1. Introduction
Asthma is a chronic inflammatory disease of the respiratory system that causes
repeated bronchoconstriction and airflow limitation [1]. The CD4 T helper
(Th) cells contribute significantly in the pathogenesis of asthma [2].
Clinically, asthma was generally divided into two categories: allergic asthma and
non-allergic asthma [3]. In recent years, with the in-depth research on the
pathogenesis of severe asthma and refractory asthma. It has been found that
neutrophilic inflammation in the airways and airway remodeling are essential
factors contributing to the occurrence of severe asthma and refractory asthma [4, 5]. IL-17, mainly produced by Th17 cells, stimulate the neutrophilic airway
inflammation [6] and airway hyperresponsiveness that induces the pathogenesis of
asthma [7, 8, 9]. Given the central role of IL-17 in the pathogenesis of asthma, the
exploration of the factors regulating Th17 cell polarization and affecting the
secretion of IL-17 by Th17 cells is crucial.
Dipeptidyl Peptidase-4 (DPP4) is a soluble glycoprotein with serine protease
activity, expressed as CD26 on the cell surface in immune cells [1]. In asthma,
CD26 is an activation marker, up-regulatedin lymphocytes, particulary in the
CD4 T cells [2]. Studies have confirmed that the expression of CD26 in
lymphocytes of asthmatic patients was significantly increased [10]. Furhermore,
the high expression of CD26 was associated by the differentiation of T
lymphocytes into Th1 and Th17 [11, 12]. Further research is needed to investigate
the potential mediation of airway neutrophil inflammation by DPP4 in asthma, by
affecting Th17/IL-17 signaling.
Airway remodeling is a common pathological feature of severe asthma, resulting
in permanent airway obstruction in up to 50% of cases and respiratory
dysfunction [13]. Airway epithelial mesenchymal transition (EMT) is a vital
mechanism of airway remodeling in asthmatic patients [14]. Previous studies have
shown that IL-17 enhanced TGF-1-induced EMT in bronchial epithelial
cells (BECs) [15]. In addition, DPP4 could promote the EMT of BECs induced by
TGF-1 and the IL-17 had a synergistic effect on TGF-1-induced
EMT [16]. Based on the latter, the central hypothesis in this paper was that DPP4
could be related to airway inflammation and airway remodeling in asthma by
promoting TGF-1-induced airway EMT and modulating the Th17/IL-17 axis.
Therefore, the effect of DPP4 on Th17 cell polarization initially investigated
in vitro. Subsequently, the TGF-1-induced bronchial epithelial
cells (BECs) model and Ovalbumin (OVA)-induced mouse asthma model were established to assess
the effect of DPP4 on airway EMT and remodeling in asthma.
2. Materials and Methods
2.1 Experimental Animal Parameters
Forty-five female C57BL/6J mice aged 6–8 weeks were obtained from Hunan SJA
Laboratory Animal CO., LTD (Changsha, China). All mice were acclimated for one
week, followed by subsequent experiments. The breeding conditions for mice were
as follows: temperature and humidity ranges of 20~26 °C and
40~70% (respectively), and light cycle of 12/12 h of
light/darkness. All mice had sustained access to food and water. Five female
C57BL/6J mice were anesthetized by intraperitoneal injection of overdose 2%
phenobarbital to obtain spleen and bronchus for isolation of CD4 T cells
and bronchial epithelial cells (BECs). Th17 cells were induced by 2 µg/mL
anti-CD3 (100340, Biolegend, San Diego, CA, USA), anti-CD28 (102116, Biolegend,
USA), 10 µg/mL anti-IFN- (505701, Biolegend, USA), 10 µg/mL
anti-IL-4 (504102, Biolegend, USA), 5 ng/mL TGF-1 (763102, Biolegend,
USA), 20 ng/mL IL-6 (216-16, Peprotech, Rocky Hill, NJ, USA) and 50 ng/mL IL-23
(589002, Biolegend, USA) from CD4 T cells. The CD4 T cells and Th17
cells were identified using flow cytometry in Supplementary Fig. 1A. The
BECs were identified by immunocytochemistry in Supplementary Fig. 1B.
This experimental program presented in this paper was approved by the
Institutional Animal Care and Use Committee of Guilin Medical University (NO.
201903190).
2.2 Culture and Differentiation of CD4 T Cells
Female C57BL/6J mice (5 mice) were anesthetized by intraperitoneal injection of
overdose 2% phenobarbital to obtain spleen. CD4 T lymphocytes were
purified from the spleens of mice using a CD4 T cell isolation kit
(130-104-454, Miltenyi biotec, Bergisch Gladbach, Germany) according to the
manufacturer’s instructions. The isolated CD4 T cells were cultured in
RPMI-1640 containing 10% FBS, 2 mM L-glutamine, 5 µg/mL concanavalin A,
100 U/mL penicillin and 100 µg/mL streptomycin. Subsequently, the CD4
T cells were split into the following six groups: control group, Th17 group, 10,
50, and 100 ng/mL DPP4 group, and the DPP4 inhibitor group. Cells in control
group were treated with 2 µg/mL anti-CD3 (100340, Biolegend, USA) and
anti-CD28 (102116, Biolegend, USA). Th17 group: based on the control group, 10
µg/mL Anti-IFN- (505701, Biolegend, USA), 10 µg/mL
anti-IL-4 (504102, Biolegend, USA), 5 ng/mL TGF-1 (763102, Biolegend,
USA), 20 ng/mL IL-6 (216-16, Peprotech, USA) and 50 ng/mL IL-23 (589002,
Biolegend, USA) was added. 10, 50, and 100 ng/mL DPP4 groups: based on the Th17
group; 10, 50, 100 ng/mL of reconstituted sCD26/sDPP4 (HG-PT010074, HonorGene,
Changsha, China) were added respectively. The DPP4 inhibitor group: based on the
50 ng/mL DPP4 group, cells were initially treated with 10 µM DPP4 inhibitor
(B3941, APEXBIO, USA) for 30 min. After 5 days of culture, cells and cell
supernatants were collected for subsequent tests.
2.3 Isolation and Characterization of Mouse BECs
Female C57BL/6J mice (5 mice) were anesthetized by intraperitoneal injection of
overdose 2% phenobarbital, then the bronchus was extracted. A moderate amount of
0.05% pronase pre-cooled in 4 °C was injected into the bronchi to
cleanse the inner wall of the trachea. The entire trachea was filled with
digestive enzymes and infiltrated in DMEM/F12 after being ligated. After
incubating at 4 °C for overnight, the digestion was collected. The red
blood cells were lysed by red blood cell lysis buffer (R1010, Solarbio, Beijing,
China). BECs were obtained by removing the adherent fibroblasts after 1 h of
incubation in complete medium.
2.4 EMT Induction of BECs
BECs were divided into the following four groups: Normal group (no
intervention), TGF-1 group (10 ng/mL TGF-1), DPP4 group and the
DPP4 inhibitor group. DPP4 group: 50 ng/mL of reconstituted sCD26/sDPP4
(HG-PT010074, HonorGene, China) were added to the TGF-1 group. The DPP4
inhibitor group, based on the DPP4 group, cells were first treated with 10
µM DPP4 inhibitor (B3941, APEXBIO, Houton, TX, USA) for 30 min. After the
cells were cultured for 72 h, the cell morphology changes were evaluated by a
microscope (BA210T, Motic, Xiamen, China).
2.5 Co-Culture of Mouse BECs and Th17 Cells
BECs and Th17 cells were co-cultured at a ratio of 1:1, 1:5, and 1:10 in a
complete medium with 10 ng/mL TGF-1 (763102, Biolegend, USA). Cell
morphology evolutions were assessed usinga microscope (BA210T, Motic, China). The
co-culture experiments’ results were used for the selection ofthe optimal ratio of
BECs and Th17 for subsequent experiments. BEC cells were divided into the
following three groups: Th17 + BECgroup, Th17+ DPP4 group, and Th17 + DPP4
inhibitor group. Th17 + BEC group: BECs and Th17 cells were co-cultured at the
optimal ratio in a complete medium with 10 ng/mL TGF-1. Th17 + DPP4
group: Based on the Th17 + BEC group, 50 ng/mL reconstituted sCD26/sDPP4
(HG-PT010074, HonorGene, China) was added. Th17 + DPP4 inhibitor group: Based on
the Th17 + DPP4 group, BECs and Th17 cells were first treated with 10 µM
DPP4 inhibitor (B3941, APEXBIO, USA) for 30 min.Cells and cell supernatants were
collected for subsequent detection after being cultured for 72 h.
2.6 Experimental Grouping and EStablishment of a Mouse Asthma Model
Forty female C57BL/6J mice were divided into four groups: control group, OVA
group, OVA + DPP4 group, and OVA + DPP4 inhibitor group. No-loaded overexpressing
lentivirus (oe-NC) and DPP4 overexpressing lentivirus (oe-DPP4, NM_010074,
HG-LV010074, HonorGene, China) were purchased from HonorGene. The mice in control
group were treated with normal saline and oe-NC. The mice in OVA group were
intraperitoneal-injected with lentivirus 30 min before OVA (A5503,
Sigma-Aldrich, Darmstadt, Germany) ultrasonic nebulization. Moreover, the mice in
OVA + DPP4 group were intraperitoneal-injected of oe-DPP4 30 min before OVA
ultrasonic nebulization. The mice in OVA+DPP4 inhibitor group were intraperitoneal
injected with 200 mg/kg DPP4 inhibitor (B3941, APEXBIO, USA) 30 min before OVA
ultrasonic nebulization. On days 0 and 12, mice were sensitized by
intraperitoneal injection of 0.2 mL of aluminum hydroxide gel, containing 10
µg of OVA. Mice in the control group received the same volume of normal
saline. Subsequently, from day 18 to day 23, all groups of mice received 5% OVA
for 30 min daily, through the airway. Afterwards, mice were exposed to 5% OVA
once every two days for 30 min until operated on day 56.
2.7 Bronchoalveolar Lavage
The airways of the mice were lavaged three times with 0.4 mL of PBS by tracheal
intubation. Subsequently, the bronchoalveolar lavage fluid (BALF) was centrifuged
at 2000 g for 5 min (4 °C) and the supernatant was collected for
subsequent experiments. The pellets were resuspended in 50 µL pre-cold PBS
and the cells were calculated by a hemocytometer.
2.8 Flow Cytometry
The CD4 T cells and lymphocytes were collected for flow cytometry assay.
Cells were fixed and permeabilized by Fixation/Permeabilization concentrate
(00-5123-43, eBiosciences, San Diego, California, USA). For Th17 cell detection,
cells were stimulated using Cell Stimulation Cocktail (00-4975-93, eBiosciences,
USA) for 4 h before fixation and permeabilization. Subsequently, cells were
labeled with FITC-conjugated CD4 antibody (11-0041-82, eBiosciences, USA) and
PE-conjugated IL-17A antibody (12-7179-42, eBiosciences, USA) or FITC-conjugated
CD4 antibody (11-0041-82, eBiosciences, USA), PE-conjugated CD25 antibody
(12-0250-42, eBiosciences, USA) and APC-conjugated Foxp3 antibody (17-5773-82,
eBiosciences, USA). Finally, the staining cells were analysed by flow cytometry
(A00-1-1102, Beckman, CA, USA).
2.9 Western Blotting (WB)
Total proteins from cells or lung tissues were extracted using RIPA lysis buffer
(AWB0136, abiowell, Changsha, China) containing protease inhibitor (583794,
Jintai Hongda, Beijing, China) and quantified using a BCA kit. Equal protein was
separated by 10% SDS-PAGE gel, and transferred to a PVDF membrane (Invitrogen,
Carlsbad, CA, USA). The membranes were incubated with a primary antibody at 4
°C overnight, including E-cadherin (1:5000, rabbit, 20874-1-AP,
Proteintech, USA), -SMA (1:5000, rabbit, 55135-1-AP, Proteintech, USA),
-actin (1:5000, mouse, 66009-1-Ig, Proteintech, USA). Then, the
membranes were incubated with a secondary HRP goat anti-rabbit IgG (1:6000,
SA00001-2, Proteintech, USA) antibody or HRP goat anti-mouse IgG (1:5000,
SA00001-1, Proteintech, USA) antibody at room temperature for 2 h. The images
were collected using a ChemiScope6100 (Clinx, Shanghai, China) and the gray
values of the protein bands were evaluated using a Bio-Rad Quantity One v4.62
(Bio-Rad, San Francisco, CA, USA).
2.10 Quantitative Real-Time PCR (qRT-PCR)
TRIzol reagent (15596026, Thermo Fisher Scientific, Waltham, MA, USA) was
utilised to extract the total RNA from cells and lung tissues. RNA samples were
subsequently applied to generate the cDNA by mRNA Reverse Transcription Kit
(CW2569, CWBIO, Beijing, China). The expression of specific RNAs was quantified
by UltraSYBR Mixture (CW2601, CWBIO, Beijing, China) in a QuantStudio1 Real-Time
PCR System (ABI, Fosters, CA, USA). -actin was used as reference gene
and the primer sequences are listed in Table 1.
Table 1.Primer sequences.
| Gene |
Sequence |
Length (bp) |
| E-cadherin |
F AGCCATTGCCAAGTACATCCTC |
155 bp |
| R CGCCTTCTGCAACGAATCCC |
| -SMA |
F GCCCCTGAAGAGCATCCGAC |
179 bp |
| R CCAGAGTCCAGCACAATACCAGT |
| -actin |
F ACATCCGTAAAGACCTCTATGCC |
223 bp |
| R TACTCCTGCTTGCTGATCCAC |
F, Forward Primer; R, Reverse Primer.
2.11 Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of IL-17 (CSB-E04608m) in the cell supernatant and DPP4
(CSB-E08520m), IL-17 (CSB-E04608m), IL-4 (CSB-E04634m), IL-5 (CSB-E04637m), IL-13
(CSB-E04602m), TGF-1 (CSB-E04726m), MMP9 (CSB-E08007m) in BALF were
detected by ELISA kit (CUSABIO, Wuhan, China).
2.12 Histological Staining
Lung tissues were fixed with 4% paraformaldehyde, embedded in paraffin, and cut
into sections. Sections were stained with hematoxylin-eosin (H&E), periodic acid
Schiff (PAS) and Masson to evaluate the inflammation, epithelial injury, and
degree of collagen deposition, respectively. Images were collected using a
microscope (BA210T, Motic, Xiamen, China).
2.13 Immunocytochemistry (ICC)
BECs slides were prepared and fixed with 4% paraformaldehyde. After quenching
with 3% HO, slides were incubated with KRT8 (17514-1-AP,
Proteintech, Chicago, IL, USA) antibody overnight at 4 °C, followed by
an incubation with anti-rabbit-IgG antibody-HRP polymer (7074P2, Cell Signaling
Technology, Boston, MA, USA) for 30 min at 37 °C. The immunoreactivity
was observed with diaminobenzidine (DAB). Yellow or tan stain was considered
positive. Images were collected using a microscope (BA210T, Motic, China) and
analysed using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
2.14 Immunofluorescence (IF)
Co-staining was conducted on lung tissue sections attached on BECs slides.
Antigen retrieval of tissue sections was performed in an electromagnetic oven
using EDTA (pH 9.0) in boiling water for 24 min. Slides and sections were
eventually incubated overnight at 4 °C with primary antibodies against
E-cadherin (20874-1-AP, PTG, USA) and -SMA (66516-1-Ig, PTG, USA).
Subsequently, CoraLite488–conjugated Affinipure Goat Anti-Rabbit IgG (H+L)
(SA00013-2, Proteintech, USA) or CoraLite488–conjugated Affinipure Goat
Anti-Mouse IgG (H+L) (SA00013-1, Proteintech, USA) were incubated with slides and
sections for 90 min at 37 °C. Nuclei were stained with DAPI for 10 min
at 37 °C. Images were collected using a microscope (BA210T, Motic).
2.15 Statistical Analysis
The data was analyzed using GraphPad Prism 8.0 (GraphPad, San Diego, CA, USA).
The results from the data presented were the form of mean standard
deviation. The normality and homogeneity of variance were tested to confirm the
normality of the data distribution and homogeneity of variance. Statistical
differences were assessed by unpaired two-tailed Student’s t test
between two groups. One-way analysis of variance (ANOVA) was applied to
statistical differences among multiple groups. Tukey’s test was used for pairwise
comparison after one-way ANOVA. Statistical significance was defined at
p 0.05.
3. Results
3.1 sCD26/sDPP4 Promoted Th17 Cell Polarization and IL-17 Secretion
CD4 T cells were successfully isolated and (Supplementary Fig.
1A), treated with sCD26/sDPP4 to explore Th17 cells polarization and IL-17
secretion. Flow cytometry detection of Th17 cells revealed that 10–100 ng/mL
sCD26/sDPP4 significantly promoted the Th17 polarization. The higher
concentrations of sCD26/sDPP4 were accompanied by a higher degree of polarization
of Th17 cells. After the addition of the DPP4 inhibitor, the effect of
sCD26/sDPP4 was inhibited (Fig. 1A,B). In addition, the change trend of IL-17
level in cell supernatant was consistent with the Th17 cells (Fig. 1C).
Therefore, sCD26/sDPP4 could promote Th17 cell polarization and IL-17 secretion
in a dose-dependent manner.
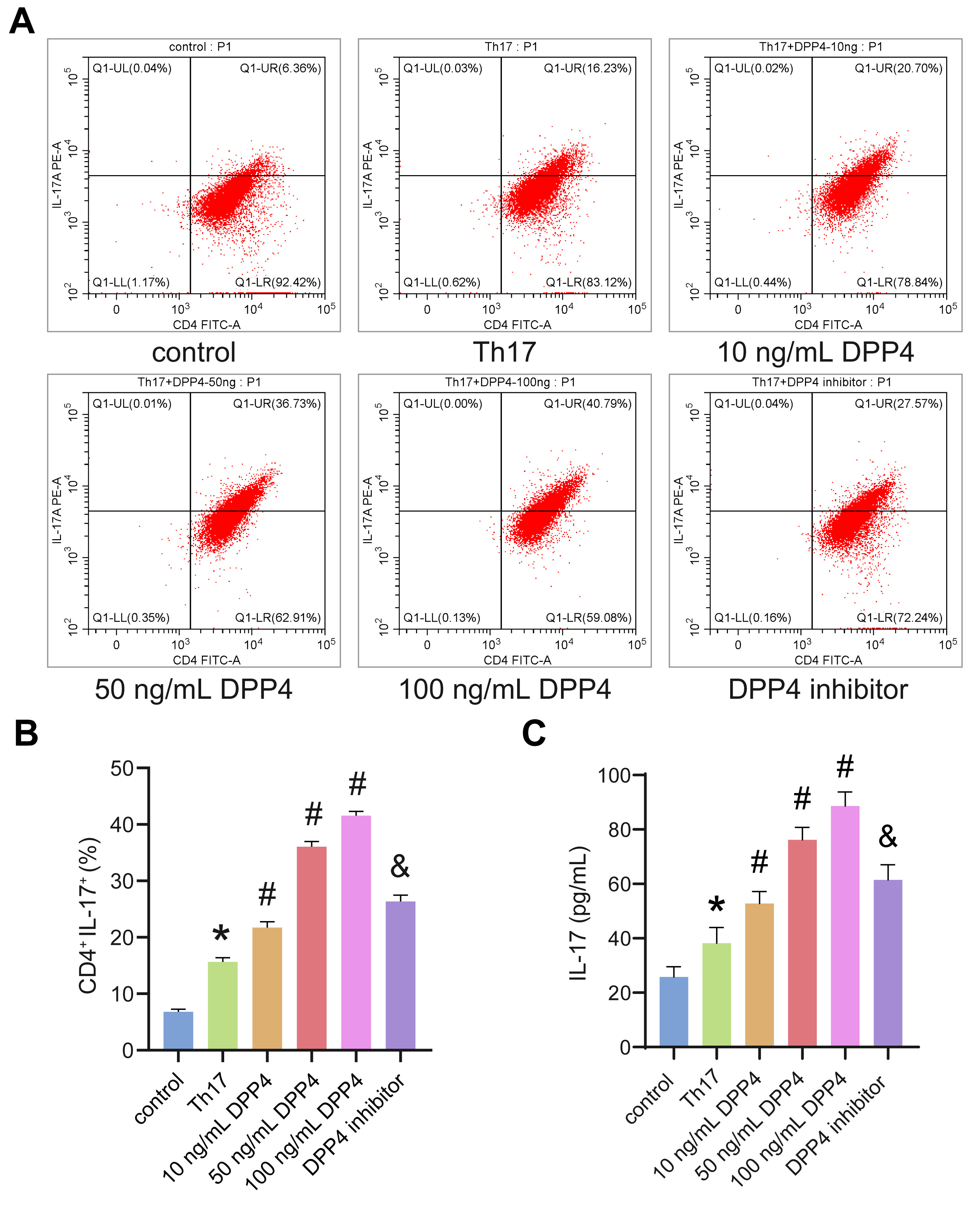 Fig. 1.
Fig. 1.
sCD26/sDPP4 promoted Th17 polarization and the secretion of
IL-17. CD4 T cells were induced differentiation into Th17 cells and
treated with sCD26/sDPP4 or dipeptidyl peptidase-4 (DPP4) inhibitor. (A,B) The
ratio of Th17 cells was detected by flow cytometry. (C) The level of IL-17 in the
cell supernatant was analyzed by enzyme-linked immunosorbent assay (ELISA). Data
was showed as the mean SD, *p 0.05 vs control, #p
0.05 vs Th17 group, &p 0.05 vs 50 ng/mL DPP4 group.
3.2 sCD26/sDPP4 Promoted EMT in TGF-1-Induced BECs
The influence of sCD26/sDPP4 on EMT in TGF-1-induced BECs was further
analysed. BECs were identified by immunocytochemical staining and the expression
of KRT8 was positive (Supplementary Fig. 1B). As shown in Fig. 2A, after
TGF-1 treatment, cell-to-cell contact was reduced, and cells became
spindle-forming fibroblasts. IF (Fig. 2B,C, Supplementary Fig.
1C,D) and WB (Fig. 2F–H) were used to evaluate the expression of E-cadherin and
-SMA in BECs. Compared with the normal group, the protein level of
E-cadherin decreased in the TGF-1 group, while the protein level of -SMA increased. sCD26/sDPP4 enhanced
TGF-1-induced down-regulation of E-cadherin and up-regulation of
-SMA, while DPP4 inhibitors alleviated the effects of sCD26/sDPP4. In
addition, the mRNA level of E-cadherin decreased in the TGF-1 group and
a further decrease following sCD26/sDPP4 treatment, while the level of
-SMA was reversed. DPP4 inhibitor reversed the effect of sCD26/sDPP4 as
expected (Fig. 2D,E). The above results indicated that sCD26/sDPP4 promoted
TGF-1-induced EMT in BECs.
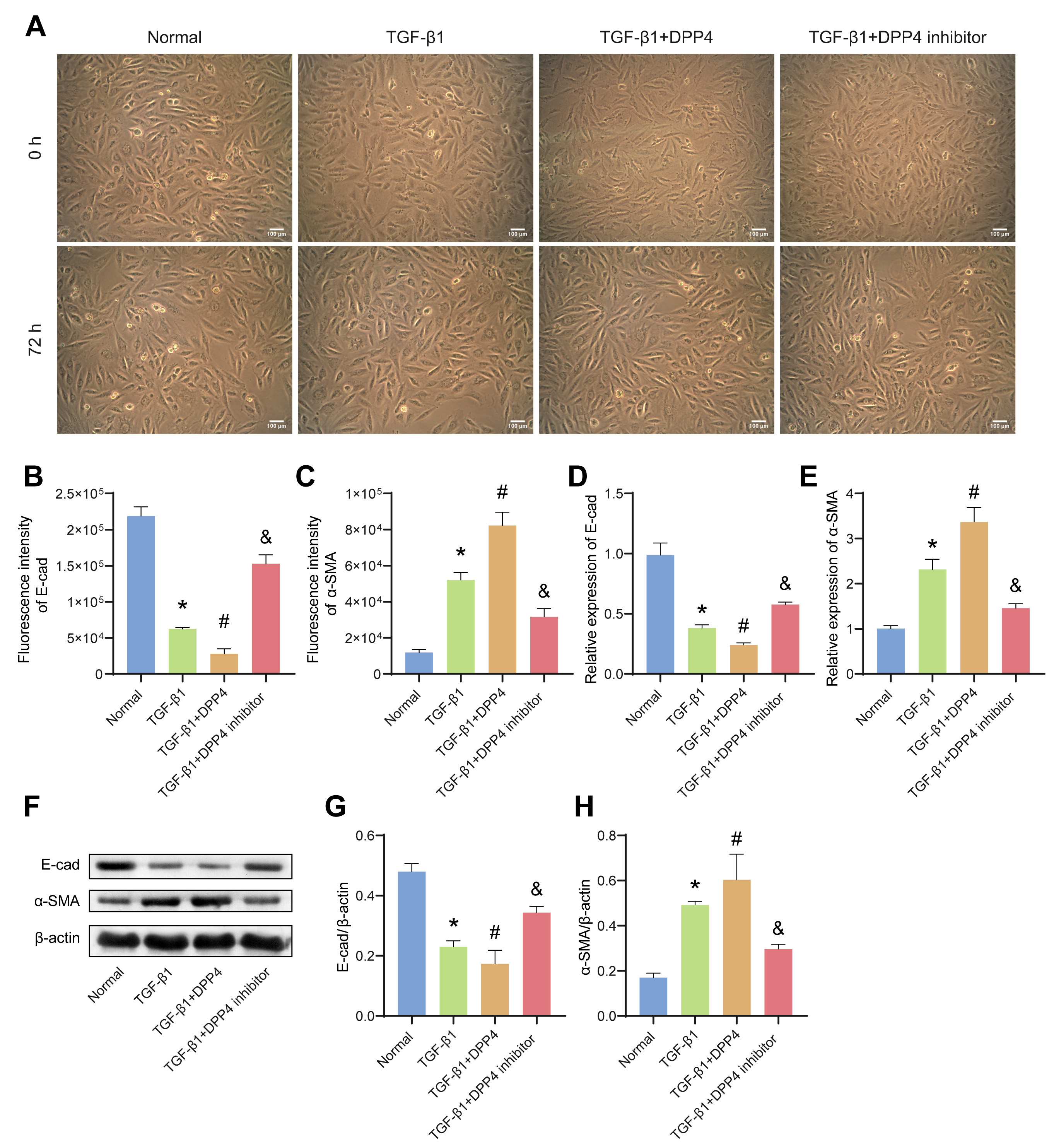 Fig. 2.
Fig. 2.
sCD26/sDPP4 promoted EMT in TGF-1-induced
bronchial epithelial cells (BECs). The BECs were treated with TGF-1,
sCD26/sDPP4 or DPP4 inhibitor. (A) The cell morphology changes of BECs were
observed by microscope. (B,C) The fluorescence intensity of E-cad and
-SMA in BECs. (D,E) The mRNA expression of E-cad and -SMA in
BECs were evaluated by qRT-PCR. (F–H) The protein levels of E-cad and
-SMA in BECs were assessed by western blotting (WB). Data was showed as
the mean SD, *p 0.05 vs Normal, #p 0.05 vs
TGF-1 group, &p 0.05 vs TGF-1+ DPP4 group. E-cad,
E-cadherin; EMT, epithelial mesenchymal transition.
3.3 sCD26/sDPP4 Regulated EMT in Mouse BECs by Modulating the
Th17/IL-17 Axis
In order to explore the interaction between BECs and Th17 cells, the latter were
successfully induced from CD4 T cells
(Supplementary Fig. 1A). BECs were
co-cultured with Th17 at the ratio of 1:1, 1:5, 1:10 for 24 h, 48 h, and 72 h.
The EMT degree of BECs was gradually deepened by the increase of the cell ratio
and the prolongation of culture time (Fig. 3A). Moreover,the level of IL-17 in
the cell supernatant was also gradually increased (Fig. 3B). Therefore, the BECs
and Th17 were co-culture at the ratio of 1:10 for 72 h for subsequent
experiments. The protein levels of E-cadherin and -SMA in BECs were
detected by IF (Fig. 3C,D, Supplementary Fig. 2A,B) and WB (Fig. 3G–I).
Compared with the Th17+BEC group, the protein level of E-cadherin was
down-regulated and protein level of -SMA was up-regulated in the
Th17+DPP4 group. However, the addition of the DPP4 inhibitor could reverse this
process. The mRNA expression of E-cadherin and -SMA was accordant with
the protein expression (Fig. 3E,F). In addition, compared with the Th17+BEC
group, the level of IL-17 in the Th17+DPP4 group was significantly increased
(Fig. 3J), suggesting that sCD26/sDPP4 could promote the secretion of IL-17. The
increase of IL-17 level was accompanied by the deepening of EMT in BEC cells,
indicating that sCD26/sDPP4 and Th17 cells had a synergistic effect on the
formation of EMT. The combination of the above results suggest that sCD26/sDPP4
promoted EMT in BECs by modulating the Th17/IL-17 axis.
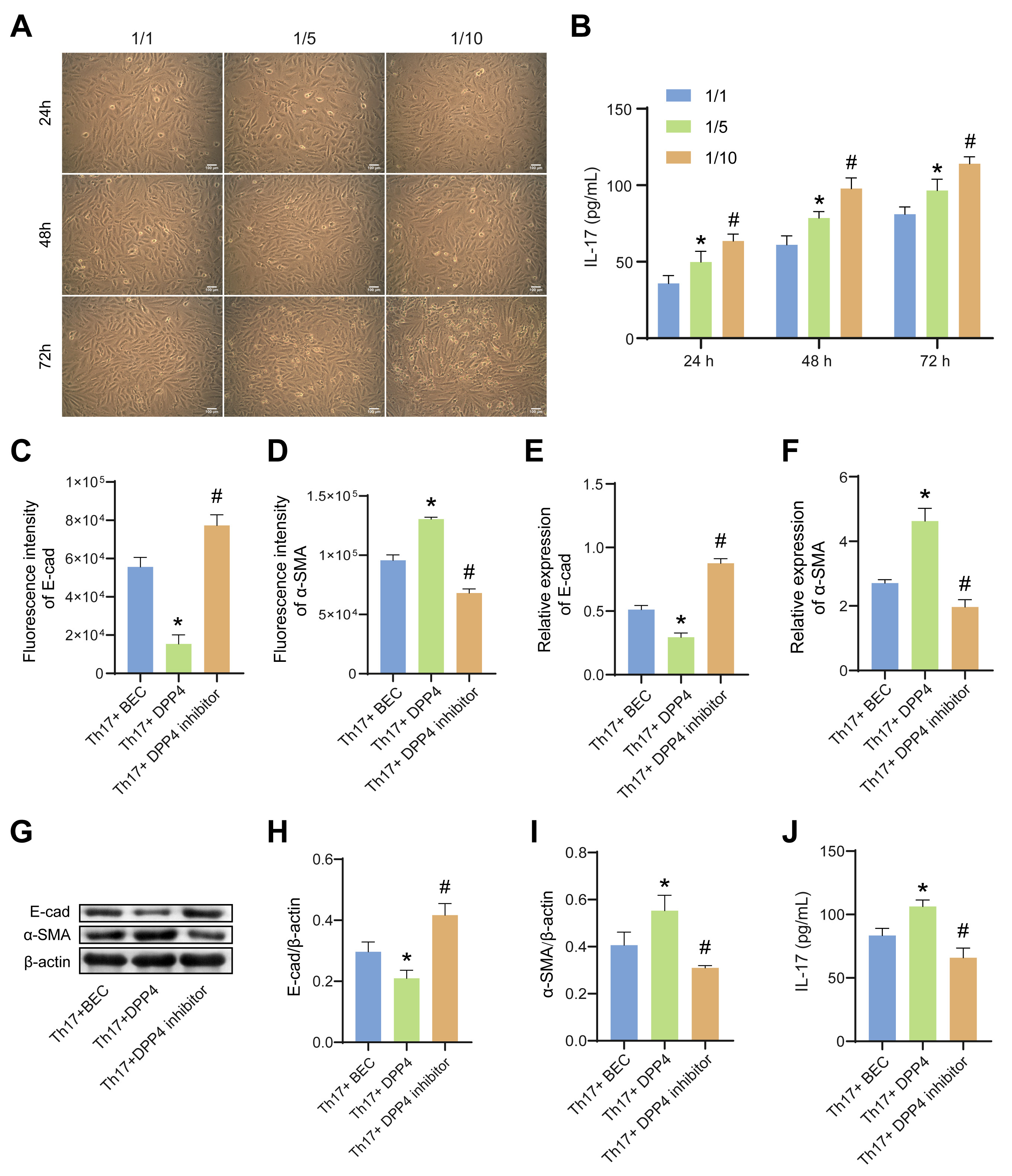 Fig. 3.
Fig. 3.
sCD26/sDPP4 promoted EMT in BECs by modulating the Th17/IL-17
axis. In (A,B), the BECs were co-cultured with Th17 at the different ratio,
while in (C–J), the BECs were co-cultured with Th17 at the ratio of 1:10 and
treated with sCD26/sDPP4 or DPP4 inhibitor. (A) Microscope estimation of the cell
morphology changes of BECs. (B) The IL-17 level in the cell supernatant of BECs
co-cultured with Th17 was analyzed by ELISA. (C,D) The fluorescence intensity of
E-cad and -SMA in BECs. (E,F) The mRNA expression of E-cad and
-SMA in BECs was assessed by qRT-PCR. (G–I) The protein levels of
E-cad and -SMA in BECs were measured by WB. (J) The level of IL-17 in
the cell supernatant of BECs co-cultured with Th17 at the ratio of 1:10 and
treated with sCD26/sDPP4 or DPP4 inhibitor was analyzed by ELISA. Data was showed
as the mean SD, in Fig. 3B, *p 0.05 vs 1/1, #p 0.05 vs 1/5; and in Fig. 3C–J, *p 0.05 vs Th17+BEC group,
#p 0.05 vs Th17+DPP4 group. E-cad, E-cadherin.
3.4 Overexpression of DPP4 Promoted Airway Inflammation in
OVA-Induced Asthmatic Mice
The role of DPP4 in OVA-induced asthmatic mice was further investigated. An
OVA-induced asthma mouse model was established to evaluate the effects of DPP4 on
asthma. Compared with the control group, the concentration of DPP4 in the BALF of
asthmatic mice increased, and the total number of leukocytes decreased (Fig. 4A,B). After oe-DPP4 treatment, the concentration of DPP4 in the BALF of
asthmatic mice was further increased, while the total number of leukocytes
decreased (Fig. 4A,B). DPP4 inhibitors has the opposite effect in OVA-induced
asthma mouse (Fig. 4A,B). HE staining indicated that oe-DPP4 could significantly
promote OVA-induced BECs shedding and inflammatory cell infiltration in
lungs-bronchi (Fig. 4C). Similarly, PAS staining demonstrated that oe-DPP4
enhanced OVA-mediated bronchial goblet cells proliferated and increased mucus
secretion in lungs-bronchi (Fig. 4D). In addition, compared with the OVA group,
the levels of IL-4, IL-5, and IL-13 in the BALF of mice in the DPP4 group were
distinctly increased (Fig. 4E–G). DPP4 inhibitor could alleviate OVA-induced
bronchial inflammation and reduce the levels of IL-4, IL-5, and IL-13 in the BALF
of asthmatic mice (Fig. 4C–G). These results confirmed that oe-DPP4 promoted
airway inflammation in asthmatic mice.
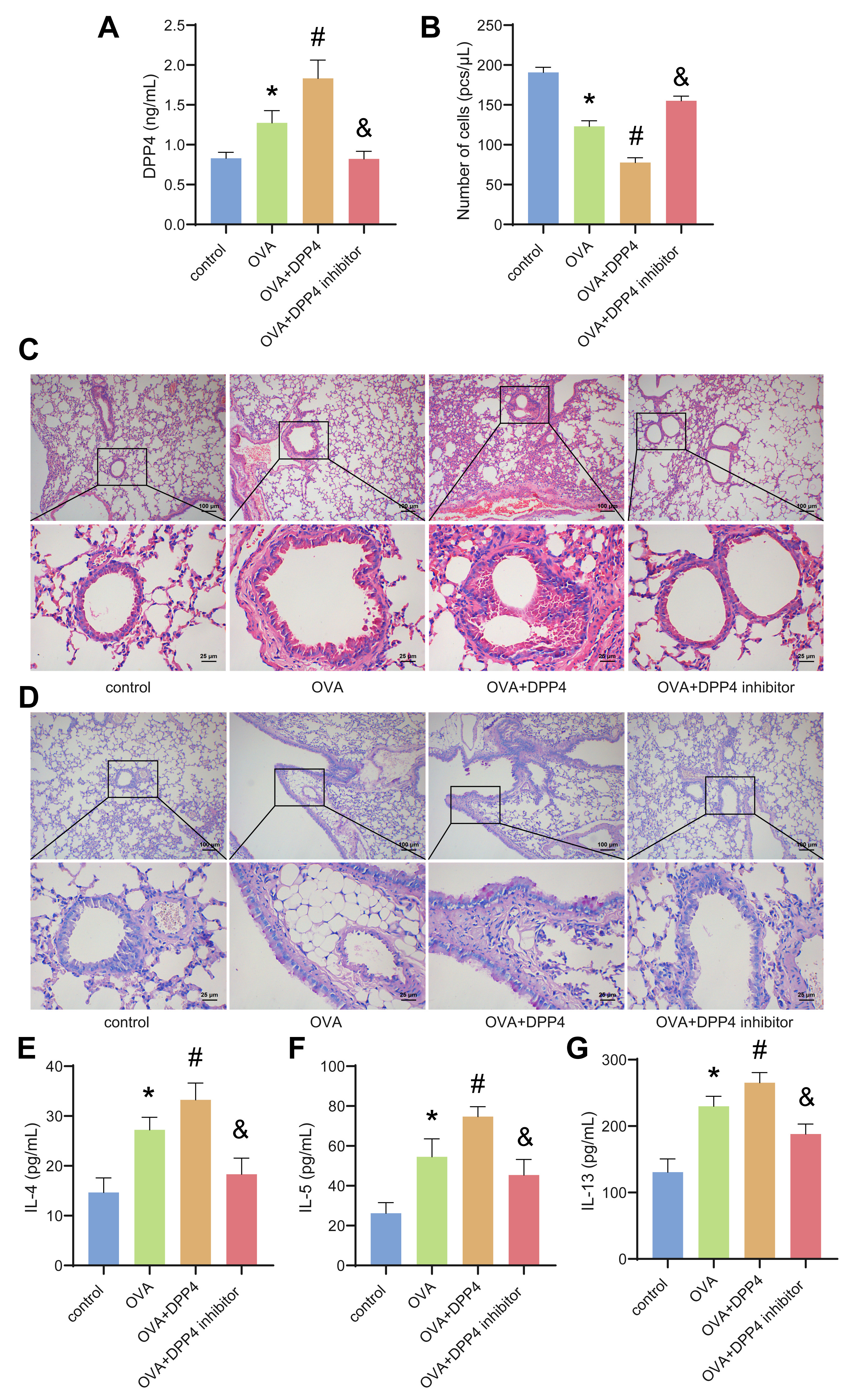 Fig. 4.
Fig. 4.
Overexpression of DPP4 promoted airway inflammation in asthmatic
mice. (A) The level of DPP4 in the bronchoalveolar lavage fluid (BALF) was
analyzed by ELISA. (B) The total number of leukocytes in the BALF was calculated
by a hemocytometer. (C) Hematoxylin-eosin (HE) staining. (D) Periodic acid Schiff
(PAS) staining. (E–G) The levels of IL-4, IL-5 and IL-13 in the BALF were
assessed by ELISA. Data was showed as the mean SD, *p 0.05
vs control group, #p 0.05 vs OVA group, &p 0.05 vs
OVA+DPP4 group.
3.5 Overexpression of DPP4 Promoted Airway EMT in OVA-Induced
Asthmatic Mice
The collagen fibers in the bronchus of the mice in the OVA group were
significantly proliferated compared with the control group (Fig. 5A).
Overexpression of enhanced OVA-induced bronchus collagen fibril deposition in
lungs-bronchi, which was significantly improved by DPP4 inhibitor (Fig. 5A). The
protein levels of E-cadherin and -SMA in the bronchial-lung tissue of
mice were assessed by IF (Fig. 5B,C, Supplementary Fig. 3A,B) and WB
(Fig. 5F). Compared with the OVA group, the mRNA and protein expressions of
E-cadherin in the bronchial-lung tissue of mice in the DPP4 group were
down-regulated, while -SMA was up-regulated. Overexpression of DPP4
significantly promoted airway EMT, whereas DPP4 inhibitors relieved airway EMT in
asthmatic mice. Evolutions of the mRNA levels of E-cadherin and -SMA
were consistent with the proteins levels (Fig. 5D,E). In addition, oe-DPP4
significantly increased the concentration of IL-17, TGF-1, and MMP9 in
the BALF of OVA-induced asthmatic mice (Fig. 5G–I). Similarly, oe-DPP4 enhanced
the OVA-mediated reduction of Treg cells while contributing to an increase in the
ratio of Th17 cells. In contrast, DPP4 inhibitors reversed the OVA-mediated
increases of Th17 and IL-17 in asthma mice (Fig. 5J,K, Supplementary Fig.
3C,D). The above results demonstrated that oe-DPP4 increased the levels of
TGF-1 in airway and regulated the Th17/IL-17 axis, thereby mediating
airway EMT and causing airway remodeling.
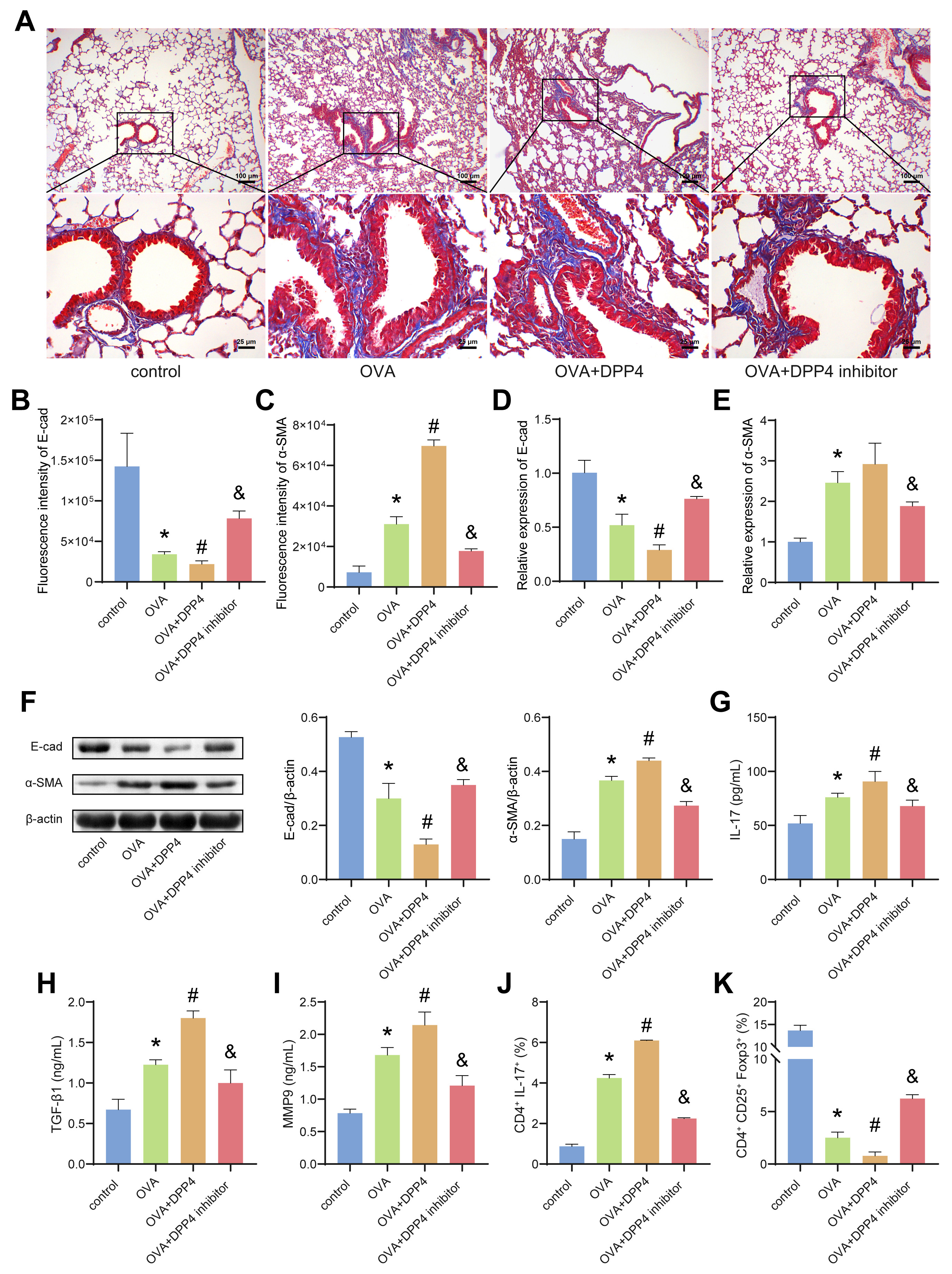 Fig. 5.
Fig. 5.
Overexpression of DPP4 promoted airway epithelial mesenchymal
transition (EMT) in OVA-induced asthmatic mice. (A) Masson staining. (B,C) The
fluorescence intensity of E-cad and -SMA in lungs-bronchi of mice.
(D,E) The mRNA expression of E-cad and -SMA in lungs-bronchi of mice
were evaluated by quantitative Real-Time PCR (qRT-PCR). (F) The protein levels of
E-cad and -SMA in lungs-bronchi of mice were estimated by WB. (G–I) The
levels of IL-17, TGF-1 and MMP9 in BALF were analyzed by ELISA. (J,K)
The ratio of Th17 and Treg cells in the airways of mice was detected by flow
cytometry. Data was showed as the mean SD, *p 0.05 vs
control, #p 0.05 vs OVA group, &p 0.05 vs OVA+DPP4
group. E-cad, E-cadherin.
4. Discussion
CD26/DPP4 is a multifunctional glycoprotein with broad distribution, which can
exist both in the form of homodimers on the surface of immune cells and in a
solubilized form in body fluids [17]. Studies have shown that CD26 was highly
expressed on the surface of Th17 cells and participated in coordinating the
immune response of Th17 cells in human inflammatory diseases [11]. Zhao
et al. [12] confirmed that the expression of CD26 was conducive to the
differentiation of CD4 T cells to Th17 cells. Meanwhile, there other
studies have demonstrated that Th17/IL-17A could induce the accumulation of
neutrophils in the airways and eventually participate in the pathogenesis of
neutrophilic asthma [5, 13, 18]. Therefore, the impacts of DPP4 on Th17 cell
polarization in vitro was initially investigated, and the results
suggested that sCD26/sDPP4 promoted Th17 cell polarization in a dose-dependent
manner, while DPP4 inhibitors could inhibit Th17 cell polarization. The change in
IL-17 concentration was consistent with the change in Th17 cell count. In
conclusion, sCD26/sDPP4 could promote the secretion of IL-17 by promoting the
polarization of Th17 cells.
EMT is a dynamic process in which epithelial cells gradually lose their
epithelial characteristics and acquire mesenchymal characteristics [19]. During
EMT, polarized bronchial epithelial markers such as cytokeratin and E-cadherin
are down-regulated, and mesenchymal-specific markers such as -SMA and
vimentin are up-regulated [20]. Studies have indicated that BECs could be
transformed into myofibroblasts after EMT, thereby promoting asthma airway
remodeling [21]. In this paper, BECs of mice were stimulated with TGF-1
for 72 h, and the cell morphology changed from goose-warm stone-like to
spindle-like and fibroblast-like morhologies. sCD26/sDPP4 significantly promoted
the EMT process induced by TGF-1. Compared with the TGF-1
group, the expression of E-cadherin was down-regulated while the expression of
-SMA was up-regulated in the DPP4 group, and DPP4 inhibitor reversed
the effects of DPP4. In addition, one of our previous studies showed that the
chronic inflammatory environment provided by IL-17 was beneficial to the
TGF-1-induced EMT in BECs [15]. And another study has shown that IL-17
and DPP4 had a synergistic effect on the formation of EMT [16]. The findings in
this paper indicated that sCD26/sDPP4 promoted Th17 cells to secrete IL-17 to
further promote EMT in BECs.
OVA, one of the most abundant glycoprotein allergens, induces IgE production and
Th2 immune responses in asthmatic patients [22]. To further explore the role of
DPP4 in asthma, an OVA-induced asthmatic mouse model was established. We observed
that DPP4 significantly aggravated airway inflammation in asthmatic mice, while
promoting mucus secretion, goblet cell hyperplasia, and collagen deposition.
Additionally, DPP4 increased the levels of Th2 cell-derived cytokines IL-4, IL-5
and IL-13 in the BALF of asthmatic mice. These cytokines not only contribute to
airway inflammation and airway hyperresponsiveness, but also induce subepithelial
fibrosis [23, 24, 25]. TGF-1 is a central mediator is involved in tissue
repair and fibrosis progression, and induces EMT in multiple organs [26]. In our
study, the concentration of TGF-1 in the BALF of DPP4 group mice was
increased compared with the OVA group. Meanwhile, oe-DPP4 significantly
down-regulated the E-cadherin expression and up-regulated the -SMA
expression in bronchial of asthmatic mice. These results suggest that oe-DPP4
could promote the asthmatic airway EMT, subsequently promoting airway remodeling
in asthmatic mice.
In addition, researches have revealed that the decreased expression of
E-cadherin in the lung tissue of asthmatic patients results in the loss of airway
barrier function, which further promotes the occurrence of airway remodeling
[27]. A Th17/Treg imbalance has been reported in acute OVA challenge or house
dust mite-induced asthmtic mouse models [28, 29]. Similar to the previous
findings, Th17/Treg imbalance was also confirmed in our OVA-induced asthmatic
mouse model. The number of Th17 cells in the airways of asthmatic mice increased,
while the number of Treg cellsdecreased. Overexpression of DPP4 promoted the
differentiation of CD4 T cells to Th17 cells but not Treg cells, therefore
further accelerated this imbalance. Meanwhile, compared with the OVA group, the
level of IL-17 in the BALF of the DPP4 group mice were also significantly
increased. High levels of IL-17 were related to the airway inflammation,
responsiveness, and remodeling in asthmatics [30, 31]. Therefore, oe-DPP4 promote
airway inflammation and remodeling in asthmatic mice by promoting airway EMT and
Th17 cell polarization.
