
molecular mechanisms. In the canonical pathway, NF-B
activation is initiated by the phosphorylation and subsequent degradation of
IB proteins, which normally sequester NF-B
in the cytoplasm. Upon stimulation by specific signals such as proinflammatory
cytokines or microbial products, the IKK (IB kinase) complex
is activated and phosphorylates IB proteins. This
phosphorylation targets IB for ubiquitination and
proteasome-mediated degradation, allowing NF-B to translocate
to the nucleus and activate the transcription of target genes [22]. The
noncanonical pathway involves a different set of signaling events and is
typically activated by specific members of the tumour necrosis factor receptor
superfamily. In this pathway, the activation of NF-B-inducing
kinase (NIK) leads to the phosphorylation and processing of the
NF-B precursor protein p100. This processing generates the
mature p52 subunit, which can form a complex with RelB to activate gene
transcription [22].
As an important regulator of immunity and inflammation, the activation of
NF-B signalling is influenced by multiple regulatory
mechanisms. Numerous post-translational modifications of p65 have been shown to
have positive or negative effects on transcriptional responses of
NF-B. In addition, NF-B signalling
components have been reported to interact with chromatin-modifying enzymes, such
as histone deacetylases or acetyltransferases, with other transcription factors,
and with phosphatases, to fine-tune the NF-B response [23, 24]. Notably, many NF-B target genes encode inhibitors of the
NF-B response thus resulting in a complicated network involved
in the regulation of vascular inflammation [24, 25].
5. Epigenetic Regulation of Vascular Inflammation
Recent evidence suggests that internal components, such as hypertension,
hyperglycaemia, growth factors, oxidant stress and inflammatory factors [26], and
external components, including diet, life habits, and environmental pollutants
[27], may alter the epigenome, the collection of epigenetic marks on cell DNA, by
modulating gene expression without altering DNA sequence [28, 29, 30]. The mechanisms
determining epigenetic modifications, which are inherited and long-lasting, but
also reversible, can be divided into four groups: DNA methylation, RNA
methylation, histone post-translational modifications, and the broad family of
epigenetic regulators made of non-coding RNAs (ncRNAs). These epigenetic
mechanisms are involved in the modulation of the intricate processes underlying
vascular inflammation, and will be described below with a focus on the expression
of endothelial adhesion molecules and endothelium-leukocyte adhesion.
5.1 DNA Methylation
DNA methylation is the covalent attachment of a methyl group to the cytosine
base of the 5-CpG-3 dinucleotide, known as 5-methylcytosine (5mC) by
DNA methyltransferases (DNMTs), divided into maintenance (DNMT1) and de novo
(DNMT3A and DNMT3B) types [31]. On the other hand, ten-eleven translocation
enzymes (TET, TET1-3) can actively cause locus-specific DNA demethylation by
catalysing the hydroxylation of the 5mC residue to 5-hydroxymethylcytosine [32].
Methylation of DNA in the enhancer or promoter region inhibits the binding of
transcription factors, thereby decreasing gene transcription [33, 34]. In
contrast, DNA demethylation or hydroxylation of the methyl group within the
enhancer or promoter region enhances gene activity and expression [34, 35]. DNA
methylation is a critical epigenetic mechanism associated with vascular
inflammation and atherosclerosis development [36, 37]. Aberrant genome
hypomethylation has been found in leukocytes from patients with vascular disease
[38] as well as in advanced and early atherosclerotic lesions [39, 40, 41]. In the
atherosclerotic process, upregulated expression of enzymes that regulate DNA
methylation has been observed [42]. DNMTs can promote DNA hypermethylation in the
promoter region of anti-inflammatory and anti-atherosclerotic factors, leading to
repression of their expression [43]. Oscillatory shear stress, a crucial factor
involved in the initiation and development of atherosclerosis, has been found to
upregulate DNMT1 expression in endothelial cells. Furthermore, in a mouse model,
blood flow disturbed by partial carotid ligation surgery upregulates DNMT1
expression in the arterial endothelium, leading to DNA hypermethylation in the
promoter of mechanosensitive transcription factors, including homeobox A5 (HoxA5)
and kruppel-like factor (KLF) 3 [43].
HoxA5 is involved in the regulation of endothelial functions such as migration,
inflammation and angiogenesis [43]. Inhibition of HoxA5 markedly increases the
attachment of monocytes to endothelial cells, indicating the essential role of
flow-mediated HoxA5 function in the regulation of endothelial inflammation [43].
Moreover, the zinc finger transcription factors KLFs play important roles in
vascular biology, being involved in counteracting cytokine-induced adhesion
molecule expression and immune cell adhesion [44]. In human endothelial cells,
haemodynamically disturbed flow upregulates DNMT3A and inhibits KLF4 expression
[45]. Moreover, ox-LDL, a major atherosclerotic risk factor, reduced the
expression of KLF2 and of the cellular repressor of E1A-stimulated genes (CREG)
by upregulating DNMT1 and DNMT3B, respectively [46]. DNMT1 upregulation has also
been observed to modulate pro-inflammatory activation of
atherosclerosis-associated macrophages and the progression of atherosclerosis
[47, 48]. DNMT1 is induced in macrophages after treatment with proinflammatory
cytokines, such as lipopolysaccharide (LPS) and interferon-gamma
(IFN-). Consistently, DNMT1 expression is elevated in atherosclerotic
plaque macrophages from human and mouse samples [47]. Increased DNMT1 expression
has been shown to promote macrophage activation by suppressing KLF4 expression,
catalysing DNA methylation of the KLF4 promoter region [47]. Furthermore,
upregulation of macrophage DNMT1 is able to suppress peroxisome proliferator
nuclear receptor (PPAR)--mediated anti-inflammatory effects. Indeed,
PPAR- has been identified as a target of DNMT1-regulated DNA
methylation [48]. Moreover, the DNMT inhibitor as well as antioxidant molecule
N-acetylcysteine can restore aberrant hypermethylation through demethylation and
significantly attenuate vascular inflammation and endothelial dysfunction [45, 46].
Studies have also indicated an important role of the hydroxylation of methyl
groups by TET2 in counteracting endothelial and macrophage activation in
atherosclerosis [49, 50]. TET2 has been found to be downregulated in
atherosclerotic lesions and involved in the progression of atherosclerosis [49].
Low shear stress downregulates expression of TET2 and decreases expression of
autophagy-related genes (Beclin1 and microtubule-associated protein1 light chain
3) by repressing endothelial cell autophagy [51]. By contrast, autophagy level
and autophagy-related gene expression are upregulated by TET2 overexpression,
which also improves endothelial function [51]. Furthermore, in ox-LDL-treated
vascular endothelial cells, autophagy and autophagic flux are improved by TET2
overexpression and decreased by TET2 silencing [52]. In ApoE mice, TET2
overexpression markedly decreases atherosclerotic lesions, by promoting autophagy
and downregulating the expression of proinflammatory factors, such as monocyte
chemoattractant protein 1 (MCP-1, also known as chemokine (CC-motif) ligand 2,
CCL2), VCAM-1, ICAM-1, and IL-1 [52].
5.2 RNA Methylation
RNA methylation is a reversible post-transcriptional modification of RNA. It can
affect the phenotype of cells by influencing transcription, splicing, stability,
trans-nuclear transport, and translation of RNA [53, 54]. RNA methylation can be
found in both coding and non-coding RNA molecules [55], and includes the addition
of either a single or a double methyl group at specific nucleotide residues in
RNA, such as N6-methyladenosine (m6A) [56]. m6A methylation is the most common
and abundant RNA molecular modification in eukaryotes [53]. It is mostly observed
at the 5 end of the terminal exon, near the stop codon; however, it may also
occur at the 3 untranslated region (UTR) and within long internal exons
[56]. m6A methylation is regulated by three groups of proteins known as
methyltransferases (writers), demethylases (erasers), and m6A binding proteins
(readers) [57]. Recent evidence shows an association between m6A methylation and
cardiovascular atherosclerotic risk factors and endothelial inflammation [56, 58, 59]. The methyltransferase-like 14 (METTL14) plays an important regulatory role
in endothelial inflammation by regulating m6A modification of forkhead box O
(FOXO) 1 mRNA [60]. Mechanistically, the protein METTL14 has a direct binding
affinity to the mRNA of the transcription factor FOXO1. This interaction leads to
an increase in the m6A modification (methylation) of FOXO1 mRNA. The m6A
modification enhances the translation of FOXO1 mRNA by facilitating its
recognition by the YTH N6-methyladenosine RNA binding protein (YTHDF)-1 [60].
Additionally, METTL14 has been found to interact with FOXO1 and directly act on
the promoter regions of VCAM-1 and ICAM-1. This action promotes the transcription
of these genes, which are involved in the inflammatory response of endothelial
cells [60]. In vivo experiments have shown that METTL14 gene
knockout significantly reduces the development of atherosclerotic plaques and the
overwhelming inflammatory response of macrophages [60, 61]. Specifically, through
m6A modification, METTL14 upregulates the expression of the adaptor protein
myeloid differentiation primary response 88 (MyD88), which affects the
transcription of IL-6 through NF-B signalling [61]. In
endothelial cells, the regulation of RNA methylation by METTL3 plays an essential
role in endothelial function and angiogenesis, potentially affecting the
processing of angiogenic microRNAs (let-7e and miR-17-92 clusters) [62]. METTL3
also acts during ox-LDL-induced monocyte inflammation, where, in cooperation with
YTHDF-2, it modifies peroxisome proliferator-activated receptor-gamma coactivator
(PGC)-1 mRNA, mediating its degradation, and thereby enhancing the
inflammatory response [63].
In addition to the methylation process, mRNA demethylation enzymes also play a
significant role in inflammation and atherosclerosis. One such enzyme is FTO (fat
mass and obesity-associated), which has been reported to be involved in the
regulation of vascular inflammation [64]. The knockdown of FTO enhances the mRNA
and protein expression of KLF2 and endothelial nitric oxide synthase (eNOS) but
attenuates TNF--induced VCAM-1 and ICAM-1 expression, as well as the
adhesion of monocytes to endothelial cells. Conversely, FTO overexpression
significantly upregulates the mRNA and protein levels of VCAM-1 and ICAM-1 as
well as downregulating those of KLF2 and eNOS [65].
5.3 Histone Post-Translational Modifications
In eukaryotic cells, epigenetic marks involve reversible histone modifications,
including methylation and acetylation, among others, which induce conformational
shifts in the protein structure allowing the docking of specific regulatory
proteins [66]. Histone methylation, one of the most important post-translational
modifications, is regulated by histone methyltransferases (HMTs) and histone
demethylases (HDMTs). HMTs transfer methyl groups to arginine (R) to form mono-
or di-methylated residues, or to lysine (K) which can accept one, two or three
methyl groups [67]. Methylation of lysine residues at the 4th site of H3 (H3K4)
is associated with gene activation, and occurs mainly in regions of active
transcription, such as the transcription start site, promoter, and enhancer
regions. In contrast, methylation of lysine residues at the 9th and 27th sites
(H3K9, and H3K27) is associated with gene silencing (Fig. 2) [67]. An
interconnection between histone and DNA methylation in atherosclerosis has been
demonstrated. The HMT enhancer of zeste homolog-2 (EZH2) induces the expression
of the DNMT1, which, in turn, increases DNA methylation of ATP-binding cassette
transporter A1 (ABCA1) promoter, inhibiting its expression and thus promoting the
formation of macrophage-derived foam cells and the development of atherosclerosis
[68]. When endothelial cells are exposed to elevated levels of LDL (resembling a
state of hypercholesterolaemia), by inducing DNMT1, LDL recruit a transcriptional
repressor complex (methyl-CpG binding protein 2, MeCP2, and EZH2) to the KLF2
promoter, which results in a shift in promoter occupancy that causes closed
chromatin and repression of KLF2 expression [46]. These studies suggest that EZH2
and DNMT1 may form a positive feedback regulatory system. On one hand, they
regulate foam cell formation by inhibiting ABCA1 expression; on the other, they
influence endothelial dysfunction by suppressing KLF2, and jointly promoting the
atherosclerotic process. Recent studies have demonstrated a link between histone
methylation and high glucose-induced vascular inflammation and accelerated
atherosclerosis. Transient hyperglycaemia has been shown to be able to induce
upregulation of the NF-B p65 subunit gene in endothelial cells
which is associated with increased methylation of H3K4 (H3K4me1) and decreased
methylation of H3K9 (H3K9me2 and H3K9me3) on the NF-B p65
promoter [69]. At the same time, in human endothelial cells treated with high
glucose, dimethylated and trimethylated H3K4 forms are enriched at the promoter
of the MCP-1 gene, and the HMTs mixed-lineage leukaemia (MLL) and
Su(var)3‑9, enhancer of zeste, Trithorax (SET) domain-containing protein 7 (SET7)
are increased, while the histone demethylase LSD1 is decreased [70]. In human
aortic cells, the HMT SET7 has also been found to mediate glucose-induced
inflammation through epigenetic regulation of the transcription factor
NF-B [71]. Knockdown of SET7 reduces the H3K4me1 mark and
abolishes NF-B-dependent inflammatory signalling [71].
Concordantly, SET7 has been observed to contribute to vascular dysfunction in
patients with type 2 diabetes mellitus (T2DM). In peripheral blood mononuclear
cells from T2DM patients, an increase of SET7 expression and SET7-dependent
monomethylation of H3K4 (H3K4me1) on the NF-B p65 promoter is
observed. This epigenetic signature is associated with upregulation of
NF-B, subsequent transcription of inflammatory genes, and
increased plasma levels of ICAM-1 and MCP-1 [71]. Moreover, epigenetic changes
have been implicated in the persistence of vascular inflammation induced by
hyperglycemia [72]. In response to hyperglycaemia, the HMT SET7 accumulates in
the nucleus of endothelial cells, promoting IL-8, ICAM-1 and CXC motif chemokine
ligand 2 (CXCL2) expression in an H3K4me1-dependent manner. SET7 also inhibits
heme oxygenase 1 (HMOX1) expression in an H3K4me1-independent fashion to regulate
insulin sensitivity and “hyperglycemic memory” [72]. In endothelial cells,
oxygen-glucose deprivation/reperfusion injury upregulates histone demethylase
Jumonji domain-containing protein 3 (JMJD3) expression, leading to greater JMJD3
interactions with NF-B and CCAAT-enhancer-binding protein at
the IL-6 gene promoter, which decreases the trimethylated form of H3K27 to
promote IL-6 expression and regulate the inflammatory response [73]. A similar
mechanism is active in endothelial cells stimulated with LPS, where increased
JMJD3 expression induces demethylation of H3K27me3 and activates the expression
of target genes by interaction with NF-B [74].
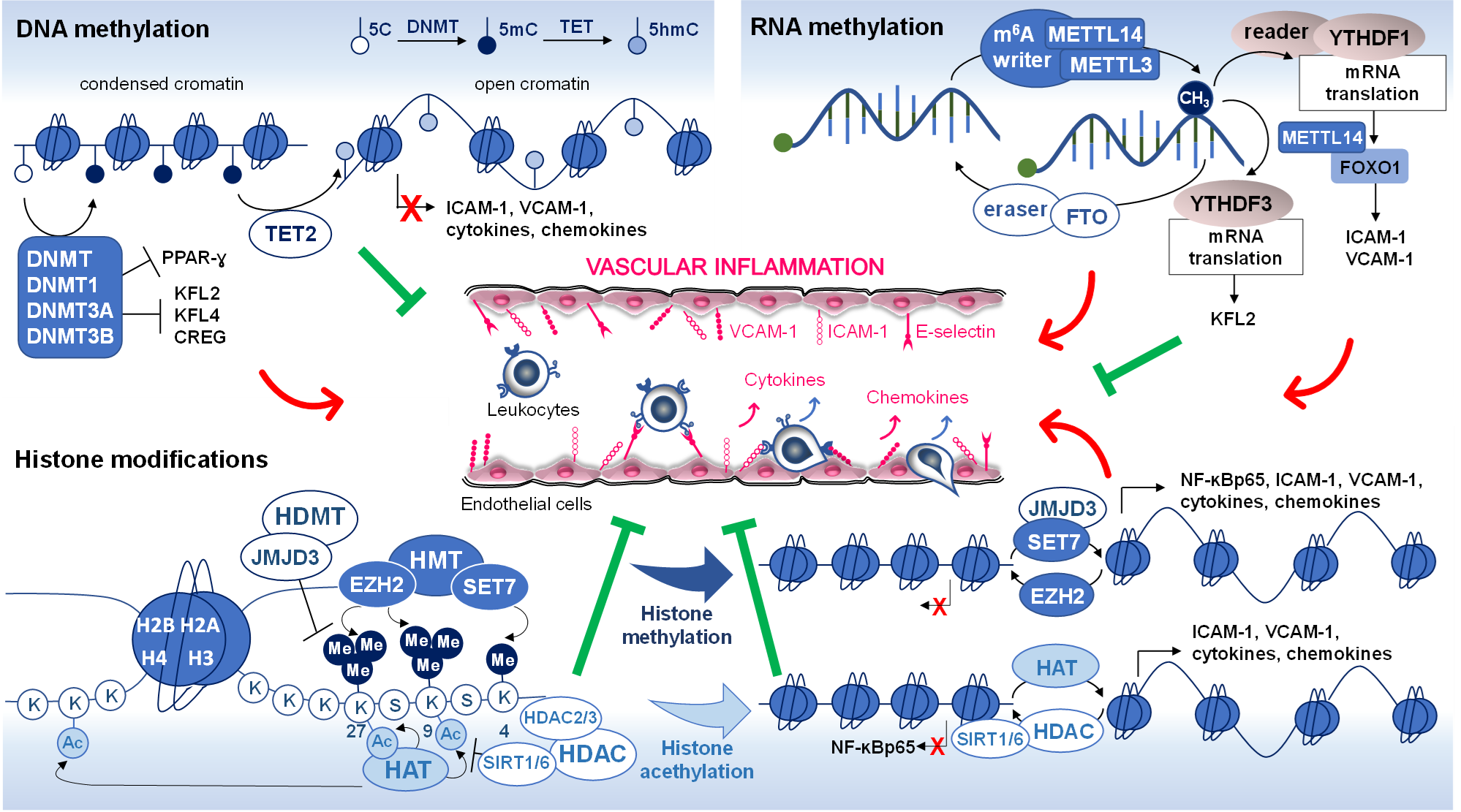 Fig. 2.
Fig. 2.
Epigenetic modifications in vascular inflammation. DNA
methylation, RNA methylation, and histone modifications (methylation and
acetylation) are common epigenetic alterations that have been associated with
changes in gene expression affecting vascular inflammation. The red arrow
indicates increased levels of expression and the green lines (block sign)
indicates reduced levels of expression. DNMT, DNA methyltransferase; TET,
ten-eleven translocation; 5C, 5-cytosine; 5mC, 5-methylcytosine 5hmC,
5-hydroxymethylcytosine; KLF2 and 4, kruppel-like factor 2 and 4; CREG, cellular
repressor of E1A-stimulated genes; PPAR-, peroxisome proliferator
nuclear receptor-; ICAM-1, intercellular adhesion molecules-1; VCAM-1,
vascular cell adhesion molecule-1; MELLT3 and 14, methyltransferase like 3 and
14; YTHDF1 and 3, YTH N6-methyladenosine RNA binding protein 1 and 3; FTO,
demethylase fat mass- and obesity-associated protein; FOXO1, forkhead box O1, and
O3; HMT, histone methyltransferase; EZH2, enhancer of zeste homolog-2; SET7,
Su(var)3‑9, enhancer of zeste, Trithorax (SET) domain-containing protein 7; HDMT,
histone demethylases; JMJD3, Jumonji domain-containing protein-3; HAT, histone
acetyltransferase; HDAC, histone deacetylase; SIRT1 and 6, sirtuin 1 and 6;
NF-B p65, nuclear factor-B p65 subunit; Ac,
acetylation; Me, methylation.
Another important post-transcriptional modification is histone acetylation,
regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs).
Histone modifications through acetylation are fundamental for remodelling
chromatin and consequently activating gene expression. The imbalance between
acetylation and deacetylation activity causes transcriptional dysregulation
associated with several disorders. HATs consist of two types: type A, mainly
localised at the nucleus, acetylates nucleosomal histones by transferring an
acetyl group from acetyl-CoA, and include p300/cyclic AMP response
element-binding protein (CBP) families; type B, located in the cytosol,
acetylates free histones or non-histone proteins [75]. Acetylation of nucleosomal
histones is generally associated with the activation of transcription, since the
addition of an acetyl group to lysine neutralizes the positive charge of lysine
residues, resulting in decreased histone-DNA interactions, with decondensation of
chromatin and increased accessibility of DNA to transcription factors (Fig. 2).
HDACs remove acetyl groups from acetylated proteins, consequently repressing gene
expression by condensing nucleosomes. They are classified into four categories:
class I (HDAC1,2,3, and 8), class II (2a: HDAC4,5,7, and 9; 2b: HDAC6, and 10),
class III (sirtuin, SIRT1-7), and class IV (HDAC11) [76]. Alterations in the
expression level of HDACs have been related to inflammatory diseases such as
atherosclerosis. Indeed, HDAC2 may be downregulated by ox-LDL, resulting in
increased oxidative stress [77]. HDAC3 seems to have a protective role for
endothelial integrity, and HDAC3 deletion has been linked to reduced endothelial
cell survival and increased atherosclerosis [78]. In human advanced plaques,
increased HDAC9 is associated with the expression of proinflammatory markers in
macrophages [79]. Macrophages from HDAC9-deficient mice are less responsive to
LPS stimulation in release of proinflammatory cytokines. In addition, HDAC9
deficiency upregulates histone H3 and H4 acetylation and increases levels of
ABCA1 and PPAR, preventing the efflux of cholesterol [80]. Thus, HDAC9
deficiency results in macrophages that are polarized towards promoting
inflammation resolution and reverse cholesterol transport, which can brake
atherosclerosis progression and promote lesion regression [80]. Sirtuins, now
known as class III KDACS (lysine deacetylases), are nicotinamide adenine
dinucleotide (NAD)-dependent HDACs and play pivotal roles in the regulation
of metabolism, stress responses, and ageing processes [81]. The major function of
sirtuins includes deacetylation of histones as well as some non-histone proteins
like NF-B, FOXOs, PPAR-, PGC1-, enzymes,
and structural proteins. SIRT1 and SIRT6 protect against atherosclerosis by
preventing endothelial dysfunction through pleiotropic effects on oxidative
stress and inflammation. SIRT1 reduces inflammation by direct deacetylation of
the NF-B p65 subunit, whereas SIRT6 reduces inflammation by
deacetylating H3K9 at NF-B target gene promoters [81]. Histone
acetylation mediates the expression and secretion of inflammatory mediators in
various infectious diseases. During the infection process, monocytes increase
secretion of IL-8 through hyperacetylation of histone H3 and H4 at the promoter
of IL-8 in addition to NF-B-activated transcription [82]. In
this context, endothelial cells increase the expression of inflammatory genes,
including IL-6, IL-8, granulocyte colony-stimulating factor (G-CSF),
granulocyte-macrophage colony-stimulating factor (GM-CSF), and IFN-,
regulated by Rho-GTPase-related acetylation of histone H3 and H4, leading to the
development of chronic vascular lesions [83]. In endothelial cells, HDAC
inhibitors are reported to markedly reduce TNF--stimulated VCAM-1
expression [84, 85], as well as, the LPS-induced expression and secretion of
pro-inflammatory genes, such as MCP-1, IL-6, and IL-1, by enhancing
histone H3 acetylation and associated upregulation of oxidative stress protective
genes, including catalase, superoxide dismutase 2 (SOD2), FOXO3A, and
PGC-1 expression [86]. In many chronic diseases, histone acetylation
plays a critical role in endothelial dysfunction associated with inflammation. In
addition to inducing DNA hypomethylation, in endothelial cells LDL promote the
acetylation of histone H3K9 and H3K14 in the promoter of p66shc, a major mediator
of oxidative stress-induced vascular dysfunction, thereby increasing endothelial
p66shc expression [87]. Furthermore, ox-LDL induces inflammatory activation of
human endothelial cells via the lectin-like oxidized LDL receptor-1 (LOX-1) and
extracellular regulated kinases (ERK1/2) signalling pathway, leading to
acetylation of histone H3 and H4 on the promoters of IL-8 and MCP-1 [88]. Histone
acetylation promotes the recruitment of NF-B p65/RelA and RNA
polymerase II to the promoters of IL-8 and MCP-1, increasing their expression.
Pre-treatment with anti-inflammatory agents such as statins prevents
ox-LDL-induced histone acetylation on the IL-8 and MCP-1 promoters, decreasing
the expression of the two inflammatory cytokines [88]. Moreover, the
proatherogenic lipid lysophosphatidylcholine induces mitochondrial ROS-dependent
H3K14 acetylation, increasing the binding of the proinflammatory transcription
factor activator protein 1 (AP-1) in the promoter of ICAM-1 and inducing ICAM-1
transcription in endothelial cells [89]. Upon long-term inflammation, high
amounts of proinflammatory cytokines affect endothelial function by
downregulating RNase1, a circulating extracellular endonuclease, regulating
vascular homeostasis of extracellular RNA and acting as a vessel- and
tissue-protective enzyme [90]. TNF-- or IL-1-challenged
endothelial cells reduce RNase1 expression by inducing hypoacetylation of histone
H3K27 and histone H4 through HDAC2 accumulation to the RNase1 promoter, while
class I HDAC specific inhibition abolishes the changes [91]. In pulmonary artery
endothelial cells, lipoxin A4 (LXA4), an endogenous lipoxygenase-derived
eicosanoid mediator, exerts potent dual anti-inflammatory and pro-resolving
effects by increasing formyl peptide receptor 2 (recently renamed ALX/FPR2) mRNA
and protein levels through the HAT p300 which restores chromatin accessibility
[92]. It is noteworthy that, small molecules of plant origin, including flavones,
are nutraceutical bioactive compounds known to interfere with HDAC class I
enzymes and to enhance acetylation, restoring cell homeostasis. This occurs
because flavones, i.e., apigenin and luteolin, can interact as ligands with HDAC1
and 2 at the active site binding pocket [93]. Regulation of HDAC activity by
dietary flavones could have important implications in developing epigenetic
therapy to regulate the cell gene expression. Furthermore, it has been shown that
the natural polyphenol resveratrol can bind SIRT1 by enhancing its interaction
with RelA/p65, leading to reduced activity of NF-B [94]. In
endothelial cells, resveratrol can inhibit the inflammatory response by
regulating the transcriptional and translational levels of SIRT2, SIRT5, and
SIRT7 [94].
Overall, current knowledge underlines an important role of nucleic acids and
histone modifications in regulating vascular inflammation and atherosclerosis and
suggests them as potential targets in the treatment of inflammatory diseases,
such as atherosclerosis.
5.4 Non-Coding RNAs
Advanced genome- and transcriptome-wide analyses have revealed that only less
than 2% of the human genome contains protein-coding transcripts, while more than
75% is transcribed into ncRNAs with no protein-coding potential [95]. Based on
their sizes, ncRNAs can be divided into two groups: the long non-coding RNAs
(lncRNAs), that are more than 200 nucleotides long, and the short ncRNAs that are
less than 200 nucleotides in length, including microRNAs (miRNAs). Generally,
ncRNAs can be categorized into housekeeping ncRNAs and regulatory ncRNAs. The
former, profusely expressed in all cell types, including ribosomal, transfer,
small nuclear, and telomerase RNAs, are necessary for cells to survive; the
latter, including lncRNAs and miRNAs, usually participate in regulation of gene
expression, acting at epigenetic, transcriptional, and post-transcriptional
levels [95]. In this section, an update of ncRNAs’ contribution to inflammation
and immunity is given.
5.4.1 Long Non-Coding RNAs
LncRNAs constitute the major portion of the non-coding component of the human
genome. A growing body of data suggests that lncRNAs may regulate genes either
in cis (on neighbouring genes) or in trans (on distant genes)
through specific interactions with proteins, DNA, and other types of RNA [96].
Numerous lncRNAs are functionally correlated with endothelial dysfunction,
vascular inflammation, and associated cardiovascular diseases [97]. It is worth
noting that several lncRNAs play a crucial role in NF-B
signalling during inflammatory responses (Fig. 3) [24, 98]. One of the first
known lncRNAs in humans was lncRNA H19, a key mediator of endothelial cell
function, which is down-regulated by proinflammatory cytokines, such as
IL-1 and TNF- [99]. More recently, Hofmann et al.
[100] found that H19 is expressed in the adult endothelium and its depletion
results in premature endothelial senescence. Furthermore, H19 loss-of-function
activates the inflammatory signalling pathway and impairs endothelial cell
function. Correspondingly, the overexpression of H19 ameliorates endothelial
function in aged aortas [100]. These results show a central role of H19 in
reducing endothelial cell dysfunction in ageing by controlling endothelial cell
senescence, proliferation, and inflammatory activation. However, recent evidence
also points out a contradictory role for H19, since its overexpression leads to
an increase of p38 mitogen-activated protein kinase (MAPK) activity and p65
nuclear translocation/expression in human endothelial cells, with activation of
the NF-B pathway [101, 102]. Endothelial functions are also
affected by the lncRNA named metastasis-associated lung adenocarcinoma transcript
1 (MALAT1) [103, 104]. In LPS-treated human lung microvascular endothelial cells,
MALAT1 upregulates ICAM-1 expression by competitively binding to the microRNA
miR-150-5p, whereas MALAT1 silencing or miR-150-5p overexpression decreases the
expression of pro-inflammatory mediators, including IL-6, IL-1,
TNF-, and E-selectin, thus alleviating vascular injury [105].
Furthermore, Zhao et al. [106] showed that MALAT1 regulates the
LPS-induced inflammatory response through its interaction with
NF-B (Fig. 3). Mechanistically, MALAT1 interacts with
NF-B subunits p65 (RelA) and p50 to inhibit
NF-B DNA binding activity and production of the
proinflammatory cytokines TNF and IL-6 in macrophages. These findings
suggest that MALAT1 may function as an auto-negative feedback regulator of
NF-B to help fine-tune innate immune responses. The lncRNA
Lethe (named after the mythological river of forgetfulness for its role in
negative feedback) was one of the first lncRNAs demonstrated to be involved in
modulating NF-B signalling [107]. Lethe, a
chromatin-associated lncRNA, is selectively induced by proinflammatory cytokines
via NF-B. Specifically, Lethe physically associates with RelA
(p65) to block the DNA binding activity of NF-B. Therefore,
Lethe, which is induced in a p65-dependent fashion, appears to act as a negative
feedback regulator of NF-B [108]. Lethe levels decrease with
ageing, a physiological state associated with increased NF-B
activity. Lethe is expressed in mouse embryonic fibroblasts upon exposure to
TNF- and, IL-1, but is not responsive to toll-like receptor
agonists, indicating that it may have a function in inflammation, but not in
innate immunity [107]. Another important lncRNA in inflammation is Antisense
Non-coding RNA in the INK4 Locus (ANRIL) [109]. In human vascular endothelial
cells, ANRIL is remarkably induced in response to pro-inflammatory factors in an
NF-B-dependent manner. Elevated ANRIL affects the expression
of a large portion of inflammatory genes downstream of NF-B,
such as IL-6 and IL-8 [110]. Mechanistic studies indicate that ANRIL forms a
functional complex with the transcriptional factor Yin Yang 1 to exert
transcriptional regulation on NF-B-dependent inflammatory
genes. Together, these reports suggest that lncRNAs both positively and
negatively regulate NF-B-dependent gene expression,
contributing to the fine regulation of NF-B-responsive genes.
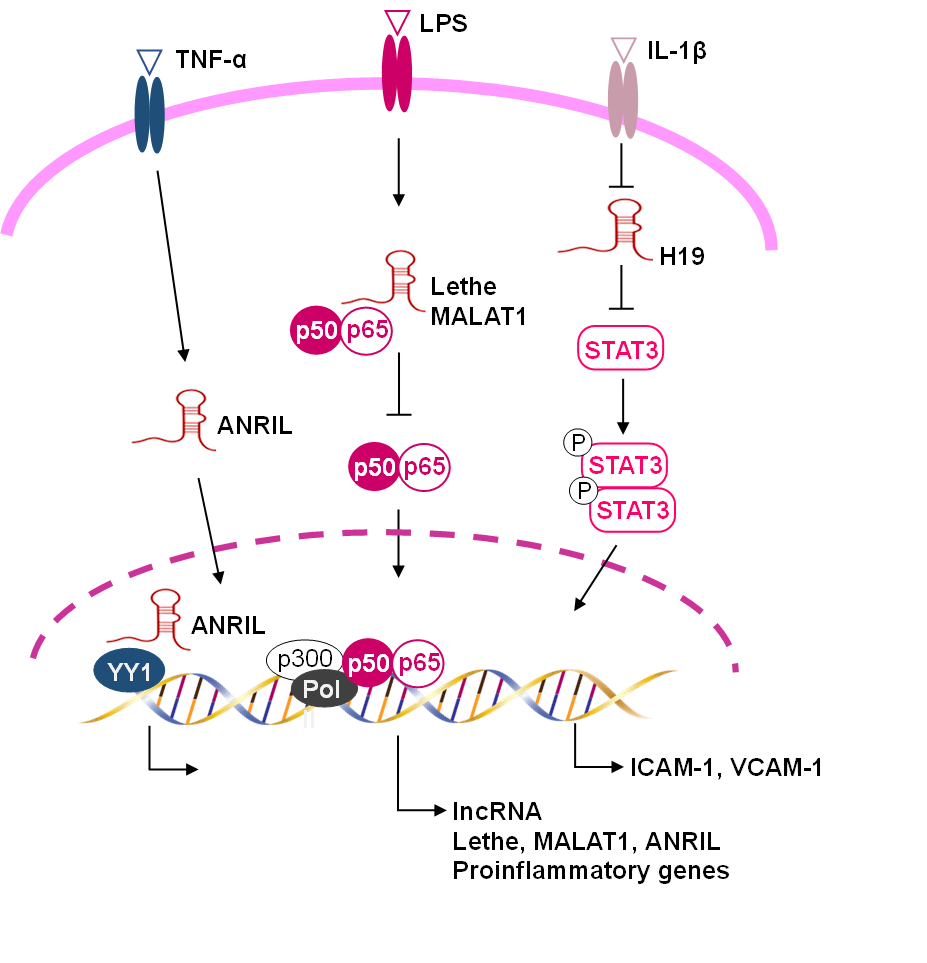 Fig. 3.
Fig. 3.
Functional lncRNAs involved in the regulation of vascular
inflammation. LncRNAs regulate, either positively or negatively, the expression
of genes involved in vascular inflammation. LncRNA Lethe and MALAT1 (metastasis
associated lung adenocarcinoma transcript 1) suppress the NF-B
signalling pathway. LncRNA H19 inhibits the phosphorylation of signal transducer
and activator of transcription 3 (STAT3) and reduces vascular cell adhesion
molecule 1 expression. LncRNA ANRIL (Antisense Non-coding RNA in the INK4 Locus)
interacts with the transcription factor yin yang 1 (YY1) to form a functional
complex that regulates proinflammatory gene expression.
5.4.2 MicroRNAs
miRNAs, a class of ncRNAs with important roles in regulating gene expression,
have emerged as key players in vascular inflammation and chronic inflammatory
diseases [111]. Usually, miRNAs are transcribed from DNA sequences into primary
miRNAs (pri-miRNAs) and processed into precursor miRNAs (pre-miRNAs) and mature
miRNAs. In most cases, miRNAs interact with the 3 UTR of target mRNAs to
suppress expression [112]. Mechanistically, miRNAs function by
post-transcriptionally regulating protein accumulation and by regulating the
transcription of other miRNAs [113]. Within a given cell type, a single miRNA can
target hundreds of mRNAs, and a single mRNA is often the target of multiple
miRNAs [112]. miRNAs can either silence the expression of the positive signalling
proteins or the inhibitors of the same pathway [112].
Several studies have pointed out miRNAs as regulators of the main
proinflammatory cytokines. The regulatory relationship between cytokines and
miRNAs seems to be reciprocal: not only do miRNAs target cytokine mRNA and
thereby regulate cytokine expression, but also the cytokine signalling likewise
has an impact on miRNA expression [114]. It has been observed that hyperglycaemia
strongly induces miR-155, which in turn can directly raise TNF- [115].
In vivo studies have confirmed that mice overexpressing miR-155 produce
more TNF- when challenged with LPS [116]. Furthermore, miR-155, as a
central regulator of the immune system, regulates the expression of MyD88 adaptor
protein by translational repression leading to the suppression of IL signalling
(Table 1, Ref. [108, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138] and Fig. 4) [117]. As
occurs with other cytokines, the expression of IL-6, a hallmark of chronic
inflammatory states, is also regulated by miRNAs [139]. Chen et al.
[140] demonstrated that IL-6 down-regulates miR-223 expression, leading
secondarily to an increase in STAT3, which then drives the expression of IL-6 and
IL-1 in a positive regulatory loop. Up-regulation of miR-146a/b, another
important regulator of IL-6 metabolism, provides negative feedback in the
inflammatory response [118, 141]. In primary human fibroblasts, the
overexpression of miR-146a/b suppresses IL-6 and IL-8 secretion down-regulating
interleukin-1 receptor-associated kinase (IRAK)-1, a crucial component of the
IL-1 receptor signal transduction pathway, thus restraining the excessive
secretion of inflammatory cytokines, and limiting inflammation [118].
Furthermore, in response to pro-inflammatory cytokines, miR-146a/b is strongly
induced in endothelial cells and inhibits expression of endothelial adhesion
molecules and endothelial activation [142]. Notably, miR-146a/b induction is
delayed and sustained compared to the expression of leukocyte adhesion molecules,
and in fact coincides with the down-regulation of inflammatory gene expression
[142].
Table 1.Pro-inflammatory and anti-inflammatory miRNAs and their targets
and functions involved in vascular inflammation.
| Pro-inflammatory miRNAs |
| miRNAs |
Target (s) |
Functions |
References |
| miR-155 |
MyD88, κB-RAS1, NF-κB p65 |
miR-155 regulates the immune response and vascular inflammation |
[116, 117] |
| miR-92a |
KLF2, KLF4 |
miR-92a enhances the expression of endothelial adhesion molecules and endothelium-leukocyte adhesion |
[136] |
| miR-34a |
SIRT1 |
miR-34a regulates flow dependent endothelial inflammation |
[138] |
| miR-132 |
SIRT1, p300 |
miR-132 is a pleiotropic miRNA that both counteracts and promotes endothelial inflammation |
[136, 137] |
| Anti-inflammatory miRNAs |
| miRNAs |
Target (s) |
Functions |
References |
| miR-126 |
VCAM-1 |
miR-126 blocks the adhesion and infiltration of leukocytes into vascular wall |
[119, 120, 121, 122] |
| miR-221, miR-222, miRNA-141, miRNA-17-3p |
ICAM-1 |
miR-221, miR-222, miRNA-141, and miRNA-17-3p reduce leukocyte-endothelial cell adhesion |
[108, 123, 124, 125] |
| miR-31 |
E-selectin |
miR-31 inhibits leukocyte adhesion and rolling on the endothelium |
[126, 127] |
| miR-146a/b |
IRAK1, TRAF6, CARD10 |
miR-146a/b inhibits endothelial adhesion molecule expression and endothelial activation |
[118] |
| miR-100 |
mTOR |
miR-100 regulates vascular inflammation and preserves endothelial functions |
[135] |
| miR-181b |
Importin-3, CARD10 |
miR-181b inhibits vascular inflammation |
[132, 133, 134] |
| miR-125a/b |
TRAF6 |
miR125a/b decreases the accumulation of macrophages and neutrophils in the myocardium |
[128, 129] |
| miR-10a |
TAK1 and -TRC |
miR-10a regulates endothelial athero-susceptibility/protection by targeting key regulators of IκB- degradation |
[130] |
| miR-23b |
IKK, NF-κB |
miR-23b regulates inflammatory cytokine pathways |
[131] |
MyD88, myeloid differentiation primary response 88; B-RAS1,
NF-B p65 subunit, nuclear factor-B p65
subunit; KLF2 and 4, kruppel-like factor 2 and 4; SIT1, sirtuin 1; VCAM-1,
vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecules-1;
IRAK1, interleukin-1 receptor-associated kinase 1/2; TRAF6, TNF
receptor-associated factor 6; CARD10, caspase recruitment domain family member
10; mTOR, mammalian target of rapamycin; TAK1, transforming growth
factor- activated kinase 1; -TRC, -transducin
repeat-containing gene; IKK, IB kinase .
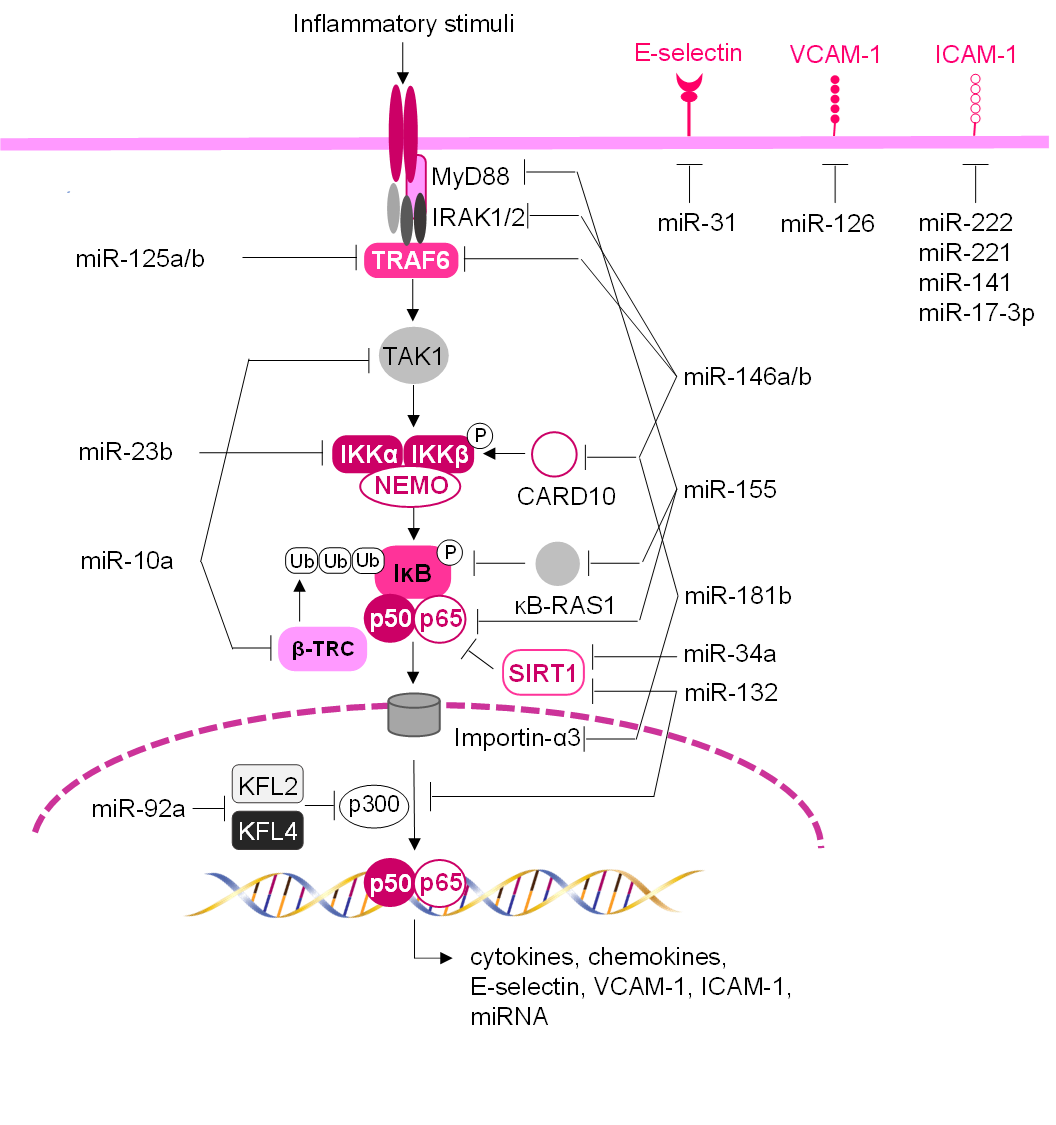 Fig. 4.
Fig. 4.
Schematic network of miRNAs regulating vascular inflammation and
adhesion molecule expression. miRNAs can regulate the endothelial inflammatory
response and leukocyte-endothelium adhesion, by directly targeting endothelial
adhesion molecule transcripts and/or by modulating NF-B
signaling pathway and subsequent transcription of pro-inflammatory genes. In
unstimulated cells, NF-B subunits (e.g., p65/p50) are
sequestered in the cytoplasm through their interaction with
inhibitor-B (IB). In response to
inflammatory stimuli, the IB kinase (IKK) complex
phosphorylates IB, which is then ubiquitylated by
-transducin repeat-containing gene (-TRC), leading to its
degradation. Then NF-B can translocate to the nucleus, where
it can bind to its transcriptional targets, which include leukocyte adhesion
molecules, chemokines, and cytokines. miRNAs can modulate the
NF-B signaling pathway to various levels thus by dampening its
activation.
Overall, various miRNAs are involved in the regulation of immune responses and
inflammatory processes and might play a role in controlling the switch from an
early proinflammatory response to the resolution phase of the inflammatory
process [143, 144]. The role of miRNAs in vascular inflammation is based not only
on regulating the inflammatory stimulus but also on the response of the vascular
endothelium to inflammatory triggers. In the following sections, the regulatory
function of miRNAs in the expression of the endothelial adhesion molecules is
described.
5.4.3 Expression of Endothelial Adhesion Molecules: Role of
miRNAs
Recent evidence supports the role of miRNAs in regulating vascular inflammation
and particularly the expression of endothelial cell adhesion molecules including
E-selectin, ICAM-1, and VCAM-1 [111, 145]. Various miRNAs can target the
expression of endothelial adhesion molecules both directly and indirectly through
modulation of the NF-B pathway (Table 1 and Fig. 4) [145]. One
of the most highly expressed miRNAs in endothelial cells is miR-126 (also
referred to as miR-126-3p), which has been associated with vascular inflammation
(Table 1 and Fig. 4) [119]. This miRNA can bind to the 3 UTR VCAM-1
transcript, inhibiting mRNA translation and protein synthesis, thereby blocking
adhesion and infiltration of leukocytes into the vascular wall. Transfection of
endothelial cells with an antisense construct targeting endogenous miR-126 allows
an increase in TNF--stimulated VCAM-1 expression [119]. The expression
of miR-126 is regulated by factors as varied as oestrogens and endotoxins. The
oestrogen E increases miR-126, which decreases VCAM-1 and monocyte adhesion
[120]. Endotoxin LPS downregulates miR-126 at the transcriptional level leading
to depression of VCAM-1, while the overexpression of miR-126 attenuates
LPS-induced vascular injury [121]. In mice, mimic-miR-126 inhibits vascular
inflammation by targeting VCAM-1 [122]. The expression of ICAM-1 is also
regulated by multiple miRNAs such as miR-221 and, miR-222 [123], which are
complementary to the ICAM-1 3 UTR region and modulate ICAM-1 expression at
the post-transcriptional level leading to a significant reduction in
leukocyte-endothelium adhesion [108]. A similar effect has been shown for
miR-141and miR-17-3p (a passenger miRNA of miR-17), other miRNAs targeting ICAM-1
[124, 125]. Moreover, miR-31 directly downregulates E-selectin expression,
induced by the pro-inflammatory cytokines IL-1 and TNF-,
impairing leukocyte adhesion and rolling on the endothelium [126, 127].
Several miRNAs are indirectly involved in the regulation of endothelial cell
adhesion molecules, affecting NF-B signalling (Table 1 and
Fig. 4) [145]. miR-146a has been found to be transcriptionally induced by
NF-B in response to the activation of innate immune signalling
in monocytes [146]. miR-146a targets the adaptor proteins tumour necrosis factor
receptor-associated factor 6 (TRAF6) and IRAK1/2 and can inhibit activation of
the NF-B pathway, suggesting that miR-146a participates in a
negative feedback loop to control NF-B signalling [147, 148].
In addition to pro-inflammatory agents, dysmetabolic stimuli may also regulate
endothelial adhesion molecules by affecting miRNA activities. Ox-LDL can
‘de-repress’ NF-B activity by reducing miR-125a, counteracting
the effects of ox-LDL on inflammation, namely the expression of ICAM-1 and VCAM-1
and leukocyte adhesion [128]. Moreover, transfection of miR-125b into the heart
significantly suppresses the expression of ICAM-1 and VCAM-1, decreases the
accumulation of macrophages and neutrophils in the myocardium, and reduces serum
levels of TNF- and IL-1 in mice, by targeting TRAF6-mediated
NF-B activation [129]. Furthermore, in human aortic
endothelial cells, miR-10a impairs NF-B-mediated expression of
E-selectin, VCAM-1, MCP-1, IL-6, and IL-8 by targeting key regulators of
IB- degradation [130]. Numerous other miRNAs, such
as miR-23b and miR-181b, are also down-regulated by inflammatory stimuli to
de-repress NF-B activity, promoting endothelial activation
[131, 132]. In addition, miR-181b also targets importin-3, an importer
protein required for the nuclear translocation of NF-B, so
diminishing the downstream expression of inflammatory genes, such as VCAM-1 and
E-selectin [133]. Following research also showed that systemic delivery of
miRNA-181b inhibits NF-B activation, vascular inflammation,
and atherosclerosis in ApoEmice [134]. This study reveals the
endothelial-specific mechanisms by which miR-181b exerts its protective effect in
the vascular endothelium and provides the rationale for the potential clinical
use of miR-181b mimetics to treat chronic vascular inflammatory diseases such as
atherosclerosis [134]. A recent study by Pankratz et al. [135] has
identified miR-100 as a potent suppressor of endothelial adhesion molecule
expression, by attenuation of NF-B signalling, resulting in
decreased leukocyte-endothelium interaction in vitro and in
vivo. These findings add miR-100 to the regulatory network of anti-inflammatory
miRNAs, suggesting a critical role in the restraint of vascular inflammation and
the maintenance of an endothelial equilibrium. Furthermore, recent evidence
reports that miR-17-3p exhibits anti-inflammatory effects in endothelial cells by
inhibition of the NF-B signalling pathway and the expression
of pro-inflammatory genes. Indeed, in addition to directly targeting ICAM-1 mRNA,
miR-17-3p suppresses the LPS-induced phosphorylation of
IB and the NF-B p65 subunit
[125].
Some miRNAs, induced by proinflammatory cytokines, exert a multifunctional role.
They can target and repress several genes with complex effects on cell
physiology. miR-132 is a pleiotropic miRNA that both counteracts and promotes
endothelial inflammation. As an inflammation promoter, miR-132 increases
NF-B signalling by targeting SIRT1 to promote inflammatory
processes including ICAM-1 expression [136]. As an inflammation inhibitor,
miR-132 targets the transcriptional coactivator of NF-B p300,
which in turn modulates the transcription of miR-132. This feedback loop may
contribute to the transient expression of miR-132 [137]. Furthermore, in human
aortic endothelial cells, miR-92a boosts NF-B activity by
targeting the endothelial transcription factors KLF2 and KLF4, that inhibit
NF-B activity by competing for access to the transcriptional
coactivator p300/CBP. miR-92a thus enhances the expression of inflammatory
molecules including E-selectin and, VCAM-1 and contributes to leukocyte adhesion
[149]. Some miRNAs are associated with shear stress conditions [150]. Several
investigations have revealed that atheroprone flow (oscillatory flow) inhibits
whereas atheroprotective pulsatile blood flow increases various miRNAs including
miR-30 and miR-10a. The transcription of miR-30 is mediated by KLF2 and activated
in response to pulsatile blood flow. Then, miR-30 inhibits the activation of
endothelial cells and the expression of E-selectin, ICAM-1, and VCAM-1 [151].
miR-10a is crucial for endothelial response to different flow patterns by
regulating the expression of its direct target GATA-binding factor 6 (GATA6) and
downstream expression of VCAM-1 [152]. In contrast, miR-34a is reduced by
pulsatile blood flow and increased by oscillatory blood flow [138]. miR-34a
affects the NF-B-mediated expression of ICAM-1 and VCAM-1,
de-repressing the NF-B activity through direct regulation of
SIRT1, thus regulating the flow-dependent endothelial inflammation [138].
miR-663, another miRNA induced by oscillatory blood flow, is necessary for the
efficient transcription of E-selectin and VCAM-1 [153]. Oscillatory blood flow
also induces the c-Jun/AP-1-mediated transcription of miR-21, which targets
PPAR-, an inhibitor of the transcription factor AP-1. This feedforward
loop enables the sustained induction of miR-21, contributing to the
proinflammatory responses of the vascular endothelium, including the expression
of VCAM-1 and MCP-1, and the consequential adhesion of monocytes in
vitro [154].
5.4.4 Circulating miRNAs and Vascular Inflammation
Recent studies have reported significant levels of miRNAs in serum and other
body fluids, raising the possibility that circulating miRNAs could serve as
useful clinical biomarkers and modulators of vascular inflammation [155].
Although the traditional idea suggests that RNA molecules cannot be stable in
extracellular environments due to ubiquitous ribonucleases, miRNA has now been
shown to circulate in a stable form in various body fluids, mainly associated in
extracellular vesicles (exosomes or microvesicles, also known as microparticles)
[156, 157]. In a cohort cross-sectional study, positive correlations between
circulating miR-1185 and the expression of E-selectin and VCAM-1 have been
observed [158]. Mechanistic analysis has confirmed that miR-1185 induces a
significant increase in the VCAM-1 and E-selectin levels in human cultured
endothelial cells, suggesting a crucial role in endothelial activation and
atherosclerosis development [158]. Another study that measured levels of miR-122
in blood samples after ischaemic stroke showed a correlation between reduced
levels of miR-122 and increased expression of target genes such as VCAM-1 and
ICAM-1 in the brain [159]. A clinical trial has revealed that circulating levels
of miR-126, miR-92a, and miR-155 are significantly reduced in patients with
coronary artery disease compared with healthy subjects [160]. A recent clinical
study has also shown that circulating miR-505 is elevated in patients with
hypertension [161]. In an animal model of hypertension, miR-505 modulates the
levels of endothelial activation markers, VCAM-1, and E-selectin, as well as
monocyte-endothelium adhesion [162]. These findings linking miR-505 to
endothelial dysfunction and inflammation under hypertensive conditions support
the translational value of miR-505 as a biomarker of hypertension-associated
endothelial impairment and inflammatory injuries [162].
In addition to their potential as diagnostic biomarkers, circulating miRNAs can
be delivered to endothelial cells and regulate inflammatory responses [156, 157].
In the vascular system, microvesicles are the major form of miRNA delivery, and
the endothelium is one of the primary targets of circulating microvesicles [156, 157]. Lipoxin LXA stimulates endothelial miR-126-5p expression and its
transfer via microvesicles, thus enhancing endothelial repair functions [163].
Jansen et al. [164] demonstrated that
miR-222 is transported into recipient endothelial cells by endothelial
microparticles and functionally regulates expression of its target protein ICAM-1
in vitro and in vivo. After simulating diabetic conditions,
endothelial microparticles derived from glucose-treated endothelial cells contain
significantly lower amounts of miR-222 and show reduced anti-inflammatory
capacity in vitro and in vivo [164]. Finally, a lowered
circulating miR-222 level has also been confirmed in patients with coronary
artery disease compared to healthy subjects [164]. Moreover, Li et al.
[165] found that thrombin-activated platelet-derived exosomes inhibit endothelial
cell expression of ICAM-1 via miR-223, through regulation of
NF-B and MAPK pathways, during the thrombosis-inflammation
response [165]. It has been observed that high-density lipoprotein (HDL)
particles, purified from plasma, can transfer functional miRNAs, such as miR-223,
suppressing the expression of ICAM-1 [166].
Overall, miRNAs are emerging as new markers and potential targets and
therapeutic tools for the treatment of diseases associated with vascular
inflammation. Further studies are needed to fully understand the
interrelationships between the different epigenetic mechanisms and the miRNA
network involved in the regulation of vascular inflammation.






 , Marika Massaro 1
, Marika Massaro 1