








1 Department of Biochemistry, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt
2 Department of Biochemistry and Molecular Biology, Faculty of Pharmacy, Deraya University, 61111 Minia, Egypt
3 Department of Medicinal Chemistry, Faculty of Pharmacy, Deraya University, 61111 Minia, Egypt
4 IBMM, CNRS, ENSCM, Université de Montpellier, 34095 Montpellier, France
5 Medicinal Chemistry Department, Faculty of Pharmacy, Port said University, 42526 Port said, Egypt
6 Basic Health Sciences Department, College of Medicine, Princess Nourah bint Abdulrahman University, 11671 Riyadh, Saudi Arabia
7 Department of Regenerative Medicine, Graduate School of Medicine and Pharmaceutical Sciences, University of Toyama, 930-0194 Toyama, Japan
†These authors contributed equally.
Abstract
Background: Breast cancer is the most predominant tumor in women. Even
though current medications for distinct breast cancer subtypes are available, the
non-specificity of chemotherapeutics and chemoresistance imposes major obstacles
in breast cancer treatment. Although combretastatin A-4 (CA-4) has been
well-reported to have potential anticancer activity, in vivo studies of
CA-4 reveal a decrease in its activity. In this respect, a series of CA-4
analogues have been designed, from which one analog
[(1-(3-chloro-4-fluorophenyl)-N-(2methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazole-3-carboxamide,
C
Graphical Abstract
Keywords
- breast cancer
- combretastatin A-4
- anticancer
- MAPK/ERK
- PI3K/AKT
- apoptosis
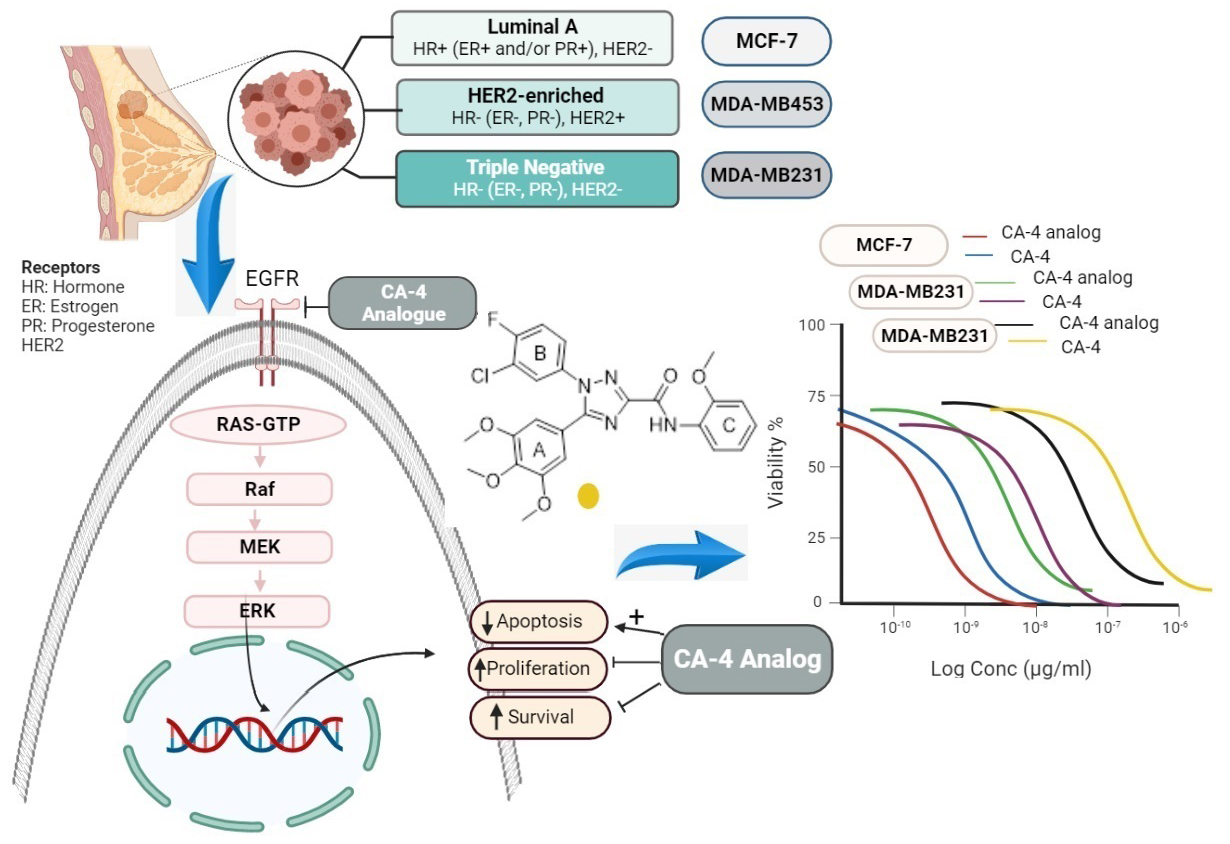
Breast cancer is among the most predominant tumors in females globally, with alarmingly increasing incidence rates. In 2020, more than 2.3 million women were newly diagnosed with breast cancer, and 680,000 reported death [1]. The intricate nature of breast cancer is largely attributed to the presence of malignant lesions in the ductal epithelium of the breast, creating a diverse array of distinct subtypes [2]. The literature extensively classifies various forms of breast cancer into distinct groups, based on the expression of key receptors such as the estrogen receptor (ER), progesterone receptor (PR), and human epithelial receptor 2 (HER2) [3]. Breast cancers can be hormonally dependent, HER2 positive, or triple-negative breast cancer (TNBC) [4]. Each of these subtypes has distinct risk factors, treatment responses, and genetic traits. The complexity and diversity of these subtypes underline the challenges involved in diagnosing and treating breast cancer [5, 6].
In recent years, remarkable progress has been made in the field of cancer treatment, offering numerous options for those fighting breast cancer, depending on the kind and severity of the disease. Although this undoubtedly has led to improved patient outcomes, the effectiveness of current treatments and patient compliance are severely hindered by high treatment costs, developed resistance, and off-target toxicity [7, 8]. The presence of multi-drug resistance and the non-specificity of chemotherapeutics present major obstacles in cancer treatment [9, 10, 11]. In light of these limitations, there is a dire need to identify or develop innovative and effective modalities for breast cancer therapy [4].
The mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways govern critical cellular events such as cell proliferation, regeneration, survival, and invasion, which play a key role in breast cancer progression [12]. Early research reveals that those pathways are constitutively active in breast tumors through various mechanisms [13]. Sustained activation of these pathways not only prevents breast cancer cell apoptosis but also contributes significantly to chemoresistance. In preclinical models, suppressing these pathways has been shown to increase the susceptibility of breast cancerous cells toward chemotherapeutics. However, manipulating only one of these pathways is insufficient for successful management of breast cancer [14]. Therefore, concurrent targeting of the PI3K/AKT and MAPK pathways might be a potential therapeutic approach for breast carcinoma.
Recently, there has been increased interest in screening new alternative
activities for natural [15] or synthetic [16] candidates that modulate pathways
involved in carcinogenesis [17]. Combretastatin A-4 (CA-4) is an alkaloid
extracted from the willow tree, Combretum caffrum, which shows
robust cytotoxicity against numerous human cancer cells. Its cytotoxic activity
is attributed to the inhibition of tubulin polymerization by binding to the
In a previous study, we synthesized a novel CA-4 analogue, (1-(3-chloro-4-fluorophenyl)-N-(2methoxyphenyl)-5-(3,4,5-trimethoxyphenyl)-1H-1,2,4-triazole-3-carboxamide, C25H22ClFN4O5, with a molecular weight of 512.92 Da), using 1,2,4-triazole to link ring-A (3,4,5-trimethoxy phenyl) and ring-B (3Cl-4-F-phenyl) [24] (Fig. 1). This study demonstrated that the CA-4 analogue exhibited potent antiproliferative activity against several human cancer cells, such as MCF-7 breast cancer, with enhanced activity and stability compared to CA-4. These results motivated us to further examine the cytotoxic activity of this novel analogue on different breast cancer subtypes (hormonally dependent, HER2+, and TNBC). Additionally, we aimed to illustrate the molecular mechanism beyond its anticancer potential against these breast carcinoma cell lines by investigating apoptosis, MAPK/ERK, and PI3K/AKT pathways.
 Fig. 1.
Fig. 1.Chemical structure of combretastatin A-4 (CA-4) and its analogue.
Sigma-Aldrich (Sigma-Aldrich, Inc., St. Louis, MO, USA) provided phosphate buffered saline (PBS),
L-glutamine, DMEM, Tween 20, and DNase I. Invitrogen ((Invitrogen, Grand Island,
NY, USA) provided penicillin-streptomycin mixture, while Biosolutions
International provided the fetal bovine serum (FBS, Biosolutions International,
Melbourne, Australia). Roche (Roche, Mannheim, Baden-Württemberg, Germany)
provided the protease inhibitor cocktail. Therrmo Fisher Scientific Life Sciences
(Waltham, MA, USA) provided TRizol® reagent, Diethylpyrocarbonate (DEPC), RNA Later, the
Revert aid RNA reverse transcription kit, and the maxima SYBR green qPCR kit.
Santa Cruz Biotechnology (Santa Cruz, CA, USA) provided the primary antibodies
against AKT, PI3K, ERK1/2, MEK1/2, p-AKT, p-PI3K, extracellular signal-regulated kinase1/2 (p-ERK1/2), mitogen-activated protein kinase kinases 1/2 (p-MEK1/2),
and
The examined compound, the CA-4 analogue, was prepared using the Schotten Baumann reaction and Sawdey rearrangement as per the instructions provided in our previous work. As previously reported, the molecule was characterized using 1H-NMR and 13C-NMR [24].
MCF7 (ER+/PR+/HER2-), MDA-MB231 (ER-/PR-/HER2-), and MDA-MB453 (ER-/PR-/HER2+) breast cancer cell lines were obtained from ATCC (CCL-2, Manassas, VA, USA). Cells were grown in fresh DMEM supplemented with L-glutamine (10 g/L), 10% FBS, and a penicillin-streptomycin solution (10 g/L). The authenticity of cell lines used in the study was verified through short tandem repeat profiling. Cells were routinely tested for mycoplasma contamination using the 4’,6-diamidino-2-phenylindole (DAPI) staining method. All cell lines tested negative for mycoplasma.
The cells were cultivated at 37 °C in a 5% CO
Cell survival was evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl-2H-tetrazolium bromide (MTT) assay. The cells were cultivated (1
Cellular apoptosis was detected using flow cytometry following the
manufacturer’s instructions (Immunotech, Marseille, France) and using the
Apoptosis detection kit. In triplicate, cells (5
The molecular docking and visualization processes were conducted on the binding site of Epidermal growth factor receptor (EGFR) using Molecular Operating Environment (MOE) 2019.0102. RCSB Protein Data Bank was used to retrieve the co-crystal structure. The CA-4 analogue was prepared using the standard protocol in MOE, and the energy of the docked compound was reduced with a gradient RMS of 0.0001 kcal/mol. The structure of the protein was generated using the QuickPrep protocol in MOE. The co-crystallized ligand was redocked using the same set of variables mentioned earlier to validate the docking study at the active site [29, 30, 31]. The best-docked pose exhibited a root mean square deviation (RMSD) value of 0.9472 Å, confirming the accuracy of the docking study with MOE software 2019.01 (Chemical Computing Group, Montreal, QC, Canada). The CA-4 analogue was docked into the EGFR binding site using the alpha triangle placement method with the Amber10: EHT forcefield. The refinement was done with the Forcefield, and the docking results were scored using the Affinity dG scoring system.
A total of 5
As per the guidelines provided by the manufacturer, a high-capacity reverse
transcriptase kit was utilized to reverse transcribe the mRNA pool using random
hexamer primers. The reverse transcription reaction was carried out with a cycle
of ten minutes at 25 °C, followed by incubation for 120 minutes at 37
°C, and finally incubation for 5 minutes at 85 °C to ensure
completion. For quantitative real time-polymerase chain reaction (qRT-PCR), the
resultant cDNA was used with the Maxima SYBR Green qPCR master mix (Thermo
Scientific, USA). The protocol included a 10-minute initial denaturation phase at
95 °C, followed by 30 amplification cycles consisting of 15 seconds at
95 °C, 30 seconds at 60 °C, and 30 seconds at 72 °C. A
final 10-minute extension step at 72 °C was included. Following the
manufacturer’s protocol, a Step One Real-Time PCR System (Thermo Fisher, USA) was
used to perform the amplification [34, 35]. All analyses were conducted in
triplicate, and the housekeeping gene Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference in
all experiments. The obtained qRT-PCR data were evaluated using the
| Primer | Sequence | |
| GAPDH | Forward | 5 |
| Reverse | 5 | |
| Bcl-2-associated X protein (Bax) | Forward | 5 |
| Reverse | 5 | |
| B-cell lymphoma 2 (Bcl2) | Forward | 5 |
| Reverse | 5 | |
| P53 | Forward | 5 |
| Reverse | 5 | |
Protein expression was assessed using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting techniques.
Cells were cultured in triplicate at an average number of 2
Data were demonstrated as mean
The potential anticancer activity of CA-4 and its analogue were investigated in
three distinct breast cancer cell lines: MCF-7, MDA MB-231, and MDA MB-453. MTT
assays were performed to assess cell growth suppression after a 48-hour
incubation period. Both CA-4 and its analogue exhibited dose-dependent
attenuation of cell proliferation in all three cell lines. Notably, the results
highlighted the anticancer potential of CA-4 against MCF-7, MDA MB-231, and MDA
MB-453 cells, with respective IC
 Fig. 2.
Fig. 2.Cell viability assay. Impact of CA-4 and CA-4 analogue on the viability of MCF-7, MDA-MB231, and MDA-MB453 cells following treating the cells for 48 hours. The cell survival rates are calculated as a percent of untreated cells, and results are calculated from three independent experiments.
| Cell line | CA-4 analogue | CA-4 |
| MCF-7 | 0.55 * | 1.44 |
| MDA-MB231 | 2.09 ƒ | 3.25 |
| MDA-MB453 | 2.6 # | 4.93 |
*: p-value
The results presented in Fig. 3 demonstrate the remarkable ability of CA-4 and
its analogous compounds to induce apoptosis in MCF-7, MDA-MB231, and MDA-MB453
cells. After 48-hour treatment with CA-4, the percentage of cellular apoptosis
was 27.89
 Fig. 3.
Fig. 3.Impact of CA-4 and its analogue on apoptosis in MCF-7,
MDA-MB231, and MDA-MB453 cancer cell lines was assessed using Annexin V assay.
(A) The dot plots depict the MCF-7, MDA-MB231, and MDA-MB453 cancer cell lines.
(B) The proportion of apoptotic cells in breast cancer cell lines was determined
after 48 hours of treatment with CA-4 or its derivative (IC50). The
presented data are expressed as mean
To further rationalize the potent inhibitory activity of CA-4 analogue on EGFR
kinase, molecular docking was performed on the crystal structure of the EGFR
kinase domain (PDB code: 1M17) in complex with erlotinib [38]. CA-4 analogue
displayed several essential interactions within the active site of EGFR and
showed better interaction energy than the native ligand erlotinib; the carbonyl
oxygen formed a hydrogen bond with the key amino acid Met769, the p-methoxy group
showed two hydrogen bonds with Met742 and Glu738, and the m
| Compound | Energy score (S) (kcal/mol) | Ligand-receptor interactions | ||
| Residue | Type | Length (Ǻ) | ||
| Erlotinib | –8.21 | Met769 | Hydrogen bond | 4.03 |
| Met742 | Hydrogen bond | 4.25 | ||
| Leu768 | Hydrogen bond | 4.03 | ||
| Gln767 | Hydrogen bond | 3.34 | ||
| Leu694 | π..H | 3.92 | ||
| Val702 | π..H | 7.78 | ||
| CA-4 analogue | –8.89 | Met769 | Hydrogen bond | 3.57 |
| Met742 | Hydrogen bond | 3.64 | ||
| Asn818 | Hydrogen bond | 3.15 | ||
| Leu694 | π..H | 3.75 | ||
| Val702 | π..H | 4.47 | ||
| Leu820 | π..H | 3.64 | ||
| CA4 | –6.87 | Glu738 | Hydrogen bond | 3.46 |
| Leu694 | π..H | 4.70 | ||
| Leu694 | π..H | 4.57 | ||
| Val702 | π..H | 4.58 | ||
| Gly772 | π..H | 4.83 | ||
 Fig. 4.
Fig. 4.3D representation and interactions of (a) CA-4 analogue, (b) erlotinib, and (c) CA-4 within the EGFR active site.
Treatment of MCF-7 cells with CA-4 analogue for 24 and 48 hours led to a
significant time-dependent decrease (p
 Fig. 5.
Fig. 5.Effect of CA-4 and CA-4 analogue on PI3k/AKT pathway in MCF-7,
MDA-MB231, and MDA-MB453 cells. (A) Representative immunoblots of p-PI3k, tPI3k,
p-AKT, and t-AKT in MCF-7, MDA-MB231, and MDA-MB453 cells treated with the
IC50 concentration of CA-4 or CA-4 analogue for 24 or 48 hours.
Furthermore, following CA-4 analogue treatment, MDA-MB 231 cells exhibited
dramatically lowered p-PI3k and p-AKT levels relative to t-PI3k and t-AKT
proteins in a time-dependent manner in comparison with untreated cells. Treatment
of MDA-MB231 cells with the CA-4 analogue for 24 or 48 hours resulted in the
attenuation of p-PI3k (0.6 and 0.2) (p
Furthermore, MDA-MB453 cells displayed significantly lower levels of p-PI3K
(0.71 and 0.4) and p-AKT (0.59 and 0.26) after 24 or 48 hours of CA-4 analogue
treatment (p
We then evaluated the in vitro effect of the tested substances on the MAPK/ERK pathway by treating various breast cancer cells with the tested compounds at IC50 concentrations for 24 and 48 hours. Fig. 6 shows representative immunoblots of protein expression (t-MEK1/2, t-ERK1/2, p-MEK1/2, and p-ERK1/2) in MCF-7, MDA-MB-231, and MDA-MB-453 cells.
 Fig. 6.
Fig. 6.Impact of CA-4 and CA-4 analogue on MAPK/ERK pathway MCF-7,
MDA-MB231, and MDA-MB453 cells. (A) Representative blots of p-MEK1/2, tMEK1/2,
p-ERK1/2, and t-ERK1/2 in MCF-7, MDA-MB231, and MDA-MB453 cells treated with the
IC50 concentration of CA-4 or CA-4 analogue for 24 or 48 hours.
The findings revealed that CA-4 analogue significantly decreased
p-MEK1/2/t-MEK1/2 ratio and p-ERK1/2/t-ERK1/2 ratio (p
In MDA-MB-231 cells, CA-4 analogue significantly (p
The MDA-MB453 cells exhibited markedly low levels of p-MEK1/2 (0.81 and 0.26)
and p-ERK1/2 (0.54 and 0.4) after CA-4 analogue treatment for 24 and 48 hours,
respectively. Meanwhile, CA-4 significantly attenuated p-MEK1/2 (0.85 and 0.43)
and p-ERK1/2 (0.77 and 0.42) after 24 and 48 hours of treatment, respectively.
The suppression of ERK1/2 and MEK1/2 phosphorylation was shown to be
time-dependent (Fig. 6). The suppression was statistically significant between
CA-4- and CA-4 analogue-treated cells at 24 hours (p-ERK: p
The MCF-7, MDA MB-231, and MDA MB453 cell lines were administered the IC
The Bax mRNA expression was upregulated, while the Bcl2 levels were reduced in the tested breast cancer cells after CA-4 analogue treatment for 24 or 48 hours. Overall, our experimental data demonstrated that the CA-4 analogue may promote apoptosis in breast cancer cells in a time-dependent approach. Furthermore, gene expression analyses conducted on MCF-7, MDA-MB231, and MDA-MB453 cells revealed that both CA-4 and the CA-4 analogue dramatically elevated the ratio of Bax to Bcl2. The impact was particularly pronounced in cells treated with the CA-4 analogue compared to those treated with CA-4 alone (Fig. 7).
 Fig. 7.
Fig. 7.Impact of CA-4 and CA-4 analogue on (A): Bax, (B):
Bcl2, and (C): P53 genes’ expression in MCF-7, MDA-MB-231
MDA-MB453 cells. Relative gene expression in all cells treated with the IC50
concentration of CA-4 or CA-4 analogue for 24 or 48 hours relative to untreated
cells. Expression was normalized to GAPDH housekeeping gene expression. Bars
represent mean
Breast cancer is the most predominant form of female malignant tumors. Extensive research has been conducted to explore innovative treatment options [39]. Despite the availability of current treatments for various breast cancer subtypes, their effectiveness is limited due to resistance and unsatisfactory outcomes. Consequently, overall patient survival rates have not shown improvement [4, 40]. This necessitates identifying new therapeutic candidates overcoming treatment failure for optimizing effective therapy and improving survival rate. Despite the in vitro cytotoxic effect of the parent CA-4 on multiple cancer cell lines, in vivo studies have revealed a dramatic decline in its antiproliferative activity due to spontaneous isomerization to the more stable trans-isomer.
Several CA-4 derivatives have been designed and assessed for their anticancer activity. Among them, one derivative has shown prominent cytotoxic activity against MCF-7 breast cancer cells [24]. These findings have motivated us to investigate further the potential cytotoxic effects of this promising compound in different types of breast cancer cell lines and to explore the plausible molecular mechanism responsible for its remarkable activity.
The current results demonstrate that the CA-4 analogue exhibits a remarkable
dose-dependent inhibitory impact on the survival of breast cancer cells, with
IC
Apoptosis induction has been proposed as an effective strategy for cancer therapy [43, 44, 45]. The extrinsic death receptor pathway, as well as the mitochondrial intrinsic pathway, were both engaged in the triggering of cellular apoptosis [46]. Numerous defense mechanisms have been established by cancer cells to combat apoptotic cell death. One such mechanism is the overexpression of antiapoptotic Bcl2, which promotes enhanced tumor cell proliferation and inhibits apoptosis [44]. Conversely, elevated expression of Bax promotes cell death and hence eradicates tumor cells [47, 48].
During the last ten years, there has been increasing interest in targeting P53, a crucial protein involved in suppressing tumor growth, as a potential strategy for cancer treatment. This interest stems from the ability of P53 to control different cellular mechanisms such as DNA repair, cellular apoptosis, and cell cycle arrest [49]. Activation of the P53 protein can increase the sensitivity of cancer cells to DNA damage, preventing their replication and promoting apoptosis [50]. In our study, we observed that the novel CA-4 derivative induced upregulation of p53 expression in MCF-7, MDA MB231, and MDA MB453 cells. Additionally, the derivative enhanced apoptosis by elevating the expression of Bax, while decreasing that of Bcl2. This led to an increased Bax/Bcl2 ratio, which can trigger the collapse of the mitochondrial membrane, the release of cytochrome c, and ultimately cellular apoptosis [25, 48]. Our investigation revealed that treatment with the CA-4 analogue resulted in a time-dependent increase in the ratio of Bax/Bcl2 after 24 and 48 hours, suggesting that the CA-4 analogue induces apoptosis through the mitochondrial pathway. This observation was consistent with the evident elevation in the apoptotic cell population, as demonstrated by annexin V/PI dual staining. The percentage of apoptotic cells (Annexin-V positive) was highest in MCF-7 cells relative to MDA-MB-231 and MDA-MB453 cells.
To better understand the mechanism of action of the new CA-4 analogue, in vitro molecular docking was conducted on EGFR binding sites. The upregulation of the EGFR signaling pathway has been associated with reduced apoptosis, which directly contributes to cancer development, tumorigenesis, increased cell proliferation, and angiogenesis [51]. The EGFR gene is expressed in approximately 14–91% of breast carcinomas, and its dysregulation is associated with more invasive breast tumor subtypes and poorer prognoses [52]. Moreover, EGFR can activate signaling cascades such as the MAPK/ERK and PI3K/AKT pathways to transmit its signal, making it a potential target for controlling cancer cell proliferation. Through in vitro molecular docking, the CA-4 analogue demonstrated potent inhibitory activity against EGFR kinase. The CA-4 analogue exhibited several critical interactions within the active site of EGFR. This observed potent inhibitory activity of CA-4 analogue explains its cytotoxic activity towards tested breast cancer cells [42].
The PI3K/AKT signaling pathway is frequently associated with breast carcinoma [53]. Cell survival, proliferation, and protein synthesis are regulated by the PI3K/AKT/mTOR pathway [54, 55, 56]. Stimulation of the PI3K/AKT/mTOR pathway has been linked to tumor development and reduced patient survival [57]. Recent studies on PI3K/AKT inhibitors suggest that recruiting the PI3K/AKT pathway is a potential strategy for treating breast cancer [58]. In humans, there are three primary MAPK pathways, but the pathway concerning ERK1/2 is the most relevant to breast cancer [59]. Genes involved in the ERK1/2 pathway directly regulate various fundamental biological functions, including differentiation, apoptosis, cellular proliferation, and survival [60]. Dysregulation of the MAPK/ERK cascade is a hallmark of robust carcinogenesis and several components of this pathway are mutated or exhibit aberrant expression in breast cancer [61, 62]. To further elucidate the molecular mechanism behind the inhibitory action of the CA-4 analogue on malignant breast cancer, we investigated whether the CA-4 analogue modulates the PI3K/AKT and MAPK/ERK signaling pathways. Treatment with the CA-4 analogue resulted in the suppression of the PI3K/AKT and MAPK/ERK pathways, as evidenced by the reduction in phosphorylation levels of PI3K, AKT, MEK1/2, and ERK1/2 in breast cancer cells. Notably, the suppressive effect of CA-4 analogue on the PI3K/AKT and MAPK/ERK signaling was greater than that of CA-4 parent drug.
Moreover, several studies have revealed that the PI3K/AKT and MAPK/ERK cascades may interloop with each other and exhibit a synergistic effect in driving breast cancer proliferation [63, 64]. Taken together, the CA-4 analogue could be a more potent anticancer drug against malignant breast cancer cells by simultaneously targeting the PI3K/AKT/mTOR and MAPK pathways [63, 65].
The current research revealed that the CA-4 analogue exhibited superior suppression of cell proliferation compared to the parent CA-4 in breast cancer cells, such as hormonal-dependent breast cancer (MCF-7), HER-2 enriched breast cancer (MDA-MB453), and triple-negative breast cancer (MDA-MB231). The enhanced anti-proliferative activity of the CA-4 analogue may be attributed to its ability to induce apoptosis and effectively inhibit two key signaling cascades, namely PI3K/AKT and MAPK/ERK, which play critical roles in chemoresistance and treatment failure. Overall, the CA-4 analogue can be considered a possible anticancer agent for the management of numerous genetic breast cancer types.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
DHA, MHN and MF conceived the idea and designed the research study. OMA and MAE performed the conceptualization. DHA, MHN and MF performed the research. OMA, MHN, MF and MAE provided help and advice on data analysis. MM, DHA, MHN, AAKE and MF analyzed the data. DHA, MHN, AAKE and MF wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
The current work was supported by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R91), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.