
- Academic Editors
†These authors contributed equally.
Background: Hepatocellular carcinoma (HCC/LIHC) is the most common type of primary liver cancer, which is a leading cause of cancer death worldwide. Patients with HCC have a short survival time after diagnosis. Unfortunately, there are no effective treatments for advanced or aggressive HCC. Thus, the rapid development of new therapeutic drugs or treatment methods for HCC is urgently needed. Methods: Bioinformatic tools and computer-aided predictions advance the processes of drug development. In this study, we incorporated bioinformatic analyses and computer-aided drug development processes to investigate the potential application of bilobetin, a bioactive compound of bioflavonoid, as a therapeutic agent for HCC treatment. Results: Our results revealed that 4 out of 20 predicted hub target genes of bilobetin displayed functional importance in cancer-related signaling pathways in different cancers, including HCC. Importantly, the mRNA expression levels of these four key hub genes (VEGFA, SRC, MMP9, and CDK1) were significantly different between normal and HCC tumor samples. Their expression levels were significantly associated with the clinical survival outcomes of HCC patients, as well as the immune cell infiltration levels in the HCC tumor microenvironment. In addition, these four genes showed significant co-expression correlated with immune checkpoint genes, including CD274, PDCD1, CTLA4, and CD47. Furthermore, we used computer-aided approaches to investigate the binding affinity and potential binding mechanisms between bilobetin and target proteins encoded by four key hub genes. Conclusions: In conclusion, our study shed light on the potential application of the bioactive bioflavonoid molecule bilobetin in LIHC treatment by regulating four key hub genes.
The most prevalent type of primary liver cancer is hepatocellular carcinoma (HCC/LIHC), one of the leading causes of cancer death worldwide [1, 2]. The number of HCC cases is increasing [3]. It has been estimated that there will be more than 1 million cases per year globally by 2025 [4]. HCC can be induced by many factors, such as extensive alcohol consumption, non-alcoholic fatty liver disease (NAFLD), liver fibrosis or cirrhosis. The incidence of NAFLD-associated HCC is increasing, which is also commonly associated with many other metabolic diseases such as obesity, type 2 diabetes mellitus, and cardiovascular disease [5, 6, 7]. Most HCC cases are diagnosed at a late stage; however, current therapies for advanced HCC are not promising [8, 9]. Therefore, the exploration of early diagnostic biomarkers and development of effective treatment drugs or strategies for HCC are urgently needed [10].
Research studies and clinical trials have demonstrated that Ginkgo biloba extract displays effective activities in the treatment of metabolic diseases, including cancers (e.g., brain and breast cancers) [11]. Bilobetin is a bioactive molecule isolated from the leaves of the Ginkgo biloba. As the bioflavonoid class of phytochemicals, bilobetin exhibits the pharmacological characteristics of Ginkgo biloba extract [12]. The important pharmacological properties of bilobetin include antioxidant, anti-inflammatory, anti-hyperlipidemic, and anti-proliferative activities [12, 13, 14]. Studies have also revealed that bilobetin possesses potent anti-cancer activity [11, 14, 15]. For example, bilobetin displays cytotoxic effects against several human cancer cell lines [14], including cervical carcinoma (HeLa), large cell lung cancer (NCI-H460), lymphoma (Daudi), myelogenous leukemia (K562), ovarian adenocarcinoma (SKOV3), pancreatic carcinoma (MIAPaca-2), and breast carcinoma (MCF-7). However, the action of bilobetin in liver cancer treatment remain to be studied.
In this study, we investigated the activity of bilobetin in liver cancer treatment. First, we applied a computer-aided drug design tool, SwissTargetPrediction, to predict molecular targets of bilobetin based on the data generated from both laboratory experiments (binding assay) and the calculation of similarities between 2D/3D structures of query molecules and known molecules from online databases [16, 17]. Then, we analyzed protein-protein interaction (PPI), KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway, and gene ontology (GO) pathway to examine the functions of target genes, including biological processes (BP), cellular components (CC), and molecular functions (MF). Among the target genes, we first identified 20 hub genes using Cytoscape, and then further revealed the functions and enriched signaling pathways of the hub genes based on the analyses of KEGG and GO pathways [18, 19]. We subsequently applied bioinformatic tools, such as UALCAN (the University of Alabama at Birmingham CANcer Data Analysis Portal) [20, 21], to analyze the expression levels of hub genes in clinical LIHC/HCC tumor and non-tumor control samples from the Cancer Genome Atlas (TCGA) database. Meanwhile, the association of hub genes with the prognostic outcomes of LIHC patients was also examined [22, 23]. Interestingly, we found that four key genes out of 20 hub genes showed significantly different mRNA expression levels between clinical tumor and non-tumor samples, and their mRNA expression levels were significantly associated with the clinical prognostic outcomes (survival rates) of LIHC patients. In addition, we analyzed the proteomic expression levels of these four key genes between normal and primary LIHC tumor samples [24, 25].
Furthermore, we evaluate the correlation between the expression levels of four key genes with levels of immune cell infiltration [26, 27] and the expression of immune checkpoints such as CD274, PDCD1, CTLA4, and CD47 in the HCC tumor microenvironment [28, 29]. Finally, computer-aided programming was used to explore the underlying molecular mechanism of bilobetin as a therapeutic agent for LIHC therapy by regulating four key genes. The binding affinities and modes of bilobetin with four target proteins were tested using computer-based molecular docking methods [30]. The key amino acids that were involved in the interactions between the bilobetin molecule and proteins encoded by four key genes were analyzed, including the hydrophobic interaction and formation of hydrogen bonds [31, 32].
SwissTargetPrediction is a commonly used bioinformatic tool to predict the targets of bioactive small molecules (e.g., bilobetin) in humans and other vertebrates. It is a useful approach for understanding molecular mechanisms, rationalizing possible side-effects, and predicting off-targets of known molecules [14]. In this study, it was used to investigate the molecular targets of bilobetin by selecting Homo sapiens genes.
We applied a bioinformatic database, STRING (https://string-db.org/), for the construction of a protein-protein interaction (PPI) network. This tool allows the construction of an interaction network based on the data obtained from both experiments and online databases. The cross-interaction of multiple queried proteins can be predicted using this tool [33]. For parameters used in this study, Homo sapiens genes were selected, and 0.4 was set as the default threshold, which was considered significant for the minimum required interaction specificity score.
The Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were used to investigate the functional enrichment and signaling pathway enrichment of bilobetin-targeted genes and 20 hub genes. The categories of the GO enrichment analysis included biological processes (BP), cellular components (CC), and molecular functions (MF). We applied DAVID bioinformatics resources for the analysis of results and used the platform (http://www.bioinformatics.com.cn) for result plotting and visualization [34, 35].
The Cytoscape (https://cytoscape.org/) platform was applied for the analysis and visualization of the complex networks including the connecting nodes and molecular interactions, as well as the identification of the key hub genes from bilobetin-targeted genes [36].
The interactive bioinformatics tool GEPIA (http://gepia.cancer-pku.cn/) [37] was
applied to compare the gene expression levels of target genes between normal
samples and LIHC tumor samples and to analyze the survival outcomes of LIHC
patients. In this study, to compare the expression levels of target genes in the
normal and tumor samples from the database, the matched normal data included the
TCGA normal data and GTEx data. ANOVA method was applied with the default
parameters (log
TIMER is a resource with multiple modules allowing for a comprehensive and
systematic examination of immune cell infiltration levels in clinical samples for
different cancer types [38, 39, 40]. In this study, we applied the gene module to
investigate the correlation between the expression levels of identified key hub
genes and the immune cell infiltration levels in LIHC. The correlation module was
applied to analyze and generate the scatterplots of the co-expression levels
between the identified key genes and the immune checkpoint-associated genes in
LIHC. The scatterplots were generated and presented based on the purity-corrected
partial Spearman’s Rho value, and p
Molecular docking was performed using Cavity-detection guided Blind Docking (CB-DOCK), the open-source program AutoDock Vina v.1.2.0. (http:/www.vina.scripps.edu/) [41], for testing the binding affinity and simulation of the binding mechanism. The crystal structures of proteins encoded by the identified genes used for docking were obtained from the PDB database, which were well-prepared for the docking program, including VEGFA (PDB ID: 5T89); SRC (PDB ID: 2H8H); MMP9 (PDB ID: 4H2E); CDK1 (PDB ID: 5HQ0). Pymol software was used for the visualization and presentation of protein-ligand interaction. PDBsum [42] and Discovery Studio Visualizer v21.1.0.20298 (BIOVIA Dassault Systemes, San Diego, CA, USA) were used for the 2D interaction analysis and presentation.
The pharmacokinetics, drug-likeness, and medicinal chemistry of bilobetin were evaluated using the SwissADME bioinformatics tool [43].
The mRNA expression levels of immune checkpoint genes PDCD1,
CD274, CD47, and CTLA4 were analyzed using UALCAN
web-based tool. The required gene expression level in LIHC was analyzed to
compare the normal and primary tumor groups using Student’s t-test, and
p
The Human Protein Atlas (https://www.proteinatlas.org/, version 23.0) [44, 45] was applied for the generation of a representative IHC-staining panel to demonstrate the pathologic effects associated with different protein expression levels of key genes VEGFA, SRC, MMP9, and CDK1. This database was also used to validate the association between the expression levels (low and high) of four key genes and the survival outcomes of LIHC patients.
Student’s t-test (considering unequal variance) was used to compare
mRNA expression levels of key genes in normal samples and LIHC tumor samples, and
p
To investigate the drug targets of bilobetin, we applied one of the computer-aided drug discovery tools, SwissTargetPrediction, which can predict drug targets of a small molecule based on the datasets generated from well-designed experimental binding assays and the online database. Homo sapiens was defined as a species for the identification of target proteins. The obtained results showed that among the predicted targets, 26% were kinases, 23% were enzymes, 10% belonged to family A of G protein-coupled receptors, 6% were proteases, whereas 5% were oxidoreductases and 5% were lyases (Fig. 1A). Other targets include cytochrome P450, hydrolases, isomerases, nuclear receptors, cytosolic proteins, phosphatases, and secreted proteins (2% for each class category), electrochemical transporters, erasers, ligand-gated ion channel proteins, other ion channel proteins, phosphodiesterases, primary active transporters, and transcription factors (1% for each class category) (Fig. 1A).
 Fig. 1.
Fig. 1.Predicted drug targets of bilobetin and the protein-protein interaction (PPI) network of targets. (A) The pie chart shows the bioactivity categories of bilobetin-targeted proteins and the percentages of each category. (B) The constructed PPI network of all target proteins of bilobetin.
To further examine the properties of the bilobetin-targeted proteins and their
interaction partners, we applied the bioinformatic analysis tool, STRING, to
construct a PPI network of these targets (Fig. 1B). The constructed network
contained 98 nodes and 386 edges, with an average node degree of 7.88 and an
average local clustering coefficient value of 0.54. The PPI enrichment
p-value was less than 1.0
To determine the biological and functional enrichment of target genes of bilobetin, we performed GO enrichment analysis. The GO enrichment analysis results for biological processes revealed that the target genes play important roles in protein autophosphorylation, apoptotic process, mitogen-activated protein (MAP) kinase activity, metabolic processes of chemotherapy drugs such as doxorubicin and daunorubicin, vascular endothelial growth factor receptor, and metabolic regulation of reactive oxygen species (Fig. 2). For the cellular component, the results showed these genes were mostly involved in the cytosol, cyclin-dependent protein kinase holoenzyme complex, cytoplasm, plasma membrane, glutamatergic synapse, neuron projection, perinuclear region of cytoplasm, axon, and others (Fig. 2). The results of the molecular function analysis identified that the key roles of these genes were enriched for protein kinase activity, protein serine/threonine kinase activity, ATP binding, bile acid binding, oxidoreductase activity, tyrosine kinase activity, and others (Fig. 2).
 Fig. 2.
Fig. 2.The gene ontology (GO) enrichment analysis results. GO analysis results display the roles of bilobetin-targeted genes in biological processes (orange), molecular function (green), and cellular components (blue).
To better understand the major roles of these targeted genes in signaling pathways and diseases, we further performed a KEGG analysis and the top 20 enriched signaling pathways are shown (Fig. 3). The results showed that these genes played critical roles in chemical carcinogenesis-reactive oxygen species, pathways in cancer, acute myeloid leukemia, bladder cancer, and proteoglycan in cancer. The enriched signaling pathways of target genes were the RAS signaling pathway, PI3K-AKT signaling pathway, VEGF signaling pathway, and EGFR tyrosine kinase inhibitor resistance.
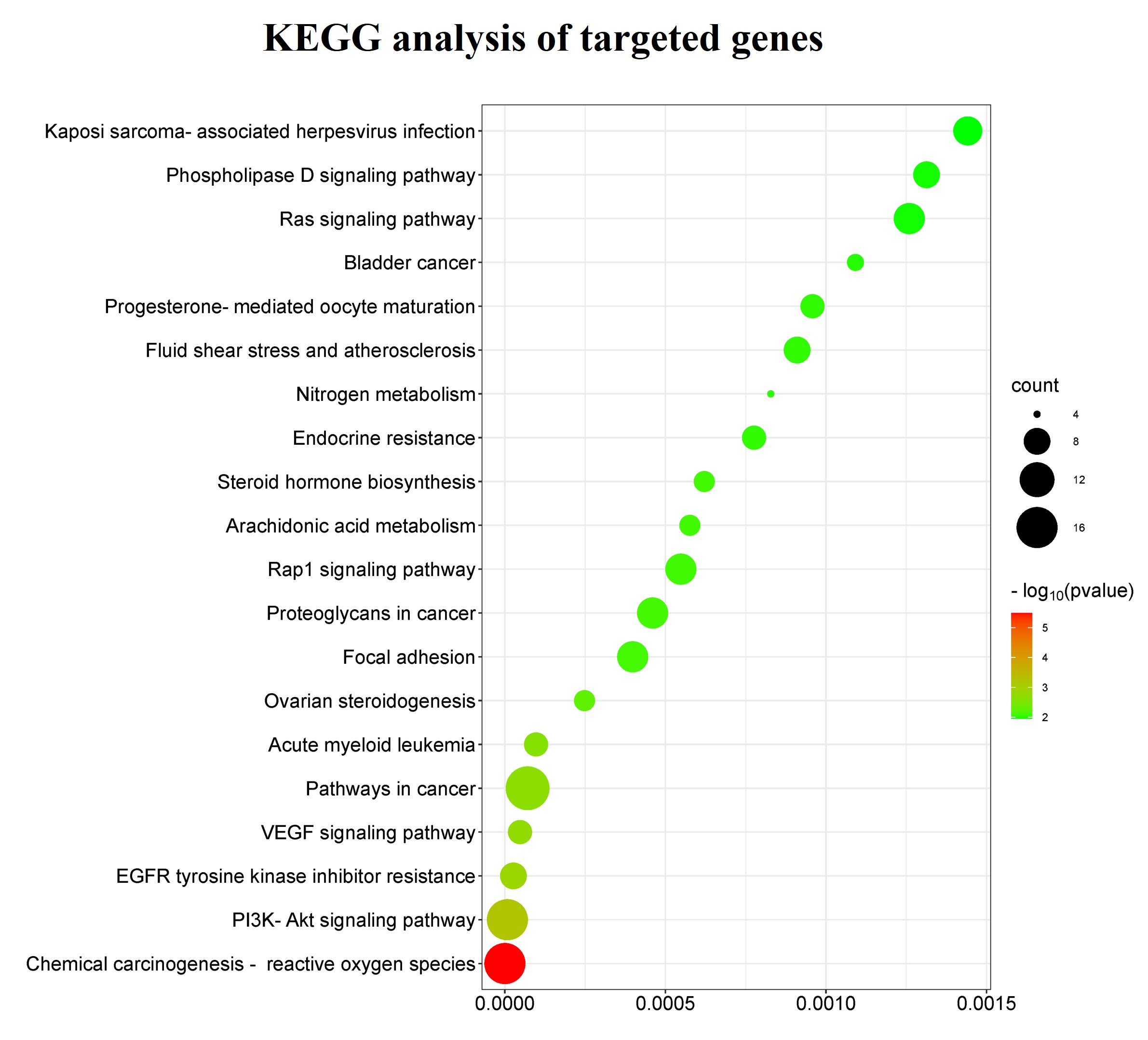 Fig. 3.
Fig. 3.The Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analysis of bilobetin-targeted genes. The top 20 signaling pathways, functional information, and related diseases are presented in the bubble plot. The size of the bubbles represents the gene count, and the color of the bubbles represents the values of –log10 (p-value).
To further identify linkage genes from bilobetin-targeted genes, we analyzed hub genes based on the previously constructed protein-protein interaction (PPI) results using the software Cytoscape. The top 20 hub genes were MET, MMP2, PARP1, CDK1, PTPN1, GSK3B, CDK6, SRC, VEGFA, PTGS2, MCL1, KIT, MPO, AKT1, MAPT, PIK3R1, KDR, PTK2, ESR2, and MMP9, and were identified to have a complex interaction (Fig. 4).
 Fig. 4.
Fig. 4.Identification of the top 20 hub genes using Cytoscape based on
the results of protein-protein interaction (PPI) network of bilobetin-targeted
genes. The PPI network contains 20 nodes and 113 edges. The average node degree
is 11.3, and the average local clustering coefficient is 0.735. The PPI
enrichment p-value is less than 6.1
To further test whether the top 20 hub genes represent the main functions of target genes, the examination of functional enrichment and signaling pathways of these hub genes was carried out. The results of GO functional enrichment analysis revealed that, for biological processes, 20 hub genes were highly enriched in the following pathways including negative regulation of the apoptotic process, positive regulation of cell migration, protein autophosphorylation, positive regulation of phosphatidylinositol 3-kinase signaling pathway, negative regulation of the intrinsic apoptotic pathway, vascular endothelial growth factor receptor pathway, positive regulation of protein localization to nucleus, cellular response to reactive oxygen species, transmembrane receptor protein tyrosine kinase pathway, and peptidyl-tyrosine phosphorylation (Fig. 5). Regarding the cellular component, these hub genes were found to have greater enrichment in the mitochondrion, cytoplasm, the extrinsic component of cytoplasmic membrane, nucleus, macromolecular complex, sorting endosome, extracellular region, plasma membrane, cell-cell junction, cytosol, etc. (Fig. 5). The results of the molecular function demonstrated that the hub genes were highly enriched in the function of enzyme binding, protein tyrosine kinase activity, transmembrane receptor protein tyrosine kinase, protein kinase activity, ATP binding, protein kinase binding, insulin receptor binding, protein binding, protein phosphatase 2A binding, protein homodimerization activity, and other molecular function processes (Fig. 5).
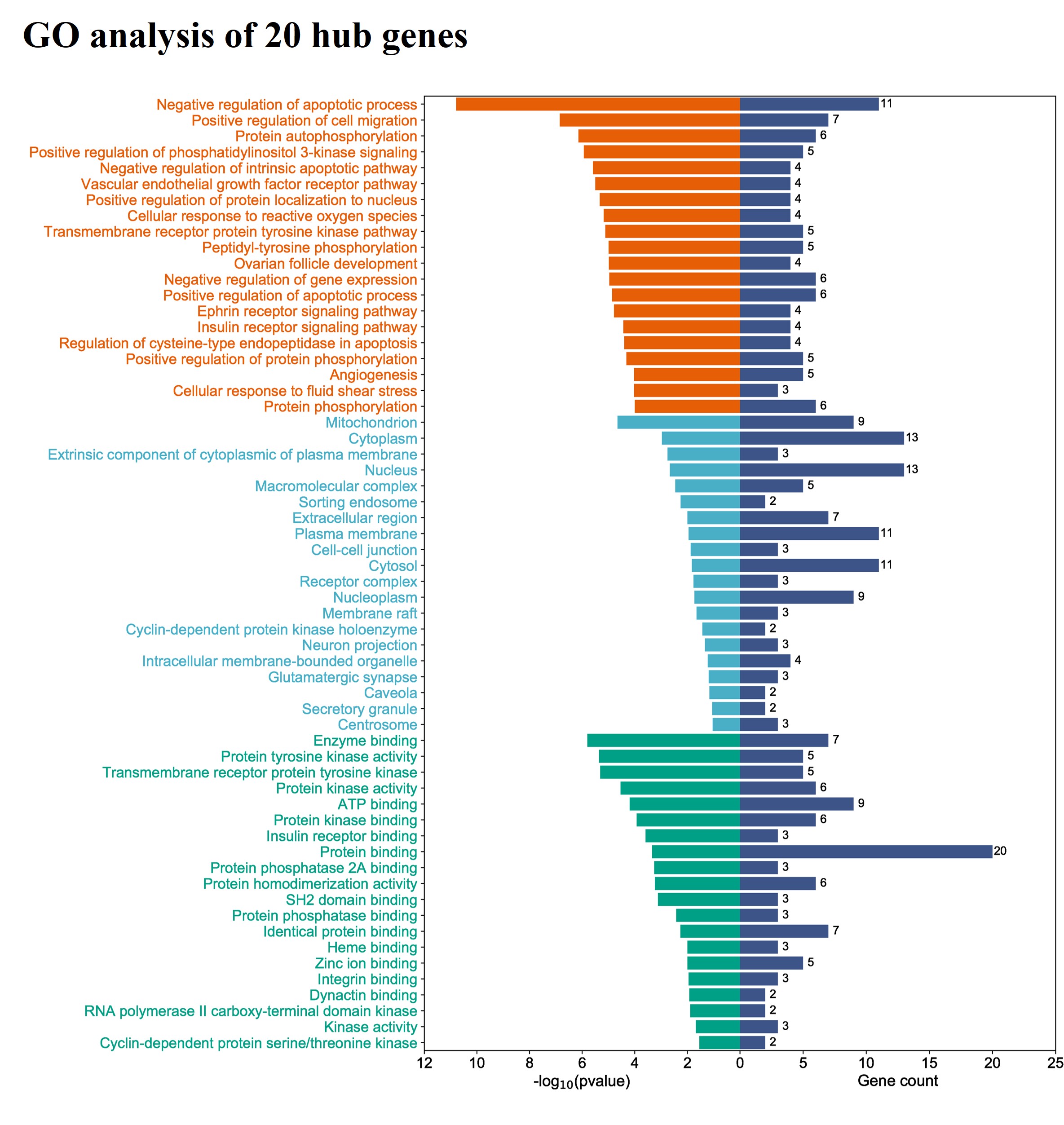 Fig. 5.
Fig. 5.The gene ontology (GO) enrichment analysis results of 20 hub genes. The top 20 functionality enrichment items for each category were presented, including biological processes (orange), molecular functions (green), and cellular components (blue).
Most importantly, the subsequent analysis of KEGG signaling pathways uncovered that 20 hub genes were highly enriched in and contributed to the important signaling pathways in cancer biology, such as the VEGF signaling pathway, pathways and proteoglycans in cancer, microRNAs in cancer, EGFR tyrosine kinase inhibitor resistance, PI3K-AKT signaling pathway, chemical carcinogenesis-reactive oxygen species, ErbB signaling pathway, small cell lung cancer, breast cancer, bladder cancer, and RAS signaling pathway. The top 25 signaling pathways and related diseases are shown (Fig. 6).
 Fig. 6.
Fig. 6.The Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling pathway enrichment analysis of 20 hub genes. The top 25 signaling pathways, associated diseases, and other functional information were presented in the bubble plot. The size of the bubbles represents gene counts, and the color of the bubbles represents the value of –log10 (p-value).
Due to the important roles of bilobetin-targeted hub genes in cancer, we extended our study by examining the mRNA expression levels of these hub genes in normal and liver tumor samples using a clinical database. Among the 20 hub genes, we identified 6 genes that showed significant differences in mRNA expression levels between the normal/non-tumor (n = 160) and LIHC tumor samples (n = 369). The expression level of VEGFA was significantly decreased in LIHC tumor samples compared with normal samples, whereas the expression levels of SRC, MMP9, PARP1, CDK1, and PTK2 were significantly increased in LIHC tumor samples compared with the normal samples (Fig. 7).
 Fig. 7.
Fig. 7.Analysis of mRNA expression levels of hub genes between
non-tumor control and liver hepatocellular carcinoma (LIHC) tumor samples. Among the 20 hub genes,
VEGFA, SRC, MMP9, PARP1, CDK1, and
PTK2 showed significantly different expression levels between non-tumor
and tumor samples. n = 160 (non-tumor samples) and n = 369 (tumor samples). ANOVA
was applied for statistical analysis, and *p
To further explore the clinical prognosis values of hub genes, we examined the association between the clinical outcomes of LIHC patients and the expression levels of 20 hub genes using the bioinformatics tool GEPIA. The results showed that, among 20 hub genes, the expression levels of eight genes (VEGFA, SRC, MMP9, GSK3B, CDK1, KIT, MAPT, PTPN1) were significantly associated with the clinical survival outcomes of LIHC patients. The expression levels of these eight genes were significantly and negatively associated with the survival outcomes of LIHC patients (Fig. 8).
 Fig. 8.
Fig. 8.Association of the mRNA expression levels of 20 hub genes with the overall survival outcomes of LIHC patients analyzed by comparing high and low gene expression groups. Among 20 hub genes, the gene expression levels of VEGFA, SRC, MMP9, GSK3B, CDK1, KIT, MAPT, and PTPN1 were significantly associated with the clinical outcomes in terms of overall survival times (months) of LIHC patients. The hazard ratio was calculated based on the Cox PH Model according to the default parameters, as well as the 95% confidence interval (CI).
Given the mRNA expression levels of four hub genes (VEGFA, SRC, MMP9, and CDK1) and their association with the survival outcomes of LIHC patients, we then focused on these four key hub genes in the following study.
To analyze the proteomic expression levels of four genes, including VEGFA, SRC, MMP9, and CDK1, we used the Clinical Proteomic Tumor Analysis Consortium (CPTAC) database to compare their proteomic expression levels between normal samples and primary LIHC tumor samples. Our results showed that the proteomic expression levels of VEGFA, MMP9, and CDK1 have significantly different. However, there was no statistically significant difference in the proteomic expression levels of SRC between normal and LIHC tumor samples (Fig. 9).
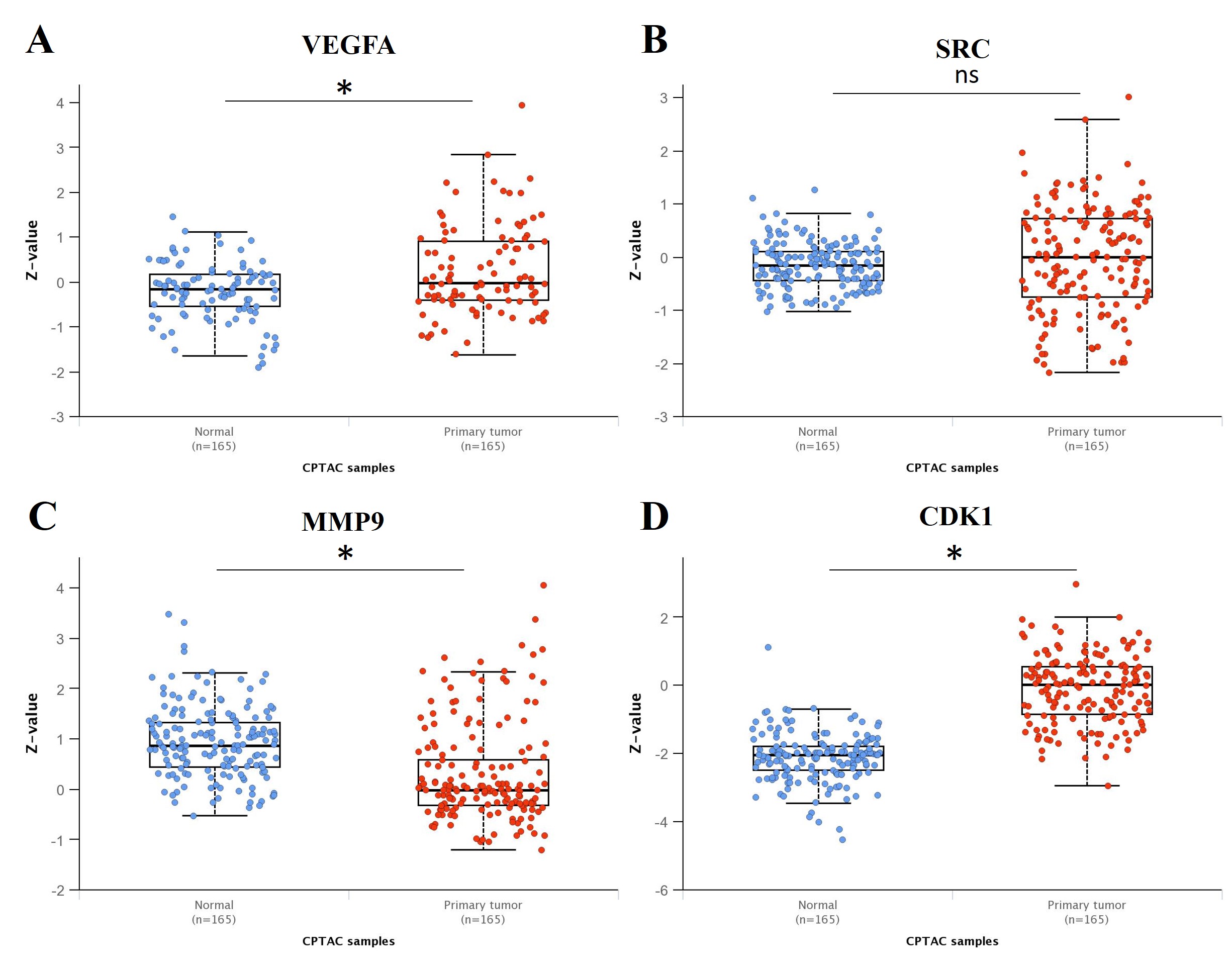 Fig. 9.
Fig. 9.Analysis of the proteomic expression levels of four genes
between the normal samples and primary LIHC tumor samples. (A) VEGFA; (B) SRC;
(C) MMP9; (D) CDK1. VEGFA, MMP9, and CDK1 showed significantly different
proteomic expression levels between normal (n = 165) and primary tumor samples (n
= 165). *p
To validate the significance of the four key hub genes (VEGFA, SRC, MMP9, and CDK1) in LIHC, the protein expression staining results and the prognosis values of the four key genes in the overall survival of HCC patients were collected from the Human Protein Atlas for analysis. The prognosis values were consistent with the results shown in Fig. 8, indicating that high expression of these four genes predicted poor overall survival of HCC patients (Supplementary Fig. 1). In addition, immunohistochemical (IHC) staining showed low and high expression levels of the proteins encoded by the four key genes in clinical LIHC samples (Supplementary Fig. 1).
Due to the critical roles of tumor-infiltrating immune cells in the survival outcomes of LIHC patients, it is important to examine the correlation of the expression levels of the four key hub genes (VEGFA, SRC, MMP9, and CDK1) with the tumor-infiltrating levels of immune cells, as well as the expression of immune checkpoint-associated genes.
Firstly, we examined the correlation between the expression levels of the four
key hub genes and the tumor-infiltrating levels of several immune cells,
including B cells, CD8
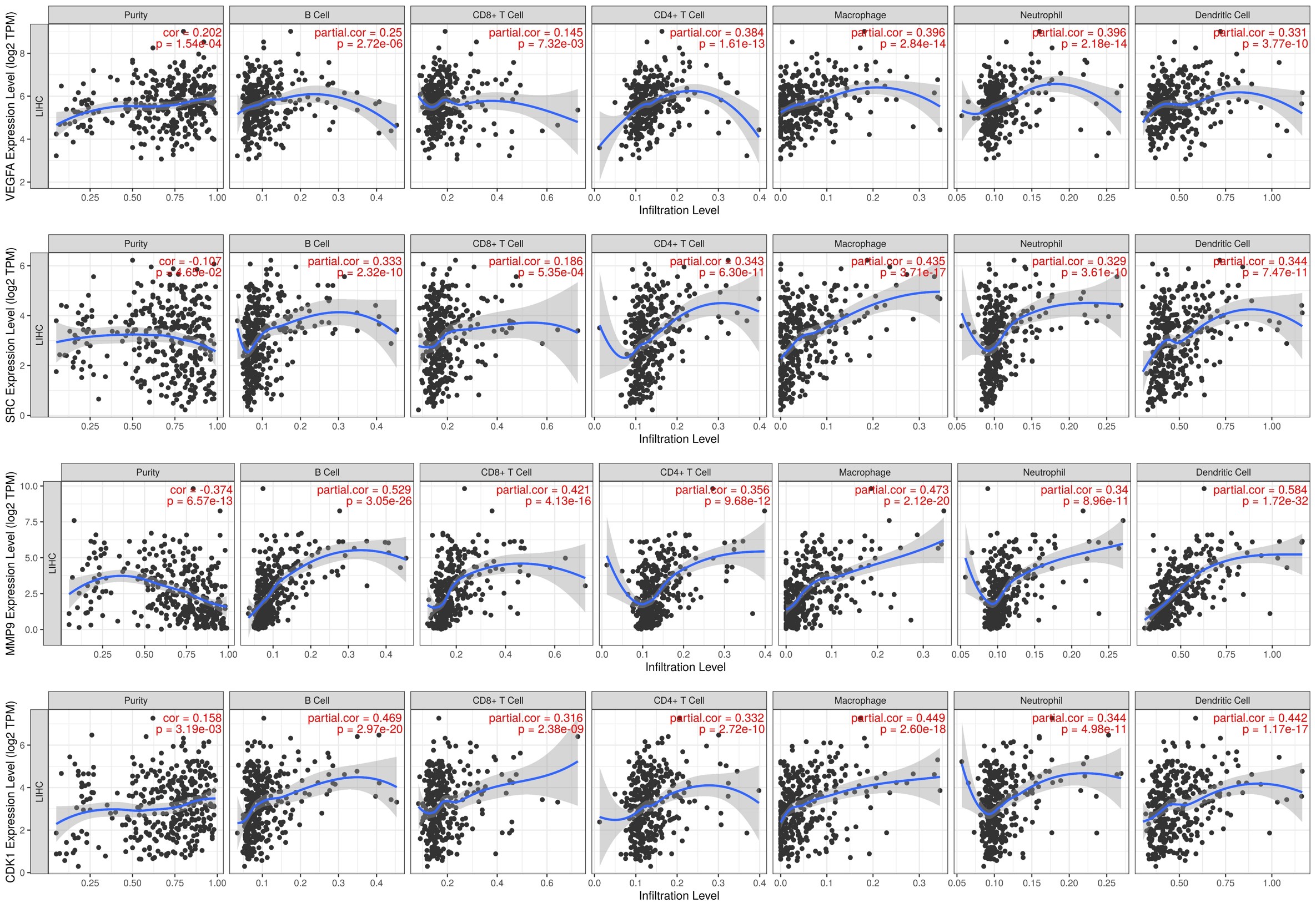 Fig. 10.
Fig. 10.The correlation of the gene expression levels and the levels of
tumor-infiltrating immune cells in LIHC. The expression levels of genes
VEGFA, SRC, MMP9, and CDK1 were significantly
correlated with the tumor-infiltrating immune cell levels of B cells, CD8
To determine the significance of the immune checkpoint markers (PDCD1,
CD274, CTLA4, and CD47) in HCC, the mRNA and proteomic
expression levels in clinic LIHC primary tumor (n = 371) and non-tumor control
samples (n = 50) were analyzed using the data from the TCGA database. The results
demonstrated that checkpoint markers PDCD1, CTLA4, and
CD47 showed significantly higher mRNA expression levels in LIHC primary
tumors compared with the non-tumor samples. In contrast, CD274 showed a
decreased trend (p = 4.95
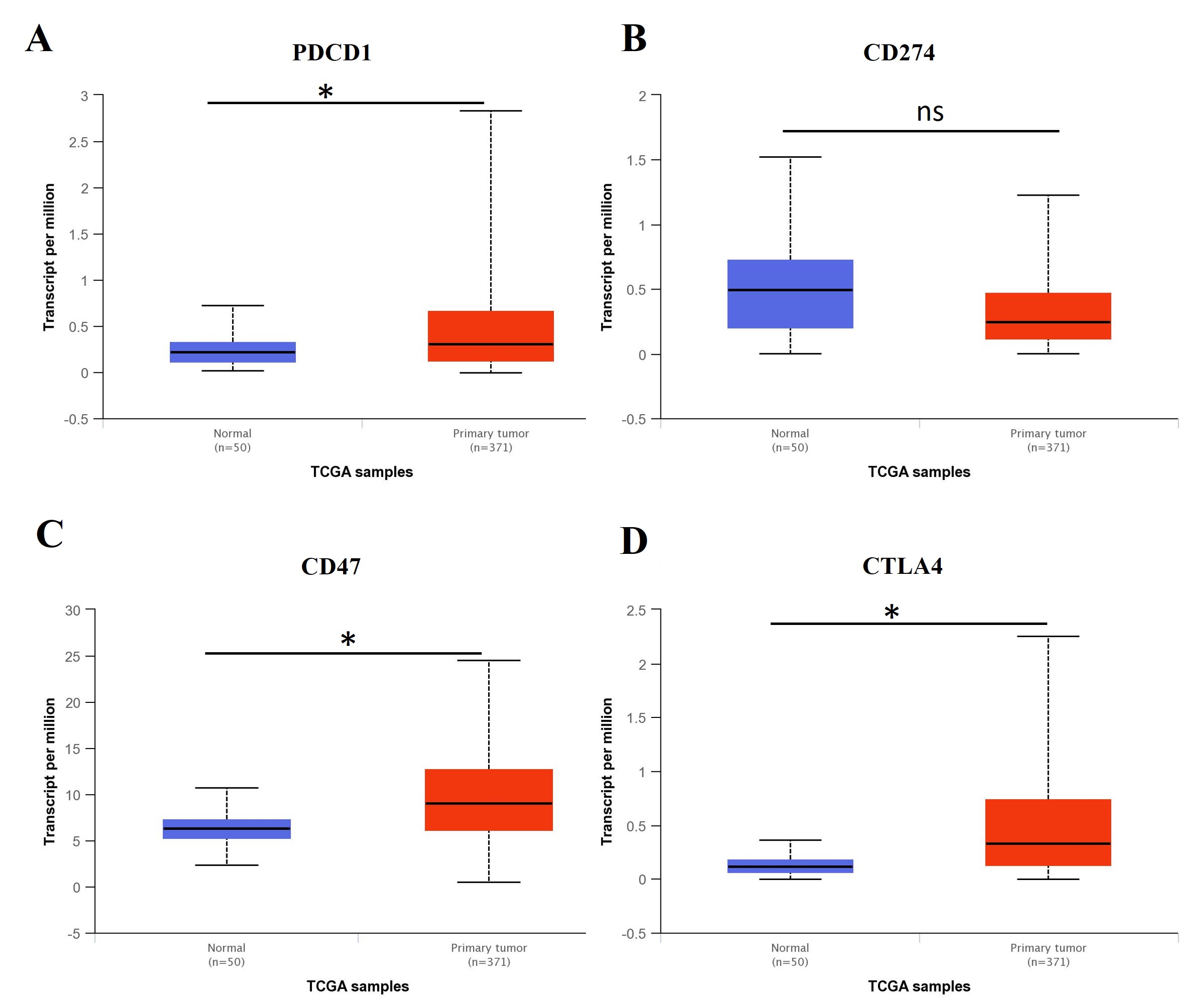 Fig. 11.
Fig. 11.The expression level of immune checkpoint biomarkers
in LIHC tumor and non-tumor control samples. The mRNA expression levels of (A)
PDCD1 (p = 3.01
 Fig. 12.
Fig. 12.The expression of proteins encoded by genes
CD274 and CD47 in the CPTAC database. The protein expression
level results revealed that both (A) CD274 and (B) CD47 proteins showed
significantly lower expression levels in LIHC primary tumors (n = 165) compared
with non-tumor control samples (n = 165). *p-value
Then, we evaluated the co-expression levels of four identified genes VEGFA, SRC, MMP9, and CDK1 with the genes of well-known immune checkpoints that play an important role in immunotherapy, including CD274, PDCD1, CTLA4, and CD47. The results showed VEGFA, CDK1, MMP9, and SRC had statistically significant co-expression levels with genes CD274, PDCD1, and CD47, respectively. VEGFA, CDK1, and MMP9 showed significant co-expression levels with CTLA4. However, there was no significant co-expression level between SRC and CTLA4 (Fig. 13).
 Fig. 13.
Fig. 13.The co-expression levels of four key hub genes VEGFA,
SRC, MMP9, and CDK1 with the immune checkpoint genes
CD274, PDCD1, CTLA4, and CD47. With the
exception of the gene SRC which did not indicate significant correlation
with CTLA4, all other genes VEGFA, MMP9, and
CDK1 showed statistically significant co-expression levels with genes
CD274, PDCD1, CTLA4, and CD47, respectively.
SRC showed a significant co-expression level with genes CD274,
PDCD1, and CD47. p-value
To investigate the potential molecular mechanisms of bilobetin-targeted genes VEGFA, CDK1, MMP9, and SRC, we applied a computer-aided approach to perform the molecular docking between bilobetin and proteins encoded by four key hub genes, respectively. The results demonstrated there was an optimistic binding affinity between bilobetin and proteins VEGFA (–8.8 kcal/mol), CDK1 (–10.0 kcal/mol), MMP9 (–11.0 kcal/mol), and SRC (–10.6 kcal/mol), respectively. The detailed docking information is listed in a table (Table 1).
| Gene | PDB ID | Cavities | Volume | Center_X | Center_Y | Center_Z | Size_X | Size_Y | Size_Z | Score |
| VEGFA | 5T89 | 5 | 54 | –62.038 | 32.762 | 55.268 | 24 | 24 | 24 | –8.8 |
| SRC | 2H8H | 4 | 697 | 17.699 | 25.222 | 57.424 | 24 | 24 | 24 | –10.6 |
| MMP9 | 4H2E | 2 | 563 | –5.979 | 0.783 | 21.686 | 24 | 24 | 24 | –11.0 |
| CDK1 | 5HQ0 | 1 | 1438 | 28.971 | –65.752 | 187.146 | 24 | 24 | 24 | –10.0 |
The formation of hydrogen bonds between bilobetin and proteins VEGFA, CDK1, MMP9, and SRC were tested and displayed with the labeled distances (Fig. 14). In the bilobetin-VEGFA binding complex, bilobetin formed hydrogen bonds with residues GLU-30, LEU-32, GLN-37, and ARG-56 of the VEGFA protein. In bilobetin-SRC binding complex, residues GLY-279, GLY-276, and SER-345 of the SRC protein contributed to the formation of hydrogen bonds. Residues ALA-189, ALA-191, GLU-227, GLN-108, and PHE-107 of the MMP9 protein were involved in the formation of hydrogen bonds in the bilobetin-MMP9 binding complex. Residue TYR-15 of the CDK1 protein formed a hydrogen bond with the bilobetin molecule during the interaction (Fig. 14).
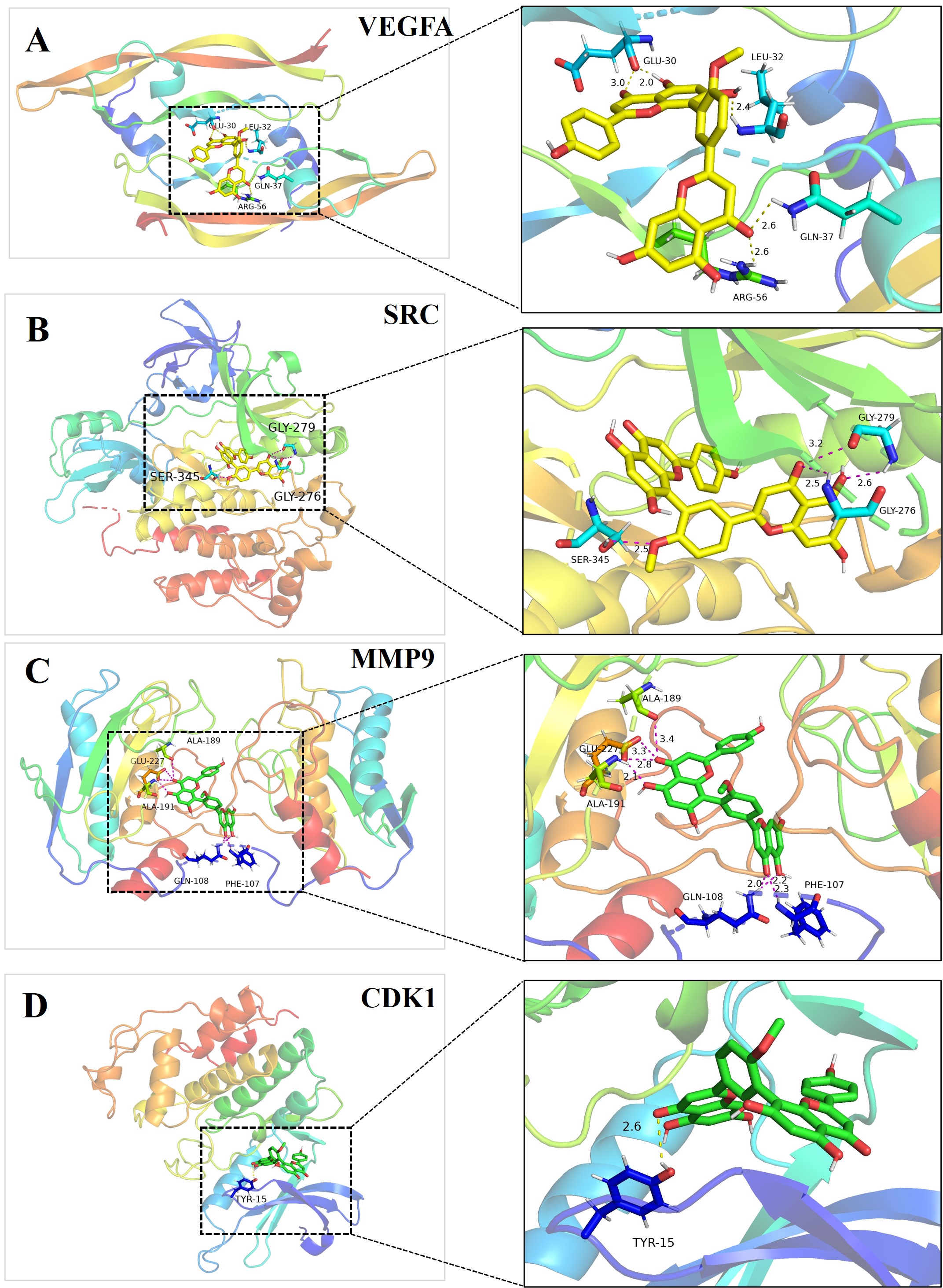 Fig. 14.
Fig. 14.The formation of hydrogen bonds in the binding complex of bilobetin and proteins encoded by target genes. The key residues that contributed to the formation of hydrogen bonds and the distance between bilobetin and residues were measured and labeled. Left: the overall structure of proteins used for molecular docking. Right: the detailed interaction between bilobetin and its target proteins. (A) VEGFA; (B) SRC; (C) MMP9; (D) CDK1.
In addition to the hydrogen bonds, the hydrophobicity interaction between bilobetin and proteins also contributed to the binding affinity and stability of the complexes (Fig. 15).
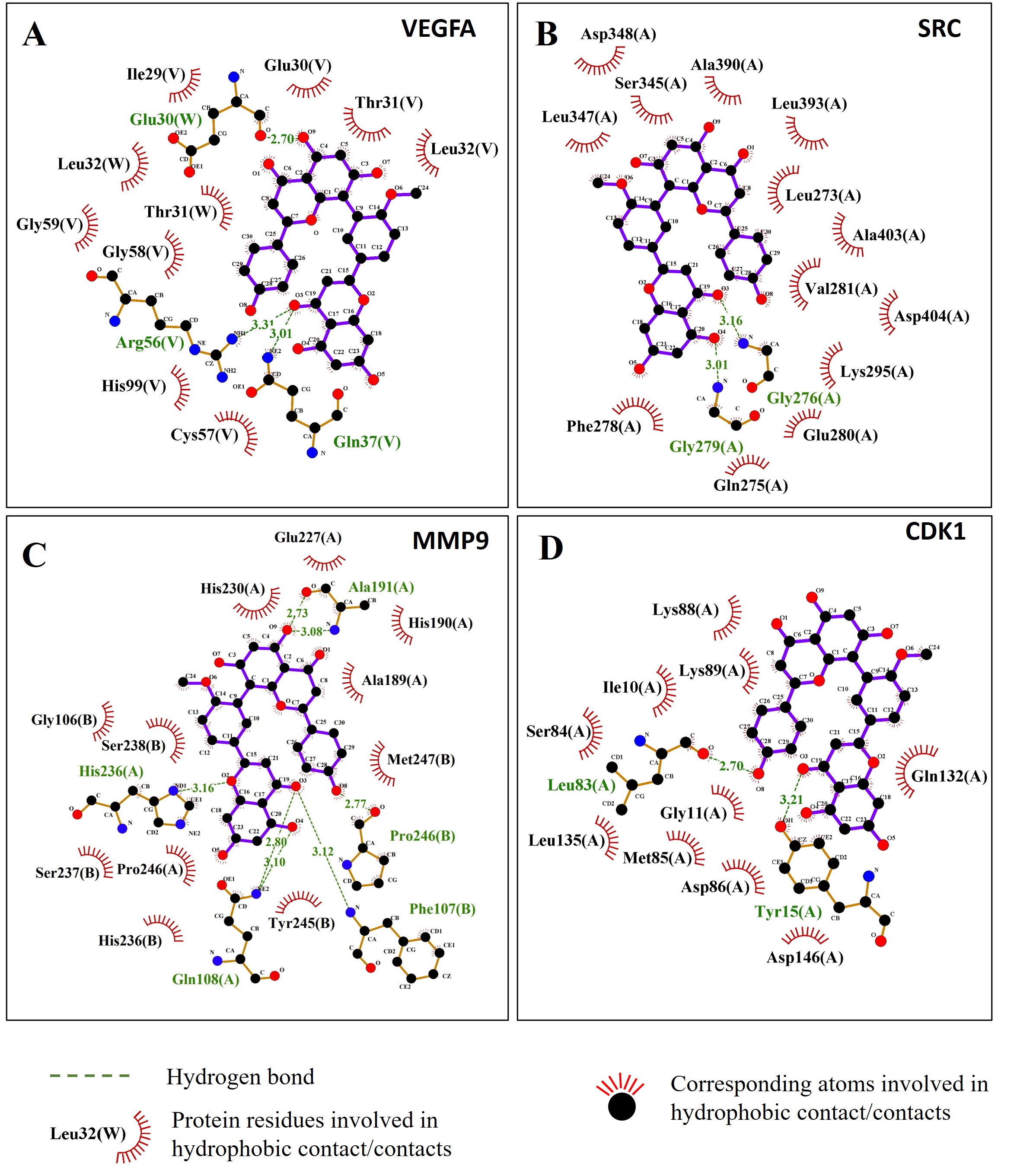 Fig. 15.
Fig. 15.The 2D interaction results showed hydrophobic interactions between bilobetin and four target proteins. (A) VEGFA; (B) SRC; (C) MMP9; (D) CDK1.
In addition, other interactions between bilobetin and target proteins include van der Waals, Pi-sigma, Pi-Pi stack, Pi-Pi t-shaped, Pi-alkyl, Pi-cation, Pi-anion, Pi-sigma, amide-Pi stack, alkyl, and covalent bonds, which contributed to the stability of the complexes (Fig. 16).
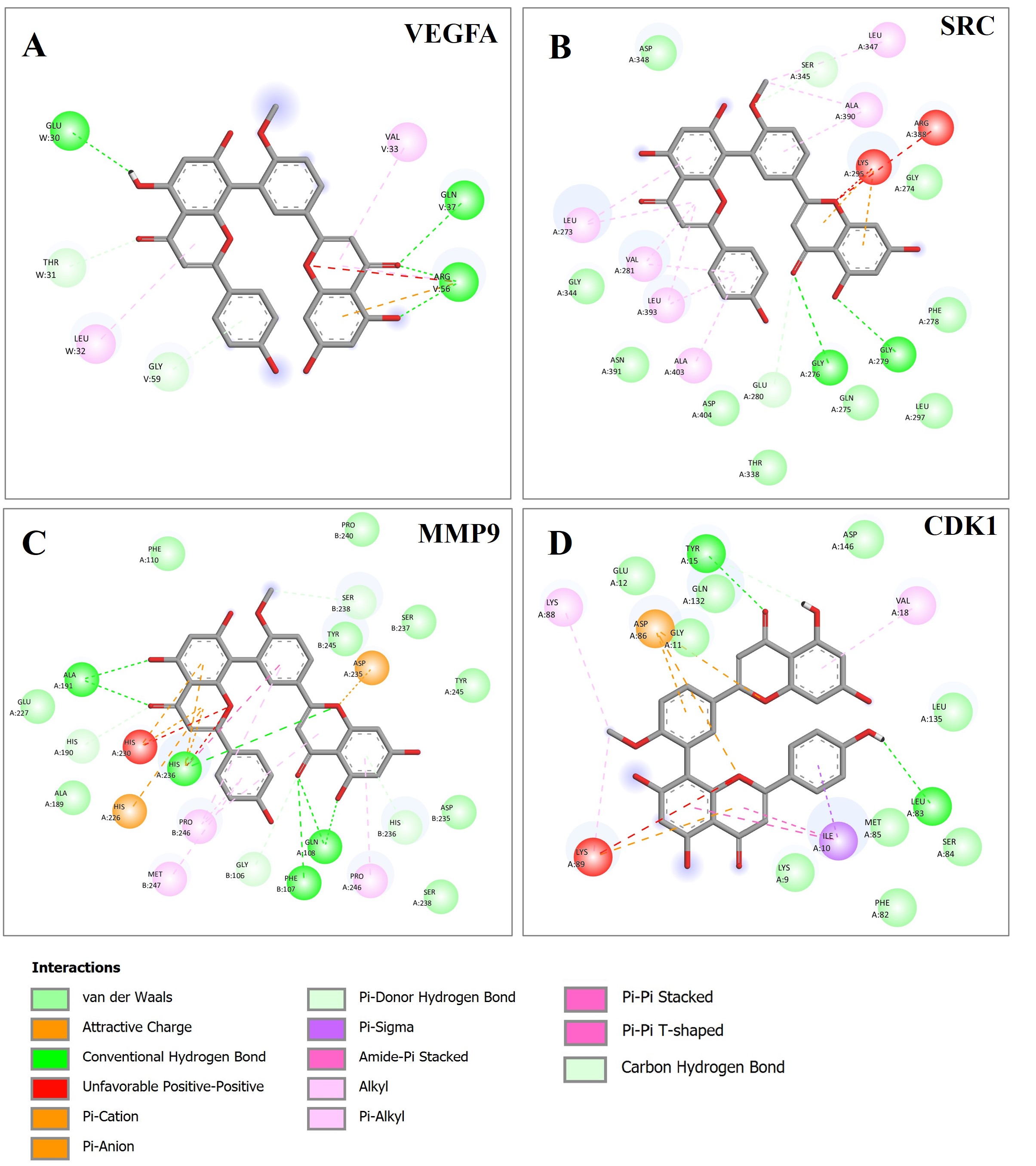 Fig. 16.
Fig. 16.The 2D interaction analysis results of bilobetin and the target proteins. The figure presents all the interactions between bilobetin and its target proteins that contribute to the formation of the protein-ligand complexes. (A) VEGFA; (B) SRC; (C) MMP9; (D) CDK1.
To further evaluate the drug-likeness of bilobetin, we performed ADME analysis using SWISSADME bioinformatic tool, a commonly used tool in drug discovery. The results suggested that the bilobetin meets the requirements of the drug-likeness according to the filter applied by the Lipinski filter (Pfizer) (Table 2).
| Parameters | Physicochemical properties |
| Number of rotatable bonds | 4 |
| Number of H-bond acceptors | 10 |
| Number of H-bond donors | 5 |
| Molar Refractivity | 151.44 |
| TPSA | 170.8 |
| Log Po/w calculation methods and lipophilicity | |
| iLOGP | 3.39 |
| XLOGP3 | 5.36 |
| WLOGP | 5.44 |
| MLOGP | 0.44 |
| Silicos-IT | 5.16 |
| Average value | 3.96 |
| Pharmacokinetics | |
| GI absorption according to the white of boiled egg | Low |
| BBB permeant according to the yolk of boiled egg | No |
| P-gp substrate | No |
| CYP1A2 inhibitor | No |
| CYP2C19 inhibitor | No |
| CYP2C9 inhibitor | Yes |
| CYP2D6 inhibitor | No |
| CYP3A4 inhibitor | No |
| log Kp (skin permeation, cm/s) | –5.86 |
| Medicinal Chemistry | |
| PAINS #alerts | 0 |
| Synthetic Accessibility | 4.35 |
| Druglikeness | |
| Lipinski Filter | Yes |
| Bioavailability Score | 0.55 |
Note: TPSA, topological polar surface area; CYP, cytochrome P450; GI, gastrointestinal; P-gp, P-glycoprotein substrate; PAINS, pan-assay interference structures.
Liver cancer ranks at the top of cancer-associated deaths with no effective therapeutic strategies. Early diagnostic biomarkers and the discovery of effective treatments for LIHC are urgently needed. Bioinformatic analysis empowers the understanding of the pathogenesis of cancers and the discovery of disease-related biomarkers [46, 47]. Computer-aided drug discovery is a favorable strategy for drug development and mechanistic investigation [48]. In this study, we comprehensively integrated these two powerful research approaches to investigate and explore potential therapeutic agents for liver cancer. Our results have demonstrated that bilobetin is a potent therapeutic target for HCC/LIHC via interaction with four key hub targets, including VEGFA, SRC, MMP9, and CDK1, which are associated with immune cell tumor infiltration and co-expression of immune checkpoints.
Among the molecular targets of bilobetin, 16 genes were identified to play
essential roles in the signaling pathway of reactive oxygen species (ROS)
(p = 2.09
VEGFA protein plays a critical role in the growth of tumor-associated blood vessels and vascular permeability, accelerating tumor cell invasion [49, 50, 51]. Studies also found that the circulating levels of VEGFA can serve as a prognostic biomarker for the prediction of survival outcomes of LIHC patients [52, 53]. In addition, VEGFA derived from hepatocytes can promote the development of liver diseases, such as non-alcoholic fatty liver disease which becomes a leading cause of HCC [54]. Currently, the investigation and development of VEGFA inhibitors as therapeutic options against cancers including LIHC are gaining much more attention [55, 56]. In this study, we found VEGFA is one of the bilobetin-targeted key hub genes. Surprisingly, our results have revealed that the mRNA expression levels of VEGFA were significantly lower in LIHC tumors compared with the normal samples. However, when we examined the proteomic data, we found the proteomic level of VEGFA was significantly increased in LIHC tumors compared with normal samples. Consistent with other studies [53, 57, 58], we also found there is a significant association between the expression levels of VEGFA and the clinical survival outcomes of LIHC patients. Therefore, targeting VEGFA using bilobetin as a potential therapeutic strategy for HCC treatment can be further investigated.
SRC plays a significant role in cancer development [59, 60, 61, 62]. In our study, we have demonstrated that SRC is one of the identified bilobetin-targeted hub genes. The mRNA expression levels of SRC were significantly different from LIHC tumor samples compared with normal samples, which were significantly associated with survival outcomes of LIHC patients. The expression levels of SRC also showed a significant association with tumor-related immune cell infiltration and the expression of immune checkpoints. However, the proteomic expression levels of SRC do not show statistical significance between normal and tumor samples, which requires further investigation. The phosphorylation of SRC proteins also plays a pivotal role in liver disease [63]. Therefore, the mRNA expression levels and phosphorylated status of SRC proteins are required to evaluate their role in LIHC.
Consistent with our previous analysis results (unpublished), MMP9 was identified as an important gene in LIHC, as there is a significant association between its mRNA and protein expression levels with the survival outcomes of LIHC patients, as well as tumor-infiltrating levels of immune cells. In this study, we have demonstrated that the bioactive compound bilobetin has the best binding affinity (–11.0 kcal/mol) with MMP9 compared with VEGFA (–8.8 kcal/mol), SRC (–10.6 kcal/mol), and CDK1 (–10.0 kcal/mol). Therefore, our study provides an optimistic insight for further evaluating bilobetin as a potential therapeutic agent for LIHC treatment by regulating the expression of MMP9, which plays a key role in cancer development [64, 65].
CDK1 is another key hub gene identified from 20 bilobetin-targeted hub genes. In line with other research findings [66, 67, 68], our results also revealed that both the mRNA and proteomic levels of gene CDK1 showed significantly higher expression levels in LIHC tumors compared with normal samples. In addition, the expression of CDK1 was significantly associated with the survival outcome of LIHC patients and tumor-associated immune cell infiltration, as well as the expression levels of immune checkpoint-related biomarkers. The molecular docking results also showed that bilobetin can be used as a compound template for designing new drugs for LIHC treatment.
Drug development is a long process, and comprehensive or integrated research approaches such as bioinformatic analysis and computer-aided methods could bring some advantages to promote the process. Overall, our study shows that bilobetin can target multiple targets that influence liver cancer development and progression, which holds great value in the improvement of treatment efficacy and reduction in the side effect.
In this study, we explored the potential of bilobetin as a LIHC therapeutic agent based on comprehensive bioinformatic analysis and computer-aided drug discovery approaches. Although one of the most important drug discovery processes, further steps such as modification, optimization, examination of toxicity or side effects, and in vitro and in vivo experiments are necessary and should be carefully evaluated. The functionality, delivery method, and treatment efficacy should also be further studied.
The data involved in this study were obtained from publicly available online database. The online sites include the following: Swiss Target Prediction: http://swisstargetprediction.ch/;GEPIA: http://gepia.cancer-pku.cn/; TCGA: https://portal.gdc.cancer.gov/; TIMER: http://timer.cistrome.org/; UALCAN: https://ualcan.path.uab.edu/; Human Protein Atlas: https://www.proteinatlas.org; PDB: https://www.rcsb.org/.
CZ and MY designed the research study. CZ, YS, SL, and MY performed the research. CZ, YS, SL, and MY analyzed the data. CZ, YS, SL, and MY wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
The authors acknowledge the use of all available public databases and software for data analysis presented in this study.
This research received no external funding.
Given his role as Guest Editor, Ming Yang had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Jian Zhang and Ricardo Jorge Pinto Araujo. The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.