


 , Zongting Gu 2,†, Ran Ni 2, Xiao Wang 2, Qitao Jiang 1, Ran Tao 2,*
, Zongting Gu 2,†, Ran Ni 2, Xiao Wang 2, Qitao Jiang 1, Ran Tao 2,*
1 Department of Surgery, Bengbu Medical College, 233030 Bengbu, AnHui, China
2 General Surgery, Cancer Center, Department of Hepatobiliary & Pancreatic Surgery and Minimally Invasive Surgery, Zhejiang Provincial People's Hospital (Affiliated People's Hospital), Hangzhou Medical College, 310000 Hangzhou, Zhejiang, China
†These authors contributed equally.
Abstract
Pancreatic cancer is the seventh leading cause of cancer-related deaths, and chemotherapy is one of the most important treatments for pancreatic cancer. Unfortunately, pancreatic cancer cells can block chemotherapy drugs from entering the tumor. This is owing to interactions between the tumor’s environment and the cancer cells. Here, we review the latest research on the mechanisms by which pancreatic cancer cells block the chemotherapy drug, gemcitabine. The results of our review can help identify potential therapeutic targets for the blocking of gemcitabine by pancreatic cancer cells and may provide new strategies to help chemotherapy drugs penetrate tumors.
Keywords
- pancreatic cancer
- chemotherapy resistance
- chemosensitization
- tumor microenvironment
- gemcitabine
Pancreatic ductal adenocarcinoma (PDAC) accounts for approximately 90% of pancreatic cancers, which have one of the worst prognoses among solid malignancies in humans. Pancreatic cancer is the seventh leading cause of cancer death, with a 5-year survival rate of only 10% [1, 2, 3]. Surgical resection is the only possible cure for early-stage PDAC; however, only 15%–20% of patients are eligible for surgery, and the overall 5-year survival rate after surgery is only 10%–25% [4, 5]. Therefore, chemotherapy remains a crucial treatment option for PDAC.
Effective delivery of gemcitabine (GEM) to tumor foci is a critical aspect of PDAC treatment. However, the presence of highly fibrotic and densely vascularized tumor tissues poses challenges to drug delivery, leading to limited delivery and difficulty in achieving adequate drug concentrations [6]. Importantly, the interaction between PDAC cells and the tumor microenvironment (TME) is known to stimulate extensive fibrous proliferation [7]. Patients with PDAC have shown poor response rates to immune checkpoint inhibitors, such as anti-programmed cell death protein 1 (PD-1), anti-programmed death ligand 1 (PD-L1), and anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), alone or in combination [8]. However, Bockorny et al. [8] recently showed that the combination of CXCR4 and PD-1 may enhance the chemotherapeutic efficacy for PDAC, and this finding was later confirmed in a subsequent randomized controlled trial (RCT). The TME in PDAC is characterized by its complexity and diverse cellular components, including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), and immune cells. These cellular elements can generate chemical signals that are associated with GEM and can, consequently, impact the efficacy of the drug [9].
GEM has been the basis of first-line chemotherapy for patients with PDAC. Although FOLFIRINOX has shown some benefits in certain studies, a recent randomized phase II clinical trial (NCT02562716) demonstrated that, compared to the AG regimen, the FOLFIRINOX regimen did not prolong survival, instead it substantially increased drug toxicity [10]. The mechanism underlying GEM cytotoxicity involves its role as a deoxycytidine nucleoside analog that competes for DNA strand incorporation and terminates its replication, ultimately leading to cell death [11]. Our team elucidated the role of metabolic remodeling in GEM-resistant pancreatic cancer chemotherapy [12]. Recently, several key developments have been uncovered regarding the involvement of TME in GEM chemotherapy resistance mechanisms in PDAC; however, few reviews are currently available. Therefore, this review aims to provide the current research progress on the mechanism of GEM chemoresistance, with the goal of identifying potential therapeutic targets for GEM resistance and providing new strategies for chemosensitization.
Extensive fibrosis and reduced blood supply vessels are among the important
features of PDAC, which lead to severe tissue hypoxia. Persistent hypoxia has
been demonstrated in pancreatic cancer, with a 17-fold decrease in oxygen
utilization in PDAC tissues compared with that of other tumors. The average
oxygenation level in the healthy pancreas is 6.8%, whereas it is only 0.4% in
pancreatic cancer [13]. Hypoxia-induced activation of hypoxia-inducible factor
(HIF) signaling is present in almost all types of malignancies [14].
Additionally, HIF-1
 Fig. 1.
Fig. 1.In the tumor microenvironment of pancreatic ductal
adenocarcinoma (PDAC), an abundance of fibrous tissue is observed, leading to a
persistent hypoxic condition. This fibrosis is also responsible for impeding
drug penetration due to its high levels. HIF, hypoxia-inducible factor; LM,
Laminin; RhoC, Ras homolog family member C; ROCK1, Rho-associated coiled-coil
containing protein kinase 1; HA, Hyaluronan; FAK, focal adhesion kinase; PLK4,
polo-like kinase4; NF-
Collagen is a major component of the TME and plays a role in forming dense
lattices in cancer fibrosis. Type I collagen, which is closely related to PDAC,
is a heterotrimer and includes many Gly-X-Y repeat sequences [20]. Type I
collagen activates focal adhesion kinase (FAK), a multifunctional non-receptor
protein tyrosine kinase, by binding to
Laminin (LM) and fibronectin are important basement membrane components involved
in blood vessel formation [28]. LM-332, composed of
Fibronectin is less abundant in the extracellular matrix (ECM), but can interact with various proteins for signal transduction, promote angiogenesis [33] and play an essential role in cell adhesion and mobility [34]. Fibronectin can resist GEM-induced apoptosis by inducing ERK1/2 phosphorylation, thereby mediating GEM drug resistance [35]. However, fibronectin was abundantly expressed in PDAC but did not affect patient survival [36]. Therefore, fibronectin may play a dual role in the development of PDAC.
Hyaluronan (HA) is a proteoglycan secreted by stromal cells highly expressed in the PDAC stroma. It has been associated with elevated interstitial fluid pressure, compression of intra-tumor vessels, decrease in tumor perfusion and impaired vascular permeability, thus, leading to increased physical barriers for chemotherapeutic drug delivery and resulting in GEM resistance [37]. In addition, HA should bind specific receptors to function, primarily CD44, a non-kinase transmembrane receptor with multiple isoforms located on the cell membrane. GEM can induce CD44 subtype switching and promote switching to the more resistant and aggressive PDAC phenotype [38]. Activation of epidermal growth factor receptor (EGFR) and insulin-like growth factor-1 receptor (IGF1R) can induce CD44 subtype switching and promote chemoresistance. However, activation of these growth factor receptors may not be required to maintain the resistant phenotype, and blocking IGF1R signaling has been shown to contribute to downregulation of GEM-induced CD44 isoform conversion, thereby inhibiting cell invasiveness and improving chemoresistance [39] (Fig. 1). Moreover, 4-methylumbelliferone (4-MU), an inhibitor of HA synthesis, has been shown to reduce HA production and PDAC cell migration. Therefore, 4-MU could be used to improve GEM sensitivity [40].
The cellular components of the TME in chemoresistance include CAFs, TAMs, immune cells, and various other types of cells. These diverse cell types play a crucial role in mediating chemoresistance. Moreover, the TME also comprises additional cell types, such as mesenchymal cells and mesenchymal stem cells that have the potential to considerably influence tumor development and chemotherapy resistance.
Activated pancreatic stellate cells (aPSCs) can interact with PDAC cells directly and through the paracrine (ECM protein) pathway, which is additionally involved in GEM resistance [41, 42, 43, 44]. In PDAC, CAFs are mainly derived from the activation of PSCs [41, 45, 46] and are induced to form two subtypes, inflammatory (iCAF) and myofibroblast (myCAF), by the direct action of PDAC cells and paracrine signaling [47]. myCAFs are naturally resistant to GEM and can induce acquired resistance through multiple mechanisms [45, 48]. iCAFs promote tumor chemoresistance through a series of cytokines and chemokines [49]. Mechanistically, iCAFs further activate the JAK-STAT signaling pathway through interleukin-1 (IL-1) stimulation to induce chemoresistance. Preclinical studies have shown that JAK inhibitors can reverse this effect [50] (Fig. 2). Unfortunately, ruxolitinib (a JAK1 and JAK2 inhibitor) did not improve the overall survival of patients with locally advanced or metastatic PDAC in two phase III RCTs (NCT02117479 and NCT02119663) [51]. Furthermore, genomic analysis has revealed that IL-6 is overexpressed in GEM-resistant iCAFs and may be a key inhibitor of immune checkpoint blockade therapy in PDAC [52]. A phase II trial (NCT02767557) is currently underway, which targets IL-6 inhibitors in combination with GEM, nab-paclitaxel, and golimumab [53]. Notably, GEM treatment has been found to induce CAFs to secrete an increased number of exosomes. CAFs present in tumors release exosomes that exhibit high expression of miR-3173-5p. This particular microRNA promotes drug tolerance by suppressing ferroptosis through the miR-3173-5p/ACSL4 pathway [54]. Ferroptosis is a newly identified form of iron-dependent programmed cell death that has been associated with GEM chemoresistance in PDAC [55]. ACSL4 plays a crucial role in regulating ferroptosis by activating fatty acid metabolism. Numerous exosome studies are ongoing [56], which may provide new insights into overcoming GEM resistance.
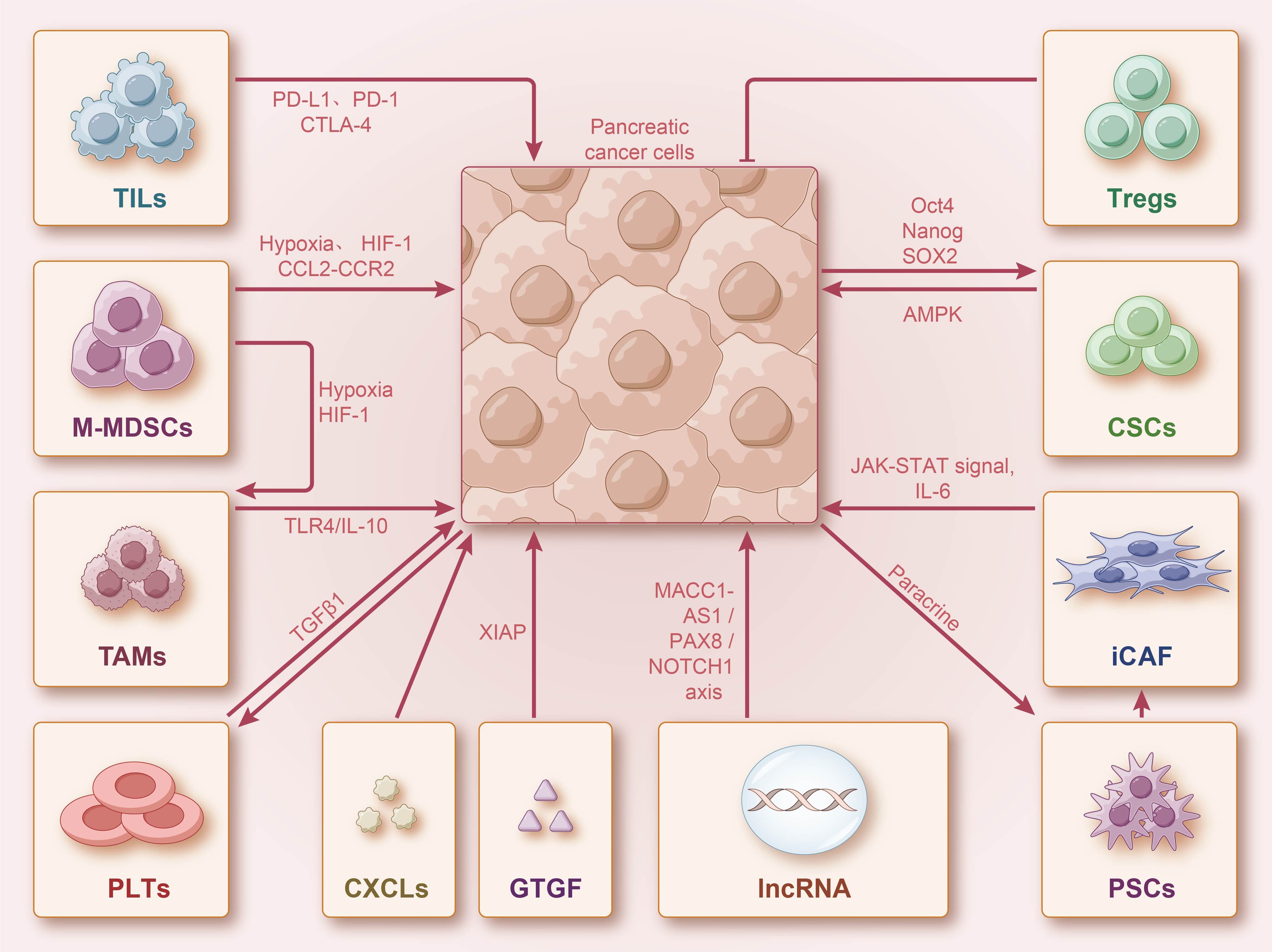 Fig. 2.
Fig. 2.PDAC cells interact with stromal cells. Stromal cells can
interact with PDAC cells through direct contact or paracrine secretion, enhancing
the efficacy of gemcitabine (GEM). Additionally, stromal and immune cells can
secrete various cytokines and chemokines, activating multiple signaling pathways
to interact with PDAC. They can also induce GEM resistance by promoting stem cell
self-renewal. Long non-coding RNA (lncRNA) can also affect PDAC cell
proliferation and induce GEM resistance through multiple signaling pathways.
PD-1, programmed death-1; PD-L1, programmed death ligand 1; CTLA-4, cytotoxic T
lymphocyte associated antigen-4; HIF, hypoxia-inducible factor; CSCs, cancer stem
cells; PSCs, pancreatic stellate cells; CAFs, cancer-associated
fibroblasts; IL, interleukin; LncRNA, long non-coding RNA; TAMs, tumor-associated
macrophages; MDSCs, myeloid-derived suppressor cells; CXCL, chemokine ligands;
TILs, tumor-infiltrating lymphocytes; PLTs, platelets; M-MDSCs,
monocytic-myeloid-derived suppressor cells; GTGF, connective tissue growth
factor; Tregs, regulatory T cells; TGF
CSCs account for approximately 0.5%–1% of pancreatic cancer cells. Furthermore, CSCs can self-renew; hence, playing a key role in the development of PDAC and chemoresistance [57, 58]. Preclinical studies have shown that methyl-CpG binding domain 3 (MBD3) is overexpressed in PDAC. The knockdown of MBD3 reduces Oct4, Nanog, and Sox2 expression; impairs the self-renewal capacity of CSCs; and inhibits the proliferation of PDAC cells [59] (Fig. 2). Similarly, inhibition of Oct4 and Nanog expression suppressed the tumorigenic phenotype of CSCs and increased the chemosensitivity of CSCs in mice [60]. Betulinic acid (BA) can downregulate the expression of Sox2, Oct4, and Nanog by activating the AMPK signaling pathway [61]. Therefore, BA is expected to be a potential GEM chemotherapy-sensitizing agent. Moreover, NR5A2 overexpressed in PDAC cells plays an important role in maintaining stemness and promoting epithelial-mesenchymal transition (EMT) in CSCs [62], suggesting that it could be a potential therapeutic target. There is growing evidence that the dysregulation of long non-coding RNAs (lncRNAs) is closely associated with the development and metastasis of PDAC. HOX transcript antisense RNA is a class of lncRNAs, which can promote proliferation and hinder apoptosis of CSCs by inhibiting miR-34a-activated JAK2/STAT3 and inducing GEM resistance [63, 64]. MACC1-AS1 was recently identified using lncRNA microarray as the most abundant lncRNA in PDAC—it promotes PDAC cell proliferation and metastasis through MACC1-AS1/PAX8/NOTCH1 axis [65, 66]. The current research on CSCs and related non-coding RNAs has received extensive attention, and it is predicted that they are highly likely to constitute another strategy for GEM sensitization.
TAMs consist of two main subtypes, i.e., M1 and M2, with the M1 type activating the immune response against the tumor and increasing chemotherapy sensitivity, contrary to the M2 type [67]. Therefore, induction of TAM reprogramming to M1 or specific inhibition of M2 conversion can help increase GEM sensitivity. CCL2/CCR2 axis activation can recruit inflammatory monocytes to the TME and achieve M2-type conversion, and CCR2 inhibitors can effectively block TAM recruitment [68]. A phase Ib clinical trial (NCT01413022) confirmed that a CCR inhibitor (PF-04136309) in combination with GEM enhances chemotherapeutic efficacy, and further RCT results are expected [69]. M2 can also promote EMT in PDAC through TLR4/IL-10 signaling and induce GEM resistance [70]. Therefore, the TLR4/IL-10 pathway may also be a promising target for chemosensitization.
Myeloid-derived suppressor cells (MDSCs) are heterogeneous immune cell
populations of immature myeloid cells that suppress T-cell immunity and promote
angiogenesis [71]. Among them, monocyte-like MDSCs (M-MDSCs) have a high
plasticity in the TME [72]. The TME can increase the number of M-MDSCs via
secretion of CCL2, CCL5, and CSF1 [73]. In addition, under hypoxic conditions
with upregulated HIF-1
Circulating neutrophils can also be recruited by tumors via chemokine action;
however, the role of TME neutrophils in chemotherapy may be bidirectional, which
may be related to distinct differentiation subtypes [77]. Inhibition of the CXCR2
axis blocks neutrophil recruitment and increases T-cell recruitment [78].
In vitro experiments have confirmed that CXCR2 inhibitors can enhance
antitumor immunity and toxic effects of GEM [77, 79]. Notably, neutrophil
extracellular trap (NET) is a newly discovered form of neutrophil activation that
has been shown to play a crucial role in innate immunity and tumor progression
[80, 81]. NET can promote EMT in PDAC cells through the IL-1
Tumor-infiltrating lymphocytes (TILs) are key cells involved in antitumor immunity and regulation of immune checkpoints. The results of several studies on PD-1/PD-L1/CTLA-4 inhibitors suggest that GEM combined with immunotherapy is a viable option, although many questions remain regarding the underlying mechanisms [83] (Fig. 2). GEM combined with anti-PD-1 antibodies can enhance the immune response of Th1 lymphocytes, resulting in benefits for patients with advanced PDAC [84]. Based on histopathological features, TME immune cells can be classified into three subtypes: immune enriched, escape, and exhaustion. Among them, the immune escape subtype is the most common and characterized by a low content of T and B cells and an enrichment of regulatory T cells (Tregs) [85]. However, Treg depletion failed to alleviate immunosuppression in PDAC tumors, but instead led to accelerated tumor progression. Thus, Tregs infiltration may have a protective role in PDAC; although the exact mechanism is unknown [86, 87]. In summary, studies on the complex mechanisms underlying the role of TILs in TME immunity are emerging and may provide new insights for the comprehensive treatment of PDAC.
The role of platelets (PLTs) in tumor development remains controversial [88].
Recently, it has been shown that PDAC cells can induce the recruitment activation
of PLTs. This further promotes EMT and induces GEM resistance through direct
contact or release of transforming growth factor
Cytokines and chemokines play a key role in mediating the interaction between PDAC cells and ECM, and in the induction of GEM resistance (Fig. 2).
Connective tissue growth factor (CTGF) can interact with other growth factors to
mediate chemoresistance. CTGF and FGF2 have been shown to jointly induce ERK
phosphorylation through fibroblast growth factor receptor 2 (FGFR2) signaling,
leading to resistance to GEM-induced cytotoxicity and promoting chemoresistance
[92]. Moreover, TGF-
Targeted anti-EGFR molecular strategies have been used to reverse the resistance
of PDAC to GEM [95]. Among them, erlotinib is one of the most studied EGFR
inhibitors. An earlier phase III clinical study showed that erlotinib combined
with GEM treatment prolonged the overall survival of patients with pancreatic
cancer [96]. Mechanistically, targeted inhibition of EGFR blocks STAT3 pathway
activation, remodels the tumor stroma, increases microvascular density, and
enhances GEM efficacy [97]. Unfortunately, a recent multicenter phase III
clinical study (NCT02694536) found no notable difference in disease-free and
overall survival for erlotinib combined with GEM compared with GEM monotherapy
for advanced PDAC [98]. Additionally, c-Kit, an important member of the tyrosine
kinase receptor protein family, is overexpressed in PDAC, and masitinib is a
novel specific c-Kit inhibitor. Masitinib reverses the efficacy of PDAC against
GEM and improves patient prognosis [99]. The IGF-1/IGF-1R pathway is upregulated
in approximately 72% of patients with PDAC [100], promoting PDAC proliferation
and inducing GEM resistance by activating the downstream PI3K/Akt/mTOR and
MEK/ERK pathways [101]. Moreover, ganitumab specifically inhibits IGF-1R, and an
earlier phase II clinical study showed that ganitumab combined with GEM prolonged
the overall survival of PDAC with a favorable safety profile [102].
Unfortunately, subsequent phase III studies showed no positive results [103].
Notably, a recent phase I/II clinical trial in PDAC (NCT00769483) has
demonstrated that the (MK-0646) IGF-1R inhibitor in combination with GEM +
erlotinib considerably improved survival compared to GEM + erlotinib [104].
Reassuringly, a recent phase 1b study showed better results with dovitinib in
combination with GEM in treating patients with advanced pancreatic cancer [105].
Dovitinib (TKI258) is a novel multi-targeted tyrosine kinase inhibitor (TKI)
targeting FGFRs, PLT-derived growth factor receptor
The CXC chemokine family (CXCL1-17) are small secreted proteins that induce GEM
resistance by binding to their receptors (CXC chemokine receptor, CXCR) to
regulate paracrine and autocrine pathways in PDAC mesenchymal cells that lead to
GEM resistance [108, 109]. A typical example is the CXCL12/CXCR4 axis that
activates FAK, ERK, and Akt pathways to promote survivin expression, thus,
inducing GEM resistance [110, 111]. A phase IIa clinical study (NCT02826486)
showed that the CXCR4 antagonist BL-8040 (motixafortide) in combination with
pembrolizumab prolonged the overall survival of patients [8]. Notably, in PDAC
cells, artemin can induce CXCR4 overexpression by activating ERK1/2 and Akt
pathways and further through NF-
In the past decade, there has been little improvement in the overall survival rate of PDAC, and the primary treatment regimen is still GEM-based chemotherapy. Overcoming chemoresistance is a key aspect of improving prognosis. Although current clinical trials targeting TME stromal components have yielded unsatisfactory results, research on the TME continues to advance, and clinical trials combining GEM with multiple immunotherapy targets have shown encouraging results, although previous immune monotherapies have mostly been ineffective [113] (Table 1, Ref. [16, 18, 19, 24, 37, 49, 50, 51, 52, 53, 59, 60, 61, 65, 66, 67, 68, 69, 74, 80, 81, 82, 83, 84, 92, 93, 94, 97, 98, 100, 101, 102, 103, 104, 108, 109, 110, 111, 112]). In addition to immunotherapy, targeted therapies, gene editing, microbial therapies, and combination therapies have exhibited promising results in the clinical management of PDAC. It is crucial to highlight that while these cutting-edge treatments hold immense potential for PDAC patients, many of them are still undergoing early-stage clinical trials, and need further research and validation to establish broader clinical applicability.
| Potential targets | Drug resistance mechanism | Drug/premedication | Clinical trials | Phases | Experimental status | Experimental result | References |
| HIF-1 | Upregulation of PLK4 and induction of differentiation of M-MDSCs into TAM | NA | NA | NA | NA | NA | [16, 74] |
| HiF-2 | Reduces macrophage chemotaxis and M2-type polarization | Belzutifan | NCT04924075 | Phase II | Recruiting | NA | [18] |
| HIF-3 | RhoC-ROCK1 signaling pathway | NA | NA | NA | NA | NA | [19] |
| FAK | Substrate physical barrier | Defactinib | NCT02546531 | Phase I | completed | Positive | [24] |
| HA | Adding IFP | NA | NA | NA | NA | NA | [37] |
| iCAFs | Activation of JAK-STAT pathway | Ruxolitinib | NCT02117479, NCT02119663, NCT02767557 | Phase III, Phase II | completed | Negative | [49, 50, 51, 52, 53] |
| CSCs | Increased Oct4, Nanog, Sox2 expression, induction of EMT | NA | NA | NA | NA | NA | [59, 60, 61] |
| lncRNA | MACC1-AS1/PAX8/NOTCH1 | NA | NA | NA | NA | NA | [65, 66] |
| TAMs | Activation of TLR4/IL-10 pathway, M2-type polarization | PF-04136309 | NCT01413022 | Phase I/II | completed | Positive | [67, 68, 69] |
| NETs | Activation of IL-1 |
NA | NA | NA | NA | NA | [80, 81, 82] |
| PD-1/PD-L1/CTLA-4 | Immune response of Th1 lymphocytes | NA | NA | NA | NA | NA | [83, 84] |
| CTGF | Phosphorylation of ERK | mAbFG-3019 | NCT02210559 | Phase II | completed | Positive | [92, 93, 94] |
| EGFR | Remodeling of tumor stroma, Activation of STAT3 pathway | Erlotinib | NCT02694536 | Phase III | completed | Negative | [97, 98] |
| IGF-1R | Activation of PI3K/Akt, mTOR, MEK/ERK pathway | MK-0646 | NCT00769483 | Phase I/II | completed | Positive | [100, 101, 102, 103, 104] |
| CXCL12/CXCR4 | Activation of FAK, ERK and Akt pathways, Promotion of |
motixafortide | NCT02826486 | Phase II | completed | Positive | [108, 109, 110, 111, 112] |
CSC, cancer stem cells; CTGF, connective tissue growth factor; EGFR, epidermal growth factor receptor; EMT, epithelial-mesenchymal transition; FAK, focal adhesion kinase; HA, hyaluronan; iCAF, inflammatory cancer-associated fibroblast; IGF-1R, insulin-like growth factor 1 receptor; lncRNA, long non-coding RNA; MDSC, myeloid-derived suppressor cells; NET, neutrophil extracellular trap; TAM, tumor-associated macrophages; NA, not available.
PDAC, pancreatic ductal adenocarcinoma; GEM, gemcitabine; TME, tumor
microenvironment; PD-1, programmed death-1; PD-L1, programmed death ligand 1;
CTLA-4, cytotoxic T lymphocyte associated antigen-4; HIF, hypoxia-inducible
factor; CA, carbonic anhydrase; PLK4, polo-like kinase4; CSCs,
cancer stem cells; RhoC, Ras homolog family member C; ROCK1, Rho-associated
coiled-coil containing protein kinase 1; FAK, focal adhesion kinase; MT1-MMP,
membrane type-1 matrix metalloproteinase; HMGA2, high mobility group protein A2;
LM, Laminin; TG2, transglutaminase 2; NF-
ZTG and RT proposed the research topic of the article, TPZ and RN collected the literature and wrote the paper. XW and QTJ performed the image design and revision. All authors participated in the editorial revision of the manuscript. All authors read and approved the final manuscript. All authors participated fully in the work, assumed public responsibility for appropriate portions of the content, and agreed to be accountable for all aspects of the work for issues related to the accuracy or completeness of the work.
Not applicable.
Not applicable.
This work was funded by This work was funded by RT’s Provincial Health Ministry Major 2020, grant number A-2019-W153.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.