



 , Eva Žerovnik 2,3,*
, Eva Žerovnik 2,3,*1 Theory Department, National Institute of Chemistry, 1000 Ljubljana, Slovenia
2 Department of Biochemistry and Molecular and Structural Biology, Jožef Stefan Institute, 1000 Ljubljana, Slovenia
3 Jožef Stefan International Postgraduate School, 1000 Ljubljana, Slovenia
Abstract
Similar to other polypeptides and electrolytes, proteins undergo phase transitions, obeying physicochemical laws. They can undergo liquid-to-gel and liquid-to-liquid phase transitions. Intrinsically disordered proteins are particularly susceptible to phase separation. After a general introduction, the principles of in vitro studies of protein folding, aggregation, and condensation are described. Numerous recent and older studies have confirmed that the process of liquid-liquid phase separation (LLPS) leads to various condensed bodies in cells, which is one way cells manage stress. We review what is known about protein aggregation and condensation in the cell, notwithstanding the protective and pathological roles of protein aggregates. This includes membrane-less organelles and cytotoxicity of the prefibrillar oligomers of amyloid-forming proteins. We then describe and evaluate bioinformatic (in silico) methods for predicting protein aggregation-prone regions of proteins that form amyloids, prions, and condensates.
Keywords
- neurodegeneration
- protein aggregation
- protein condensation
- LLPS
- membrane-less organelles
- amyloid
- intrinsically disordered proteins (IDPs)
- prion
- prediction by in silico methods
There are at least three states of proteins at the endpoint of folding, unfolding, and misfolding equilibria: the native, the unfolded, and the amyloid state. A fourth state is sometimes quoted as a metastable molten globule intermediate. However, looking at the kinetics of unfolding by several probes, even more variants of the molten-globule intermediate are detectable [1].
Protein folding, oligomerization, misfolding, and aggregation are all determined by the primary structure, the protein sequence. However, the environment and protein concentration also play an important role and influence the final folding, the kinetics of folding, and the aggregation pathway.
In general, proteins are soluble, but under some conditions, they can undergo phase separation (liquid-liquid and liquid-gel transitions), similar to other polypeptides and electrolytes. Intrinsically disordered proteins (IDPs) are especially prone to phase separation. Together or with other biomolecules, such as RNA, they form biomolecular condensates that are important for proteostasis, compartmentalization, and regulation of the cell [2].
The aim of this review is to update and summarize the current research on the condensed and aggregated states of proteins. We consider protein aggregation a normal, albeit transient, physiological process based on physicochemical laws. The forces at play in the processes of protein folding, misfolding, and aggregation are described at the beginning of this review. We describe different forms of protein aggregates and condensates as observed in cells. We then highlight the membrane-less organelles and the cytotoxicity of the prefibrillar oligomers of amyloid-forming proteins. Finally, we review and evaluate numerous in silico prediction programs for predicting the propensity of proteins to either condense or aggregate. Prions are discussed in a special subsection of amyloids.
Protein folding is a phenomenon of physical chemistry. The path that a protein
takes to build a unique three-dimensional structure starting from its amino-acid
sequence, i.e., the primary structure, is pre-defined by physical forces, such as
hydrogen bonds, electrostatic, dipole-dipole, Van der Waals, and hydrophobic
effect. Enthalpy-entropy compensation occurs when protein side chains fix and
stabilize the structure. Proteins fold by following specific routes, not
randomly. If a protein could attempt all possible conformations, it would take a
vast, unimaginable amount of time (Levinthal’s paradox [3]). Some proteins
initially undergo a hydrophobic collapse, in which a hydrophobic core forms and
others form some mobile elements of the secondary structure, following the
framework model (leading to folding intermediates of the pre-molten globule and
molten globule type and wet and dry molten globules) [4, 5, 6, 7]. The structure of
the molten globule has long remained elusive, yet recent NMR studies are more
decisive [8]. During protein folding, more specific interactions occur between
the side chains. We studied an interesting example of human stefins A and B
folding [9, 10]. We concluded that a mixed mechanism spanning between a secondary
structure governed framework model and hydrophobic collapse model with a
non-native
A large number of proteins in the human proteome, nearly 40% [11], remain intrinsically disordered (IDPs) [12], also termed natively unfolded proteins [13, 14]. Their folding is not dictated by hydrophobic collapse, but rather, it is directed by another molecular surface, serving as a template (template-like folding [11]). This type of protein forms a secondary structure along with binding [15]. Their interaction partners can be multiple and different IDPs can bind to a chosen partner [16].
Similar physical forces, as in folding and template-assisted folding, apply to
protein-protein intermolecular interactions, leading to oligomerization and
aggregation [17]. A general scheme of protein deposition (aggregation) and
condensation pathways is shown in Fig. 1 (Ref. [18]) (as adapted from Vendruscolo, M. and
Fuxreiter, M., 2022). Specifically, linear colloidal aggregation describes
the initial events in the deposition pathway of some proteins, such as yeast
Sup35, that lead to protofibrils appearing as a string of beads [19]. The
mechanism of protofibril elongation is likely due to a dipole-dipole interaction
between these beads [19], also referred to as “critical oligomers” [20].
However, the formation of amyloid fibrils is not a simple polymerization reaction
but often involves nucleation and off-pathway aggregation [21, 22]. This leads to
a lag phase during which disordered intermediates refold into non-native
secondary structures (
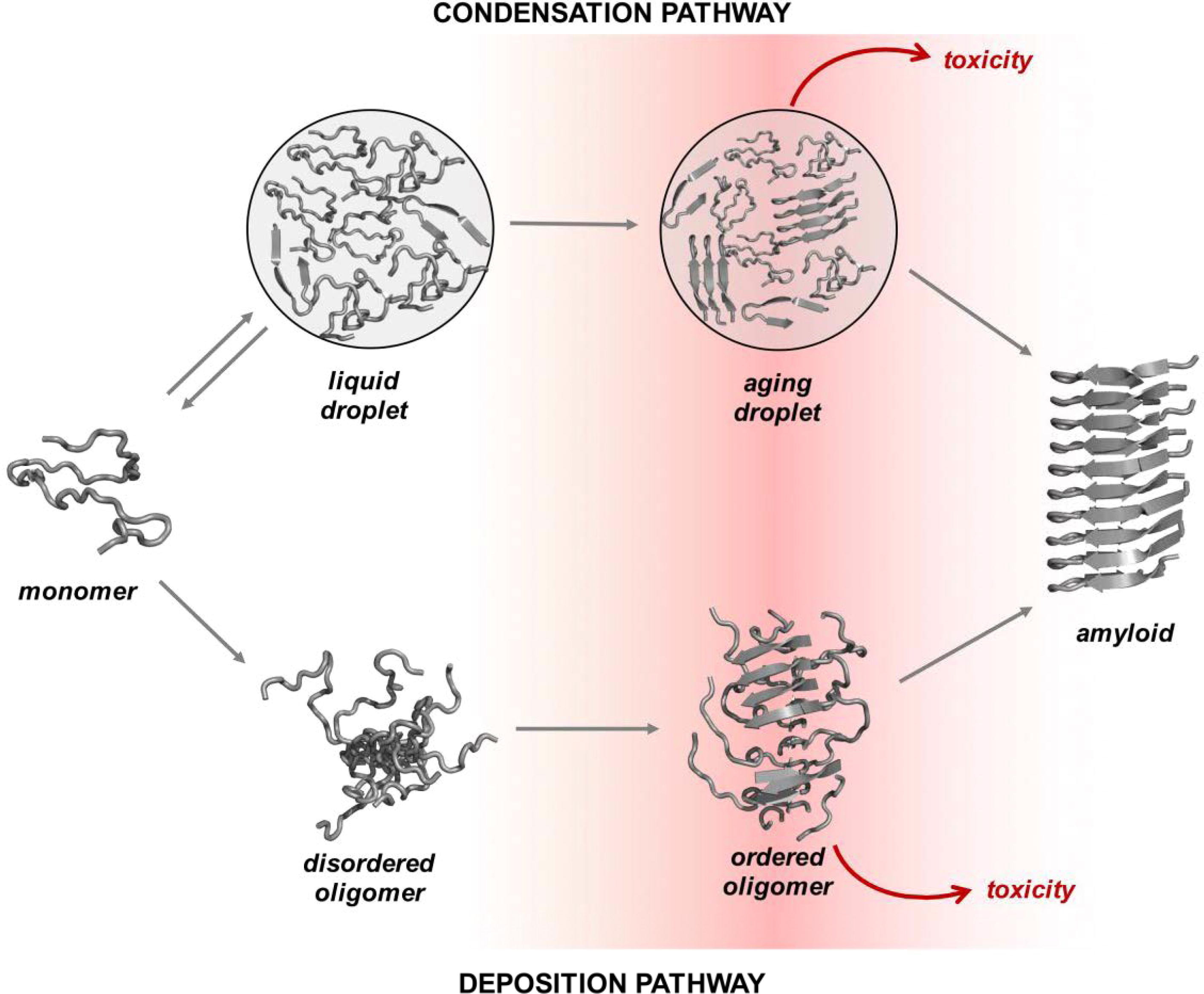 Fig. 1.
Fig. 1.Overview of the condensation and the deposition pathways for amyloid formation. Along the deposition pathway, proteins move from the native state to the amyloid state through increasingly ordered oligomer aggregates. Some of these oligomers are highly cytotoxic. Along the condensation pathway, proteins convert from the native state to the amyloid state through a dense liquid state (the droplet state). For many proteins under cellular conditions, the native and droplet states are metastable. The droplet state is functional for specific proteins, and it is stabilized by extrinsic factors, such as RNA and post-translational modifications (adapted from Vendruscolo, M. and Fuxreiter, M., 2022) [18]. This image is reproduced under the Creative Commons license: https://creativecommons.org/licenses/by/4.0/.
The mechanisms of amyloid fibril formation have been extensively studied in vitro for both pathological and non-pathological proteins. One of the authors wrote an overview of the mechanisms of amyloid fibril formation in 2002 [30]. Others have also discussed the nucleated polymerization reaction with off-pathway intermediates [21], which has been shown for our model protein, human stefin B [22]. Since then, many other reviews have appeared. Among them is a comprehensive review by Chiti and Dobson [31], in which the authors describe, among other advances, the structures of amyloid fibrils and prefibrillar oligomers and explain the mechanisms of amyloid transformation. They also discuss how cells combat the aberrations caused by protein aggregates. More reviews have recently been published [32, 33, 34].
The transition between the native and amyloid states of proteins can proceed via oligomeric intermediates as described above or via a condensation pathway involving liquid droplet intermediates generated through liquid-liquid phase separation [35]. Multivalent weak interactions between peptides are the driving force of phase separation. Proteins that undergo phase separation promote biomolecular condensate formation, which has a significant role in many biological processes. Further, these proteins can be divided into two categories according to their underlying driving force when forming condensates: self-assembling proteins (interacting with the same protein species and whose interactions are mediated mainly by intrinsically disordered regions) and partner-dependent proteins (interacting with different biomolecule species and whose interactions are mediated by multiple modular domains or motifs). Condensate proteome validation revealed that partner-dependent proteins are widespread in cells [36].
Similar to the amyloid transformation, the phase separation behavior of a protein is determined by general properties contained in the amino acid sequence and by environmental conditions, such as temperature and protein concentration, as well as post-translational modifications [37]. The IDPs are more prone to phase separation, even though condensation might be another generic property of proteins [38]. Cited from Vazquez et al. [38]: “… any sequence or conformation is susceptible to phase separation, provided that the appropriate concentration, temperature, and solvent condition are reached”.
The sequences of several RNA-binding proteins comprise prion-like LCDs (low complexity domains), which are enriched in uncharged polar amino acids. Normally, these sequences are at least 60 residues long, are predicted to be intrinsically disordered, and enable the replication of a particular protein conformation from one copy to another (a template-like mechanism). LCDs are enriched in glutamine and asparagine amino acid residues. Based on sequence prediction models, the disease-linked RNA-binding proteins usually have the greatest tendency to aggregate.
Although IDPs, including prions, are thought to be more prone to undergo LLPS, structured, globular proteins may also do so under appropriate solution conditions [39]. External factors such as temperature, pH, ionic strength, shear stress, and protein concentration strongly affect the condensation of proteins. The molecular interactions that stabilize condensates at high ionic strength are mainly aromatic, hydrophobic, and nonionic interactions, whereas electrostatic interactions play a major role at lower ionic strengths [40]. Depending on the pH and ionic strength of the solution and at sufficiently high protein concentrations, the aggregates eventually form a gel-like network.
Of interest, proteins from extremophiles show an interesting shift towards intrinsic disorder [41] and, consequently, a preference for condensation over amyloid fibrillation.
Protein misfolding in the cell occurs either in the cytoplasm or in the nucleus due to internal and external stressors, such as heat shock or oxidative stress. Misfolded proteins are prone to aggregate and are sensed by cellular defense mechanisms, such as unfolded protein response (UPR) in the endoplasmic reticulum or two degradation machineries in the cytosol. Pathological mutants, which are prone to aggregate, can exacerbate or even cause certain amyloidoses, among them neurodegenerative diseases.
Two types of protein aggregate deposits in eukaryotic (yeast) cells were previously reported, already in 2008: the IPOD and JUNQ [42, 43]. The juxtanuclear inclusions harbor misfolded, still soluble proteins, which can exchange with the cytoplasmic proteins; therefore, this compartment is called “juxtanuclear protein quality control” (JUNQ). The perivacuolar peripheral inclusion contains aggregated and insoluble proteins, hence, the term “insoluble protein deposit” (IPOD). Both compartments have certain features in common, such as the binding of the chaperone Hsp 104 and probably also Hsp 70. Only JUNQ is connected with proteasome subunits, whereas IPOD is close to autophagosomes; therefore, it is likely that the insoluble proteins get degraded by autophagy. By using electron microscopy, it has been demonstrated that JUNQ has an intranuclear localization adjacent to the nucleolus [44], and it was redefined as the INQ (intranuclear quality control compartment). The INQ serves as a deposit for both misfolded nuclear and cytosolic proteins [45]. All these aggregates are sequestered to their final location by microtubular transport, and the composition of inclusions is regulated by other regulatory proteins [42].
A third kind of inclusion exists in mammalian cells, similar to JUNQ – the aggresomes [46]. They contain fibrillar aggregates of amyloid-forming proteins, which get sequestered into the perinuclear space by the micro-tubule-organizing center (MTOC). The aggresomes are enwrapped by a shield of intermediate filament protein vimentin [47]. All the described regulated protein aggregates are thought to exert cytoprotective functions, which are vital for cell integrity and survival [45]. The escaping soluble oligomers may bind to plasma and intracellular membranes and cause more damage by perforating them, resembling pore-forming toxins [48, 49, 50]. (See section 3.2)
Apart from protein aggregates, which contain mainly one type of protein molecule
in an altered conformation, usually rich in
One of the key differences between protein aggregates and condensates is their reversibility. Protein condensates in distinction to more toxic forms of protein aggregates are at least initially reversible, as Shin et al. [52] have shown. When tagged with a light-sensitive tag, the proteins became condensed and later dissolved when the light was turned off. The gels were initially reversible, but over time and using a high-intensity light or high protein concentration, irreversible clumps formed [52], similar to those seen in neurodegenerative diseases.
Another key difference between protein condensates and aggregates is the
specificity of the molecular interactions playing a major role. Protein
aggregates of amyloid-type tend to be formed by specific interactions. The
The intracellular biomolecular condensates, i.e., the membrane-less organelles, are important for cellular compartmentalization and regulation. They are considered protective [54] because they sequester aggregation-prone proteins and prevent amyloid formation, despite the local increase in protein concentration. They can serve as reservoirs of peptide hormones and other proteins that are released when needed.
In recent years, membrane-less organelles have been discovered a-new. Such bodies in the cytoplasm and the nucleus have been known for a long time, yet their function and mechanism of formation remained unknown. Under different forms of cellular stress, proteins and RNAs undergo reversible transitions from the liquid to the gel state, forming biomolecular condensates. Different condensates can be found from bacteria to eukaryotic and mammalian cells [55]. In eukaryotic cells, such condensed bodies exist both in the nucleus and in the cytoplasm. The two best-studied stress assemblies in the cytoplasm are the RNA-based processing bodies (Pbodies) and stress granules (SGs) that form in response to oxidative, endoplasmic reticulum (ER), osmotic and nutrient stress, UV light, etc. Stress granules (SGs) are membrane-less organelles formed in the cytoplasm by liquid-liquid phase separation (LLPS) of translationally-stalled mRNA and RNA-binding proteins, such as TDP-43. P-bodies are also composed of translationally-stalled mRNAs and proteins involved in translation repression and mRNA turnover (see, Fig. 2 (Ref. [56]) for a simplified view of the biomolecular condensates in a human cell). Apart from SGs and PBs, the RNA translation initiation complex eIF2 also forms condensed bodies in the cytoplasm [57]. Furthermore, nutrient stress (starvation) leads to the formation of a variety of cytoplasmic stress assemblies, some of which do not contain RNA, such as proteasome storage granules, metabolic enzyme bodies, and Sec bodies [57]. Sec bodies are formed by Sec16, a large scaffold protein important for secretion from ER to the Golgi. The good news is that all these entities are transient–reversible and, in most cases, pro-survival [57].
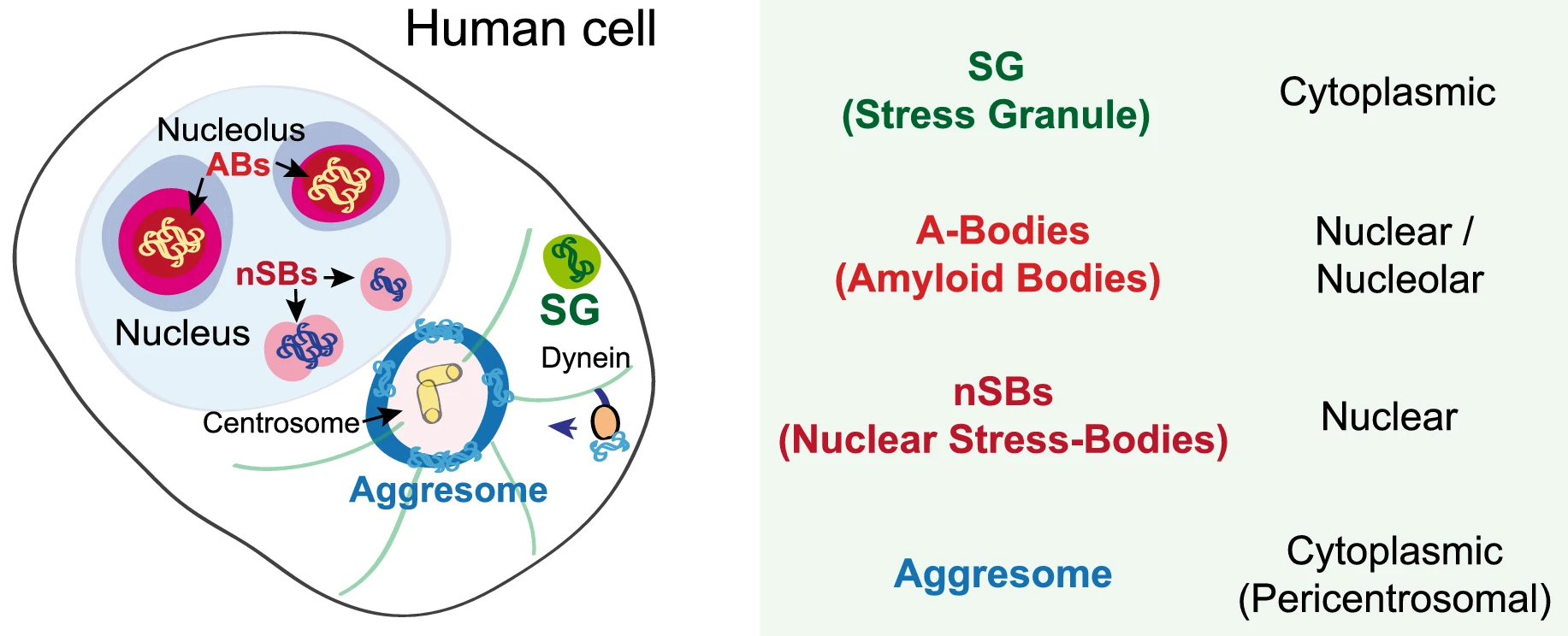 Fig. 2.
Fig. 2.Schematic presentation of various condensed bodies in a human cell adapted from [56]. ABs, amyloid bodies. This image is available via http://creativecommons.org/licenses/by/4.0/.
Reversible protein aggregation also occurs within the nuclei of stress-treated cells (for a schematic view, see Fig. 2). For example, mammalian cells protect thermosensitive nuclear proteins by their condensation into amyloid bodies (ABs). ABs assemble through the rapid accumulation of proteins and ribosomal RNA spacers and promote local nuclear translation during heat stress [56]. Nuclear stress bodies (nSBs) are also formed in nuclei from RNA and proteins upon heat shock. By sequestration of transcription factors, they inhibit RNA transcription [56].
As said, IDPs or proteins with intrinsically disordered regions (IDRs), such as
prion and
However, not only RNA-bound proteins or IDPs can form condensates upon stress.
Folded globular proteins usually do not condense, yet their unfolded states can,
forming the so-called unfolded protein deposits — UPODs [61]; proteins that are
less stable and contain many aromatic amino acids, such as Tyr and Phe, can form
UPODs [61]. Using lysozyme, a popular model protein [62], as an example, the
aggregates were found to account for ~10
The two-dimensional liquid environments provided by lipid bilayers (Fig. 3, Ref. [65]) can profoundly alter protein structure and dynamics by both specific and non-specific interactions. Kinetic and thermodynamic studies indicate that significant conformational changes can be induced in proteins encountering lipid surfaces, which can play a critical role in nucleating aggregate formation or stabilizing specific aggregation states.
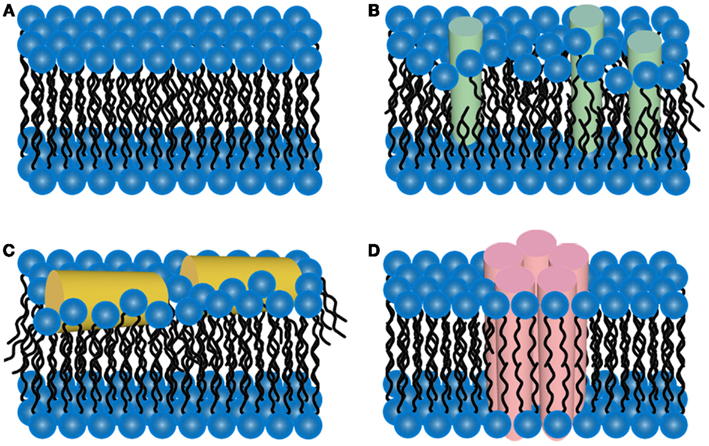 Fig. 3.
Fig. 3.Schematic representations of potential mechanisms of
amyloid/lipid association. (A) A schematic representation of simplified,
undisrupted lipid bilayer. This lipid bilayer structure can be perturbed
by (B) membrane protein insertion or (C) association of amphiphilic
Pore-forming proteins (PFP) are found in virtually all domains of life [49], and, by disrupting cell membranes, depending on the pore size, they cause ion disbalance, small molecules, or even protein efflux/influx, influencing cell signaling routes and fate. Such pore-forming proteins exist from bacteria to viruses and also shape host defense systems, including innate immunity. There is strong evidence that amyloid toxicity is also caused by prefibrillar oligomers forming amyloid pores in cellular membranes. It is believed that the prefibrillar, still soluble oligomeric intermediates would interact with cell membranes or even make the so-called “amyloid pores” [48] that exhibit structural and functional properties similar to those of pore-forming toxins.
Smaller oligomeric structures, in some cases, seem sufficient to perforate the
lipid bilayer. In amyloid-
Di Scala et al. [50] described a common molecular mechanism of
amyloid pore formation by A
The mechanism of amyloid pore formation has recently been followed by kinetic
simulations [71] and single-channel measurements [72]. Kayed et al.
[73] detected endogenous oligomeric and multimeric species in
Both plasma and mitochondrial membranes can be affected by extracellular and
intracellular prefibrillar oligomers [75]. Camilleri et al. [76]
initially observed that mito-mimetic lipid vesicles were more permeable to
oligomers of A
Based on protein sequence, environmental, and spatial factors, bioinformatic (in silico) methods can predict the propensity of proteins to transition to condensed or aggregated states; however, predicting the kinetics of aggregation remains a challenge.
Nowadays, in silico platforms provide not only single predictors but
also so-called meta-predictors that generate highly accurate predictions based on
algorithms that combine results from multiple computational models, including
predictions of condensable, amyloidogenic, or/and intrinsically disordered
regions [36]. Here, we describe and compare the state-of-the-art bioinformatic
tools for in silico prediction of condensates and aggregates. As
prediction methods are evolving rapidly, we have compiled a list of
Several computational methods are available to produce sequence-based predictions of the propensity of proteins to aggregate via the deposition pathway, but the physicochemical principles underlying condensation are known less well [18, 80]. Although prediction methods exist to estimate the propensity of proteins to undergo liquid-liquid phase separation (LLPS), it is not clear how to predict amyloid aggregation within condensates [81]. Recently, Vendruscolo and Fuxreiter [18] have provided insights into the amino acid code for protein conversion between liquid-like and solid-like condensates. As different parameters have been proposed to determine the propensity of proteins to form condensates, various in silico strategies based on machine learning models, have been developed to understand the relationship between protein sequence and protein phase behavior.
In general, most in silico tools for predicting the propensity for
condensation in two phases (liquid-liquid phase separation — LLPS) are based on
amino acid sequences. The relatively small number of experimentally validated
proteins prone to phase separation and the difficulty in detecting them remain a
bottleneck in developing accurate predictors for condensates. Despite this
limitation, new approaches are constantly being developed, and the integration of
newly updated databases into their development is increasing the accuracy of the
new predictors. PScore
(http://abragam.med.utoronto.ca/~JFKlab/Software/psp.htm) [82],
catGRANULE
(http://service.tartaglialab.com/update_submission/277133/0b8f3740ac) [83] and
LARKS (Low-complexity Aromatic-Rich Kinked Segments) [84] are
first-generation LLPS propensity predictors. Compared to the first-generation
predictors, the second-generation predictors FuzDrop
(https://fuzdrop.bio.unipd.it/predictor) [85], DeePhase
(https://deephase.ch.cam.ac.uk/) [86], PSPer (Phase Separating Protein,
https://www.bio2byte.be/b2btools/psp) [87] and PSPredictor
(http://www.pkumdl.cn/PSPredictor) [88] were developed based on larger training
datasets (LLPSDB (http://bio-comp.org.cn/llpsdb) [89], PhaSepDB
(http://db.phasep.pro/) [90], PhaSePro (https://phasepro.elte.hu/) [91]),
allowing for a broader range of LLPS protein screening. Each of the new
predictors has specific properties; DeePhase is very powerful in
distinguishing LLPS-prone proteins from structured proteins and identifying them
in the human proteome. The authors of this tool highlight that LLPS-prone
proteins are more disordered, less hydrophobic, and of lower Shannon entropy
[86]. FuzDrop (https://fuzdrop.bio.unipd.it/predictor) can identify
droplet-promoting and aggregation-promoting regions in protein sequences that
spontaneously phase separate [85]. PSPer prioritizes phase-separating
proteins among proteins with similar RNA-binding domains, intrinsically
disordered regions, and prions [87]. PSPredictor allows users to
determine the most similar proteins in the LLPSDB under experimentally validated
phase separation conditions [88]. PSAP
(https://github.com/vanheeringen-lab/psap) is a random forest classifier trained
on a set of 90 human proteins that condense with high confidence [81]. The LLPS
predictors listed above were developed based on different underlying concepts,
architectures, and training sets. This makes comparison difficult, as each method
is suitable for different applications. Nevertheless, their combined use can
significantly increase their utility, as reported by Pancsa et al. [92].
Their comparable analysis included five methods: PScore, PSPer, PLAAC,
catGRANULE, and PSPredictor. By summarizing their findings, the PLAAC performs
well in identifying prion-like LLPS proteins. PSPer and PScore show good synergy,
as PSPer mainly detects PLDs and RNA-driven phase separation, whereas PScore
detects LLPS driven by
Despite the abundance of existing computational tools, accurately predicting protein phase transitions remains a challenge, and the establishment of research and innovation hubs seems to be a possible future prospect. The PhasAGE project (https://phasage.eu/, PhasAGE – Excellence Hub on Phase Transitions in Aging and Age-Related Disorders) could be a good example of such an approach.
In recent decades, researchers have attempted to gain a better understanding of
protein aggregation and to develop computational methods to predict aggregation
propensity. It has been more difficult to predict the kinetics of aggregation.
However, AggreRATE-Pred (http://www.iitm.ac.in/bioinfo/aggrerate-pred/)
is the first tool to determine aggregation regions (APR prediction) and detect
the change in aggregation kinetics [96]. Compared to several previous
aggregation prediction models, AggreRATE-Pred considers both structural and
sequence-based properties. It also predicts the change in aggregation rate upon
point mutations. Compared with first-generation APR prediction methods such as
TANGO (http://tango.crg.es) [97], AGGRESCAN (aggregation-prone
segments in proteins, http://bioinf.uab.es/aggrescan/) [98] and GAP
(Generalized Aggregation Proneness, http://www.iitm.ac.in/bioinfo/GAP) [99],
the aggregation propensities determined by these methods do not correlate with
the aggregation rate determined by AggreRATE-Pred [96]. The old methods only
calculate the overall aggregation propensity of a polypeptide chain and do not
provide information on the growth of aggregates over time. GAP deficiency is a
small data set used in its development [99]; whereas the AGGRESCAN
algorithm is simple and fast, the last implementation of the online software was
performed in early 2023 [100]. In general, APRs are usually buried in the
hydrophobic core of the native protein and enriched with residues that favor the
formation of
SODA (Protein SOlubility based on Disorder and Aggregation, http://old.protein.bio.unipd.it/soda/) provides, in addition to aggregation propensity, information about the intrinsic disorder, hydrophobicity, and secondary structure preferences. In addition, a score to evaluate the difference in aggregation and solubility introduced by mutations can be evaluated [102]. In general, most tools for identifying APRs use amino acid residue composition and/or sequence patterns [96]. In this regard, ANuPP (Aggregation Nucleation Prediction in Peptides and Proteins, https://web.iitm.ac.in/bioinfo2/ANuPP/homeseq1/), a web meta-classifier for ARP identification, is a novelty [103]. It is unique since it is based on atom-level features and considers the diversity of aggregation mechanisms. The performance of ANuPP was evaluated on several datasets, and the results show that ANuPP is one of the best prediction methods for both the prediction of amyloidogenic hexapeptides and the identification of APRs compared with other currently available methods.
Predicting the aggregation propensity of folded proteins is a bottleneck due to the lack of known 3D structures with high resolution. Although algorithms for detecting aggregation-nucleating sequences from the primary sequences of proteins work reasonably well, many of these sequences in the folded state become part of the inner core of the protein, which does not contribute to aggregation unless the protein unfolds extensively [104]. Therefore, the development of algorithms that can detect APRs at protein surfaces is of great interest and has been under constant development in recent years. These new tools, which combine structure- and sequence-based features into integrated predictors, bear improved accuracy. Such servers for aggregation propensity prediction and protein solubility engineering based on features associated with the 3D structure of proteins are SAP (Spatial Aggregation Propensity) [105], Aggrescan3D (A3D, http://biocomp.chem.uw.edu.pl/A3D/) [106], Aggrescan3D 2.0 (A3D2, http://biocomp.chem.uw.edu.pl/A3D2/) [107], SOLart (http://babylone.ulb.ac.be/SOLART/) [108], SolubiS (https://solubis.switchlab.org/) [109], CamSol Structurally Corrected (https://www-cohsoftware.ch.cam.ac.uk/) [110], and AggScore[111].
With the advent of AlphaFold [112] and the establishment of AlphaFoldDB (https://alphafold.ebi.ac.uk/), the limitations due to the number of 3D protein structures identified are disappearing. Consequently, it is likely that in the next few years, we will foresee the development of many new tools for predicting the aggregation of protein 3D structures, which will enable new biomedical applications such as antibodies and beta-sheet-breaking peptides to treat diseases caused by protein aggregation [100]. In any case, the last decade has seen impressive innovations in ARP prediction. Several currently available algorithms enable an automated, sequence, and structure-based design strategy to improve the aggregation properties of proteins of scientific or industrial interest.
The transition of soluble proteins into insoluble amyloid fibrils is driven by
specific self-propagating short-sequence segments that can be predicted from
input sequences at the genomic level. In this regard, the propensity of different
protein sequences to aggregate into amyloids mostly depends on the stability of
the amyloid cross-
AmyLoad (http://comprec-lin.iiar.pwr.edu.pl/amyload/database/) is a web server of amyloidogenic sequence fragments (over 1480 different entries, and continues to increase). It allows users to add their sequences to the database in FASTA format and to analyze the queried sequences with implemented amyloid predictors [113]. In addition, the updated and significantly expanded database WALTZ-DB 2.0 (http://waltzdb.switchlab.org/) is now the largest freely accessible repository for determinants of amyloid fibril formation, determined experimentally based on amyloid-forming hexapeptide sequences [101].
First-generation amyloid predictors such as FoldAmyloid (http://bioinfo.protres.ru/fold-amyloid/) [114], Waltz [115], SALSA (http://amypdb.genouest.org/e107_plugins/amypdb_aggregation/db_prediction_salsa.php, are integrated into the AMYPdb database [116]. The aggregation prediction method PASTA 2.0 (http://protein.bio.unipd.it/pasta2/) [117] is complemented and enriched by other information, such as intrinsic disorder and secondary structure predictions. In this regard, the amyloid-forming regions can be correctly identified with high specificity from a larger dataset of globular protein domains [117]. Further, more advanced amyloid identification methods based on machine learning approaches are available: NetCSSP (Neural networks for calculating Contact-dependent Secondary Structure Propensity), http://cssp2.sookmyung.ac.kr/) [118], FiSH Amyloid (http://comprec-lin.iiar.pwr.edu.pl/) [119], AmyloGram (http://biongram.biotech.uni.wroc.pl/AmyloGram/) [120], APPNN (Amyloid Propensity Prediction Neural Network), https://cran.r-project.org/web/packages/appnn/index.html) [121], BAP (Budapest Amyloid Predictor https://pitgroup.org/bap/) [122] and AmyLoad (http://comprec-lin.iiar.pwr.edu.pl/amyload/database/) [113]. However, meta-predictors based on a consensus approach, which combines the strength of different individual predictors into a single predictor, exceed the accuracy of these individual predictors. Such meta-predictors are MetAmyl and AmylPred2. MetAmyl (http://metamyl.genouest.org) produces a meta-prediction of sequence amyloidogenicity based on four individual predictors: Pafig, SALSA, Waltz and FoldAmyloid [123]. AmylPred2 (http://thalis.biol.uoa.gr/AMYLPRED2/) is an improved version of the earlier amyloid propensity prediction method (http://biophysics.biol.uoa.gr/AMYLPRED/). It produces a consensus prediction based on 11 algorithms [124]. The method is useful for understanding the misfolding of disease proteins, and it also enables protein aggregation/solubility control in biotechnology.
Interestingly, in the 3D structures of most disease-related amyloid fibrils, the
structures have been shown to contain a
The first definition of prions was formulated by S.B. Prusiner as “small
proteinaceous infectious particles that are resistant to inactivation by most
procedures that modify nucleic acids” [126]. He posited two possible models for
infectious replication of prions: either by a nucleic acid, which would be hidden
and enwrapped by the protein part of the prion, or by a protein void of nucleic
acid. This protein-only hypothesis was already proposed by Griffith, J. [127] and
later substantiated by experimental evidence [128]. A broader definition of
prions encompasses the fact that the mechanism of propagation is
template-directed conformational change [129]. Prions were subsequently detected
in other organisms, e.g., Sup35NM in yeast [130], and more intriguingly, several
of the pathological amyloidogenic proteins, such as amyloid-
Since prions are a particular class of amyloids that can propagate their misfolded conformation and have unique compositional features, several bioinformatic tools capable of identifying novel pathological and functional polypeptides with prion-like properties have also been developed. Below, we discuss the features of several databases and algorithms that have been developed to study prions and prion-like proteins. ZipperDB (https://services.mbi.ucla.edu/zipperdb/) is a database that contains predictions of fibril-forming segments within proteins identified by the 3D profiling method [132]. This method is a unique approach that uses structural information to assess the likelihood of fibril formation of a given sequence [95]. Another interesting new database for predicting prion domains in complete proteomes is PrionScan (http://webapps.bifi.es/prionscan) [133], which has been used to understand the functions of prion/prionogenic protein and how their interaction networks substantially affect gene regulation, to identify regions driving LLPS [92] or proposed as a predictor of prion-like proteins capable of LLPS [134].
First-generation tools for predicting prion-like domains (PrLD) include
pWaltz (http://bioinf.uab.es/pWALTZ/) [135], PrionW
(http://bioinf.uab.cat/prionw/) [136] and PLAAC (Prion-Like Amino Acid
Composition, http://plaac.wi.mit.edu/) [94]. pWaltz was originally inspired by
the Waltz amyloid prediction strategy but used a lower detection threshold to
identify milder amyloids and used a larger sliding window for the minimum
transmissible
The formation of amyloid pores by prefibrillar oligomers shares several similarities with protein toxins and antimicrobial peptides and can also be predicted; however, this was not included in this review as it has been collected previously [139]. For space and scope reasons, we also omitted the prediction of intrinsically disordered regions of proteins, as the new meta-predictors already include these predictions in their workflows.
LLPS, liquid-liquid phase separation; IDPs, intrinsically disordered proteins; MTOC, micro-tubule-organizing center; LCDs, low complexity domains; 3D, three-dimensional.
The data underlying this article are available in the article and in its online supplementary material (Supplementary Table 1).
EŽ—Communication with the journal, Conceptualization, Writing – original draft, Writing – review & editing. KV—Methodology of in silico methods (she is responsible and corresponding author for that part of the manuscript), Writing – review & editing. Both authors read and approved the final manuscript.
Not applicable.
Authors thank for financial support to the Slovenian Research Agency: ARRS Grant Numbers P1-0140 (led by Boris Turk) and P1-017 (led by Marjana Novič).
This research received no external funding.
The authors declare no conflict of interest. Given the role as Guest Editor, EŽ had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to DC.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.