
- Academic Editor
†These authors contributed equally.
Background: Osteogenesis imperfecta (OI) is a rare genetic disorder
characterized by recurring bone fractures. Some OI patients have other clinical
manifestations such as growth retardation, dental abnormalities, blue sclera, and
hearing loss. The relationship between the phenotype and genotype of OI is
indistinct, and there is no cure for OI. Therefore, an appropriate disease model
is urgently needed to understand the pathophysiology of OI. Induced pluripotent
stem cells (iPSCs) are capable of developing into three germ layers and have the
same genetic background as the donor cells they were derived from; thus, they are
an appropriate disease model. Methods: Blood
samples collected from the proband and her affected children and one unaffected
child were used forgenotyping by whole genome
sequencing. A patient-specific iPSC line and a healthy donor iPSC line were
generated by reprogramming peripheral blood mononuclear
cells with episomal plasmids containing seven
transcription factors, namely, OCT4, SOX2, NANOG,
LIN28, cMYC, KLF4, and SV40LT.
Results: The proband and her two affected children were homozygous for
a mutation in collagen type I alpha 1 exon 10,
c.725G
Osteogenesis imperfecta (OI) is a rare and intractable heterogeneous hereditary condition characterized by bone fragility and systemic complications including bone deformity, delayed growth, dental abnormalities, blue sclera, hearing loss, cardiovascular disorders, and ligament relaxation [1, 2]. Its clinical features are heterogeneous, ranging in severity from benign to perinatal fatal [3, 4]. Despite sharing the same causative mutant genes, the clinical manifestations usually significantly differ among patients [5]. Although approximately 20 OI causative genes have been reported [6], the majority of OI cases are caused by pathogenic variants in the genes encoding collagen type I alpha-1 (COL1A1) and collagen type I alpha-2 (COL1A2), resulting in synthesis defects of type I collagen [7, 8, 9], which is the primary structural protein of bone [10]. Despite the significant progress that has been made in identifying the pathogenic genes and understanding the molecular causes of OI, the exact pathogenesis of OI remains unclear [11]. Furthermore, there is limited knowledge on the relationship between the OI phenotype and its genotype [1], and no cure is available for OI [12]. Osteoblasts from OI patients, which display OI-relevant features, are reasonable study models for OI investigation; however, obtaining osteoblast samples from OI patients is challenging. Thus, it is important to develop disease models that can, to some extent, better replicate disease-related characteristics. This will aid in understanding the pathophysiology of OI and assessing potential drug treatments.
Induced pluripotent stem cells (iPSCs) are an ideal source for developing such models [13, 14, 15], as they retain the genetic information from the donor cells and have the capacity for unlimited propagation as well as the potential to differentiate into three different cell lineages [16]. It is worth noting that iPSCs are not subject to immune rejection or ethical concerns [17] and can be constructed from a wide range of source cells [18]. Due to these advantages, many disease-specific iPSCs have been successfully established, and have become an indispensable research tool for medical, pharmaceutical, and biological research.
Here, we report the study of an OI family. The proband and her two affected
children had a heterozygous mutation, c.725G
This study received approval from the Medical Ethics Committee of Shenzhen People’s Hospital (Shenzhen, China), in accordance with the ethical guidelines outlined in the Declaration Helsinki for conducting medical research involving human subjects. All subjects included in this study gave written informed consent to participate. The permission was obtained from the participants to use their photographs. Animal studies were approved by Jinan University Laboratory Animal Ethics Committee (20210301-38). Blood samples were collected from participant donors within a family affected by OI after their informed consent (Fig. 1H), including the proband (I-1), her two affected children (II-4 and II-5) and her one unaffected child (II-1), and 100 unrelated, healthy and ancestry-matched subjects (50 males and 50 females). Human PBMCs were separated from blood samples trough density gradient centrifugation using the Ficoll gradient (Cat. No. 17-1440-03; GE Healthcare, Little Chalfont, Buckinghamshire, UK). The PBMCs were mycoplasma-free, as confirmed by a polymerase chain reaction (PCR) method. Genomic DNA was extracted with a HiPure Blood DNA Mini Kit according to the manufacturer’s instructions (Magen, Guangzhou, China). The detailed procedure is provided in the supporting information.
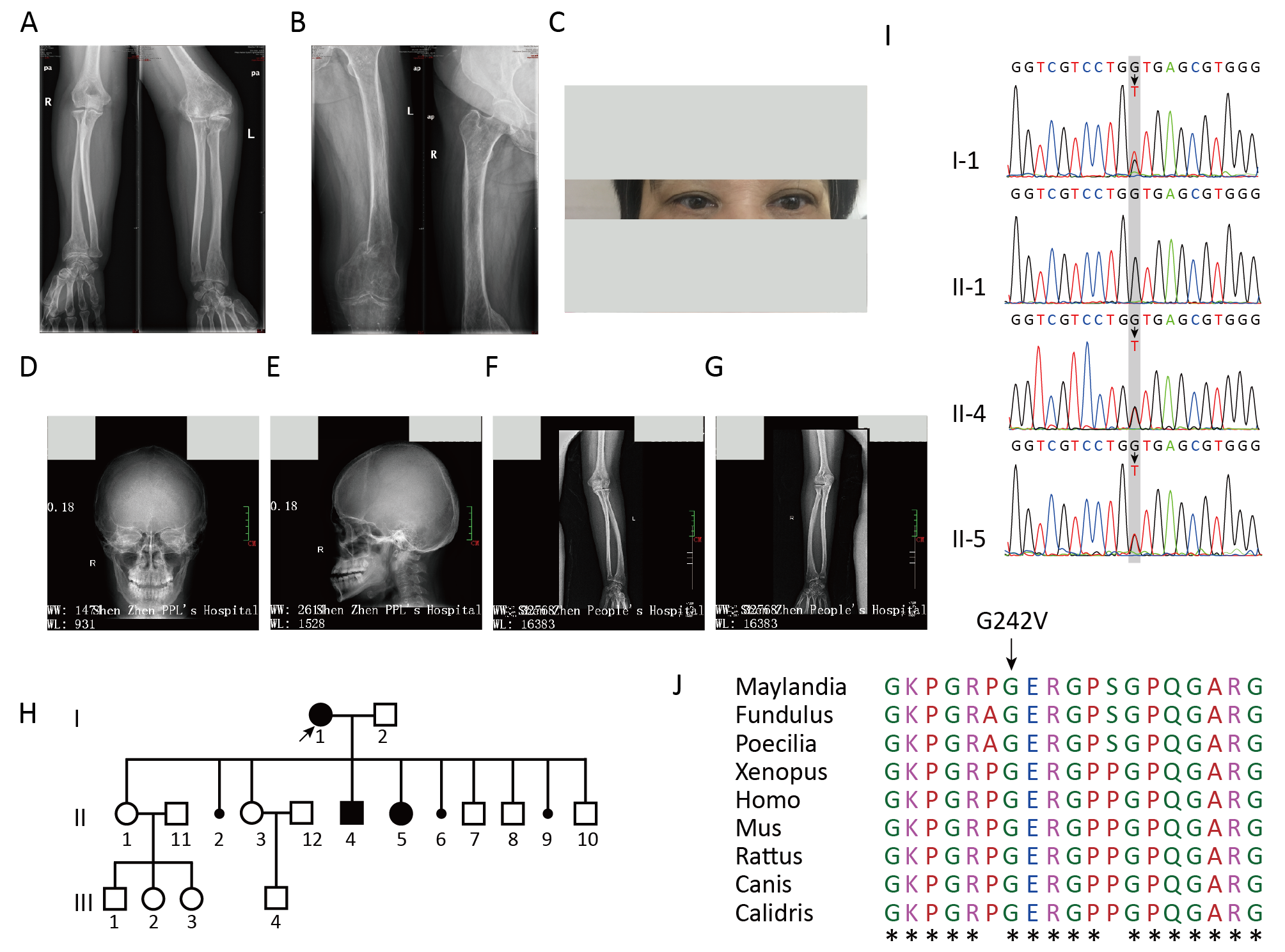 Fig. 1.
Fig. 1.Clinical and radiographic features of OI patients and genotyping
of the OI family. (A,B) Radiographs of the proband. (C) Photographs of the
proband showed blue sclera. (D,E) Radiographs of the proband’s affected son.
(F,G) Radiographs of the proband’s affected daughter. (H) Pedigree of the OI
family recruited to this study. (I) A point mutation, c.725G
Whole genome sequencing (WGS) of I-1, II-1, II-4 and II-5 was performed using the Hiseq X Ten system (Illumina, San Diego, CA, USA). The clean data were obtained after filtering and alignment to the reference human genome (GRCh37/HG19). After deduplication, barcode correction, local realignment, and base quality recalibration, the processed data were subjected to detection of single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels). The obtained variant detection results were compared with entries in databases. The candidate variants were screened using a list of OI genes (Supplementary Table 1). The detailed procedure is provided in the supporting information.
PCR amplification was conducted with the GeneAmp PCR System 9700 Thermal Cycler (Applied Biosystems, Waltham, MA, USA). After purification by agarose gel electrophoresis, the PCR products were subjected to analysis using the ABI 3730 DNA Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The results were compared to the COL1A1 gene sequence using Chromas software 2.6.6 (Technelysium, South Brisbane, QLD, Australia, http://technelysium.com.au/wp/chromas/). The detailed procedure is provided in the supporting information.
PBMCs were seeded in a 6-well plate coated with fibronectin and cultured in PBMC medium for 7 days. Then PBMCs were collected and suspended in an electroporation reaction solution, along with a plasmid mixture (pEP4EO2SEN2K, pEP4EO2SET2K, pCEP4-M2L, Supplementary Fig. 1). After electroporation using the Amaxa Nucleofector II Transfection Device (Amaxa, Cologne, Germany), the PBMCs were cultured overnight in StemSpan Serum Free Expansion Medium supplemented with cytokines in a 6-well plate coated with fibronectin and Matrigel. The next day, the culture medium was replaced with Dulbecco’s Modified Eagle Medium/F12 and supplemented with N-2, B-27, and other specified components. The medium was replaced daily. After 11 days, the culture medium was switched to NuwaCell hPSC Medium. Manually selected ESC-like colonies were cultured in a 6-well plate coated with Matrigel for expansion. The detailed procedure is provided in the supporting information.
To verify the source of the constructed iPSC lines, genomic DNAs from both iPSC lines and their parental cells were employed for short tandem repeat (STR) profiling. In brief, genomic DNAs were extracted using the Chelex 100 resin and amplified. The PCR products were electrophoresed and analyzed using the ABI 3500XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The genetic profiles were analyzed utilizing the PowerPlex® 21 PCR Amplification System (Promega, Madison, WI, USA) targeting the 21 loci markers. For a detailed description of the procedure, please refer to the supporting information.
Total RNA was extracted employing the RNAprep Pure Cell/Bacteria Kit (TIANGEN, China). Then PCR reactions were conducted with the LightCycler 96 Real Time PCR System (Roche, Basel, Switzerland), and the data were assessed utilizing LightCycler software (version 1.1.0.1320, Roche, Basel, Switzerland). The primers listed in Supplementary Table 2 were used to assess the expression of octamer-binding transcription factor 4 (OCT4, also known as POU5F1) and Nanog homeobox (NANOG). The detailed procedure is provided in the supporting information.
The anti-bodies listed in Supplementary Table 3 were used for the immunostaining of cells, and the Gallios Flow Cytometer (Beckman Coulter, Brea, CA, USA) was utilized to assess the cells.
G-banding karyotype analysis was conducted to determine the karyotype of the iPSC lines. The iPSCs were collected when they reached about 60% confluence, and chromosome G-banding analysis specimens were prepared using a standard method. A total of 20–30 metaphases were analyzed. The detailed procedure is provided in the supporting information.
The teratoma assay was conducted to analyze the pluripotency of iPSCs in vivo. After resuspending in a mixture of 50% Matrigel and phosphate-buffered saline (PBS), iPSCs were injected into the posterior thigh muscles of 4-week-old SCID Beige mice. At 8 weeks post-injection, the mice were euthanized and the tumors were collected and subjected to hematoxylin and eosin (H&E) staining. The detailed procedure is provided in the supporting information.
Mycoplasma contamination was verified through PCR analysis. The absence of pathogenic microorganism contamination (hepatitis B virus (HBV), hepatitis C virus (HCV), human immunodeficiency virus (HIV), syphilis) was verified by an accredited clinical laboratory service provider (Hefei KingMed for Clinical Laboratory, Anhui, China).
The detection of exogenous genes was performed using the quantitative PCR (qPCR)
standard curve method. Briefly, 0, 0.1, 1, 10, and 100 copies of plasmids were
mixed with pure mesenchymal stem cells (MSCs). After DNA was extracted from each
group, qPCR was performed with the LightCycler480 System (Roche, Alameda, CA,
USA). The primers are listed in Supplementary Table 4. OCT4 was
used as the endogenous control gene, and the 2
The proband (I-1), a 53-year-old Chinese woman, received a diagnosis of OI type I. At the age of 10, she had a fracture of the left elbow but took no medication, which resulted in left cubitus varus. Subsequently, bone fractures occurred more than 20 times, particularly in her hands, waist, legs and feet. Physical examination at 53 years old showed a short stature, height of 120.1 cm and weight of 50.9 kg, blue sclera, dentinogenesis imperfecta, normal hearing and vision, left cubitus varus, bending deformities of thighs, and osteoporosis (Table 1, Fig. 1C and Supplementary Fig. 2). Full-body radiographs revealed low bone mineral density in the long tubular bones, widening of the long tubular bones, slight curvature of the backbone, and multiple pathological fractures with malunion (Fig. 1A,B and Supplementary Fig. 3). The proband was married to a healthy man. She had been pregnant 10 times, with three spontaneous abortions (Fig. 1H). She had seven children (4 boys and 3 girls), of whom one boy and one girl had OI (Fig. 1H).
| Characteristics | Patients | |||
| I-1 | II-4 | II-5 | ||
| Clinical findings | ||||
| Sex | Female | Male | Female | |
| Age, years | 53 | 27 | 24 | |
| Weight, kg | 50.9 | 59.8 | 45.0 | |
| Height, cm | 120.1 | 149.8 | 139 | |
| Disease severity | Severe | Moderate | Moderate | |
| Skeletal findings | ||||
| Multiple fractures of extremities | Yes | No | No | |
| Age at first fracture, years | 10 | 15 | 6 | |
| Vertebral fractures | No | No | No | |
| Bowing of upper extremities | Yes | No | No | |
| Bowing of lower extremities | Yes | No | No | |
| Shortening of upper extremities | Yes | No | No | |
| Shortening of lower extremities | Yes | No | No | |
| Spine abnormalities | Yes | No | No | |
| Scoliosis | Yes | Yes | Yes | |
| Other findings | ||||
| Color of sclera | Blue | Blue | Blue | |
| Dentinogenesis imperfecta | Yes | No | No | |
| Hypermobility of joints | Yes | No | No | |
| Cardiac impairment | No | No | No | |
| Hearing impairment | No | Yes | No | |
| Intellectual development | Normal | Normal | Normal | |
| Therapy | ||||
| Bisphosphate treatment | Yes | No | No | |
| Right patella fixation | Yes | No | No | |
OI, osteogenesis imperfect.
The proband’s affected son (II-4), a 27-year-old male, was diagnosed with OI type I. His first brittle fracture occurred at 15 years old. He had experienced more than 10 bone fractures, particularly in his hands, waist, and legs. He exhibited blue sclerae and scoliosis with hearing loss, but no dentinogenesis imperfecta (Table 1). At 27 years old his weight was 59.8 kg and his height was 149.8 cm. X-ray films indicated a small skull base and thin facial bone, enlarged cranial bone, slight curvatures of the long tubular bones, and widening of the radius (Fig. 1D,E and Supplementary Fig. 4).
The affected daughter (II-5) is 24 years old and was also diagnosed with OI type I. Her first brittle fracture occurred at 6 years old. She exhibited blue sclera and scoliosis, with no dentinogenesis imperfecta (Table 1). Her vision and hearing were normal. At 24 years old, her weight was 45.0 kg and her height was 139 cm. The X-ray revealed a small skull base and thin facial bone, enlarged cranial bone, slight curvatures of the long tubular bones and widening of the radius (Fig. 1F,G and Supplementary Fig. 5).
Of the total 470 Gbp sequencing data, more than 100 Gbp high-quality sequencing
data with an average of 99.0% coverage were obtained (Supplementary
Table 5). The valid sequencing depth ranged from 31.6
As shown in Fig. 1I, PCR and Sanger sequencing revealed that the proband and her
two affected children carried the heterozygous G242V mutation, whereas the
healthy family member and the 100 population-matched controls did not exhibit the
mutation. One patient with OI has been reported to harbor the heterozygous
c.725G
To generate iPSCs, PBMCs taken from II-1 and II-4 were expanded and transfected with episomal vectors expressing pluripotency factors (OCT4, SOX2, NANOG, LIN28, cMYC, KLF4 and SV40LT) under feeder-free conditions [20]. Typical iPSC colonies with a morphology resembling hESCs were manually selected and expanded on Matrigel-coated plates with passages conducted every 3–4 days.
As depicted in Fig. 2A,B, OI-iPSCs showed typically hESC-like morphological features. Flow cytometry analyses revealed that OI-iPSCs were positive for the pluripotency surface markers stage-specific embryonic antigen-4 (SSEA-4) and TRA-1-81 (Fig. 2C,D). The expression of pluripotent stem cell markers (OCT4, NANOG) was also confirmed by qPCR analysis (Fig. 2E). For qPCR analysis, the MSC line P3 (MSC-P3) served as a negative control, whereas the hESC line H1 (hESC-H1) served as a positive control. Chromosome GTG band analysis showed that OI-iPSCs exhibited a normal diploid 46, XY karyotype (Fig. 2F). To assess the in vivo pluripotency of OI-iPSCs, the teratoma formation assay was conducted. OI-iPSCs were injected into SCID Beige mice, and the teratomas were harvested 8 weeks after injection. H&E staining confirmed the presence of derivatives from all three germ layers in the teratomas, such as the neural rosettes formed from the ectoderm, the cartilage differentiated from the mesoderm, and the intestinal epithelial cells developed from the endoderm (Fig. 2G–I). These results provide evidence for the establishment of OI-iPSCs and their pluripotency and ability to undergo multilineage differentiation.
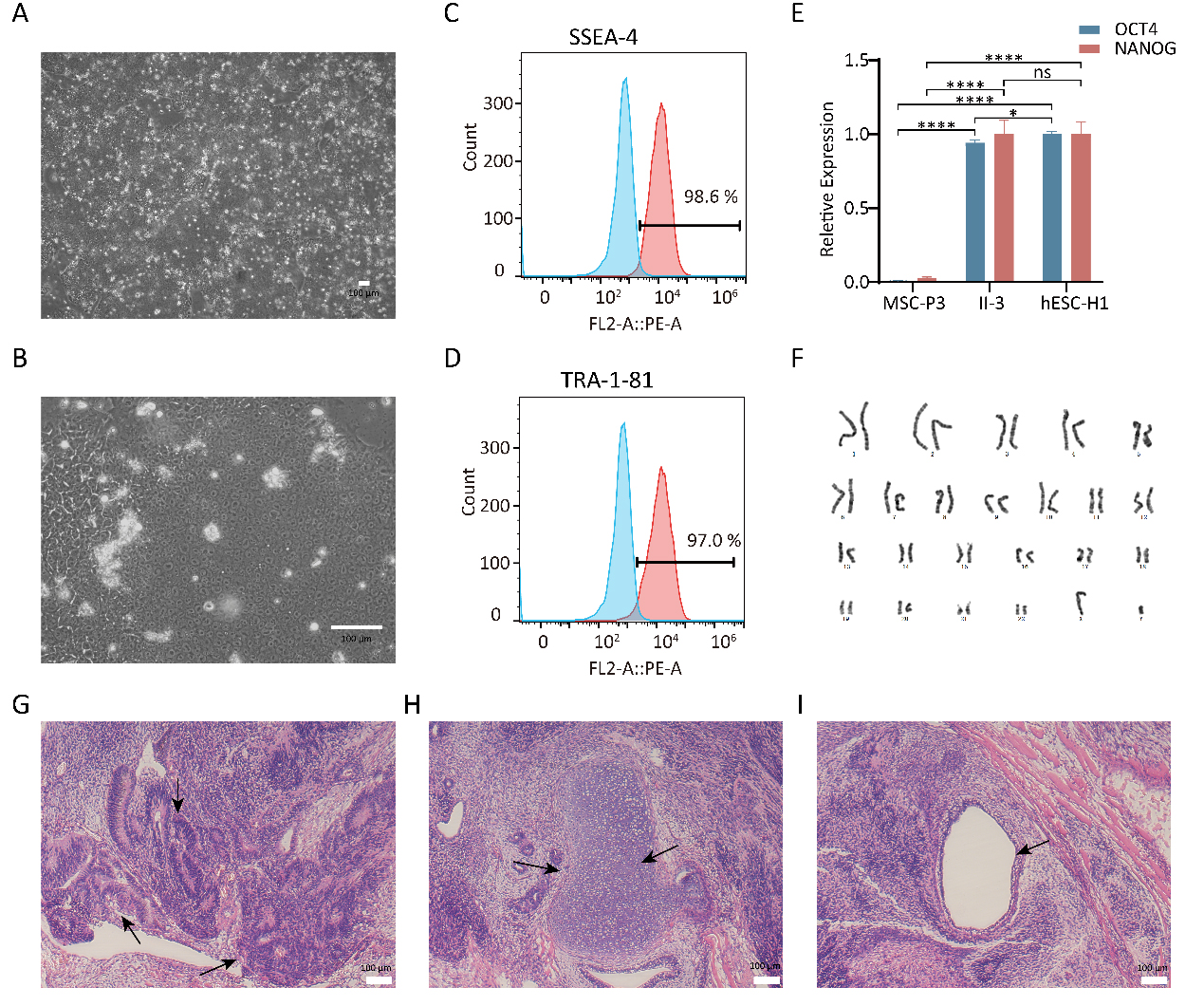 Fig. 2.
Fig. 2.Characterization of OI-iPSCs. (A,B) Images of OI-iPSC colonies
cultured on Matrigel. Scale bar: 100 µm. (C,D) Flow cytometry analysis of
pluripotency markers SSEA-4 and Tra-1-81 of OI-iPSCs. (E) Quantification via qPCR
analyses of the relative expression levels of the pluripotency markers
OCT4 and NANOG in OI-iPSCs (n = 3). (F) OI-iPSCs displayed a
normal karyotype. (G–I) H&E staining of teratomas derived from immunodeficient
mice injected with OI-iPSCs showed tissues representing all three embryonic germ
layers: ectoderm (neural rosettes), mesoderm (cartilage) and endoderm (intestinal
epithelial cells). Scale bar: 100 µm. SSEA-4, stage-specific embryonic
antigen-4; OI-iPSC, OI patient-specific iPSC line; iPSC, induced pluripotent stem cells; H&E, hematoxylin and eosin (* p
hESC-like morphology was also observed for HC-iPSCs (Fig. 3A,B). The expression of SSEA-4 and TRA-1-81 in HC-iPSCs was evaluated by flow cytometry analysis (Fig. 3C,D). The qPCR assay demonstrated that the endogenous pluripotency genes OCT4 and NANOG were activated in HC-iPSCs and hESC-H1 cells, whereas they were silenced in MSC-P3 cells (Fig. 3E). HC-iPSCs exhibited the normal karyotype of diploid 46 XX by standard G-banding chromosome analysis (Fig. 3F). The teratoma assay detailed previously was conducted to assess the pluripotency of HC-iPSCs. H&E staining verified the nature of these tumors as teratomas, as evidenced by the presence of tissues representing all three germ layers including neural epithelia (Fig. 3G), cartilage (Fig. 3H) and intestinal epithelia (Fig. 3I). These results indicated the successful generation and functional pluripotency of HC-iPSCs.
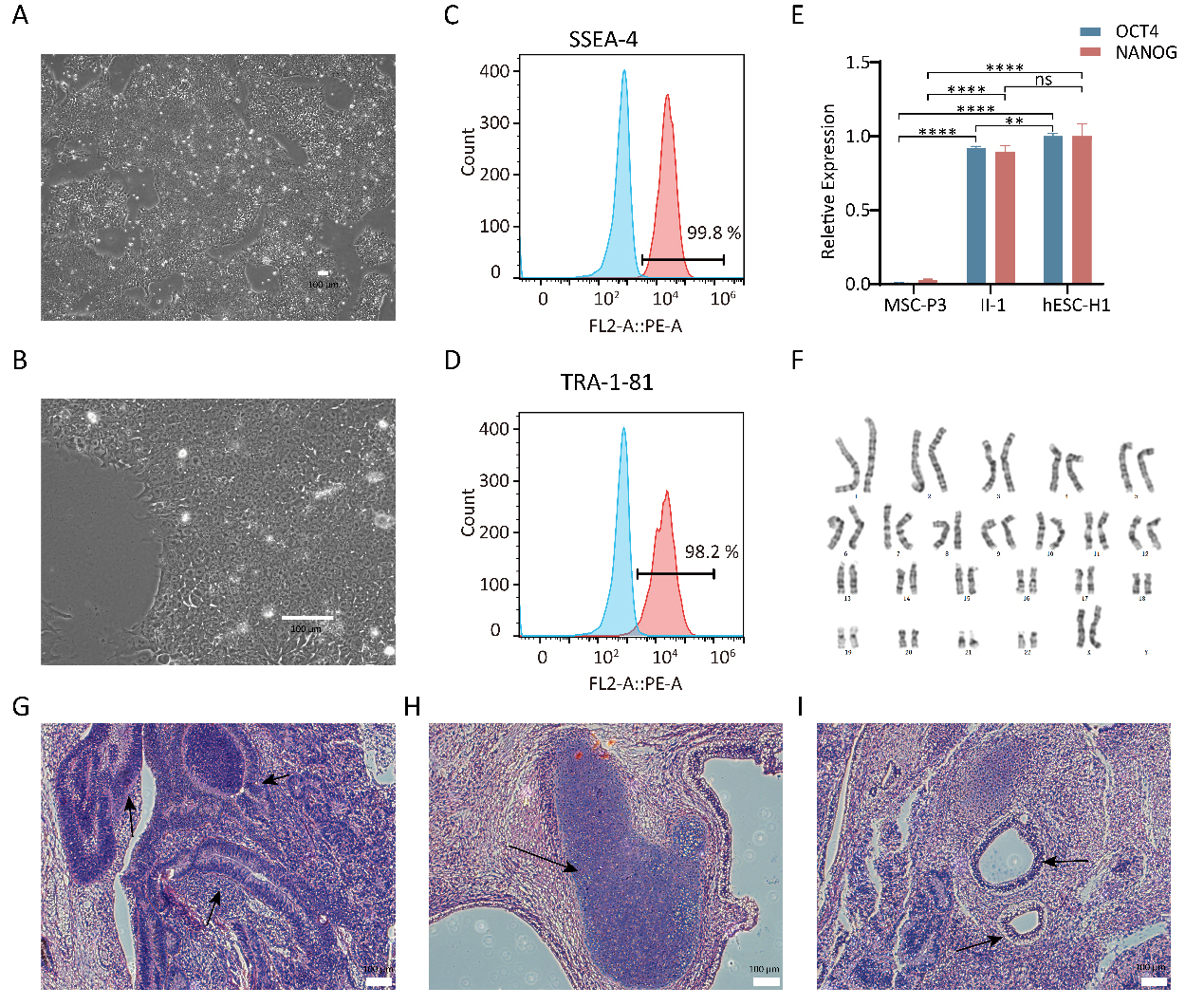 Fig. 3.
Fig. 3.Characterization of HC-iPSCs. (A,B) Images of HC-iPSC colonies
cultured on Matrigel. Scale bar: 100 µm. (C,D) Flow cytometry analysis of
pluripotency markers SSEA-4 and Tra-181 of HC-iPSCs. (E) Quantification via qPCR
analyses of the relative expression levels of the pluripotency markers
OCT4 and NANOG in HC-iPSCs (n = 3). (F) HC-iPSCs displayed a
normal karyotype. (G–I) H&E staining of teratomas derived from immunodeficient
mice injected with HC-iPSCs showed tissues representing all three embryonic germ
layers: ectoderm (neural rosettes), mesoderm (cartilage) and endoderm (intestinal
epithelial cells). Scale bar: 100 µm. HC-iPSC, healthy donor iPSC line. (** p
STR analysis demonstrated that OI-iPSCs and HC-iPSCs displayed the same 21 STR loci as their parental PBMC cells (Supplementary Tables 8,9). Mycoplasma detection confirmed the absence of mycoplasma contamination in both iPSC lines (Supplementary Fig. 6). The absence of pathogenic microorganism contamination was also confirmed for OI-iPSCs and HC-iPSCs (Supplementary Tables 10,11). The negative expression of exogenous genes in both iPSC lines was confirmed using the qPCR standard curve method (Supplementary Figs. 7,8).
In 1979, Sillence et al. [21] classified OI into four types (type 1 to
type IV) based on clinical manifestations, radiological features, and genetic
patterns; this classification is referred to as the “Sillence classification”.
Type 1 OI follows an autosomal dominant inheritance pattern and is characterized
by the presence of blue sclerae and mild bone deformities. Type II OI is used to
describe the lethal perinatal cases, often indicative of autosomal recessive
inheritance. Type III OI is associated with a progressively deforming phenotype,
where the blue sclerae tend to fade with age and autosomal recessive inheritance
is often observed. Type IV OI is characterized by white sclerae, short stature,
bone deformities, and dentin hypoplasia. In this study, we examined a Chinese
family displaying clinical characteristics of type I OI inherited in an autosomal
dominant fashion. Even carrying the same mutated gene, the three patients
exhibited different clinical features. The proband had serious OI symptoms
including multiple bone fractures, osteopenia, healing of long bones, scoliosis,
dentinogenesis imperfecta, and blue sclera. The affected proband’s son had a mild
form of OI, such as frequent bone fractures, blue sclera and hearing loss. The
affected proband’s daughter exhibited a milder form of OI. WGS and Sanger
sequencing confirmed a heterozygous mutation in COL1A1 exon 10,
c.725G
The genes COL1A1, situated on chromosome 17q21.3-22, and COL1A2, situated on chromosome 7q21.3-q22.1, encode type I collagen [22], the primary component of bone extracellular matrix, which is essential for the mechanical properties of bone [23]. Similar to other collagens, the distinctive structural characteristic of type I collagen is a rod-like trimer structure with a long continuous triple helix motif, which is composed of Gly-X-Y amino acid units [24]. Due to its smallest size, the glycine residues are positioned at the core of the triple helix domain [25]. After produced, the collagen molecules are precisely aligned to form fibers, followed by the parallel growth of hydroxyapatite crystals alongside these collagen fibrils [26]. Once glycine is replaced by a larger amino acid, such as valine, the residue no longer fits within the confined space at the core the triple helix domain, resulting in disturbed helical collagen structure [26]. The abnormal collagen I molecules can impact the mechanical characteristics of bone in two ways [1]. The abnormal collagen matrix leads to reduced bone toughness and the collagen matrix interacts with the mineral phase of bone. The damaged matrix had a dramatic effect on the integration of the mineral content, a critical factor in influencing bone strength. Type I collagen is also extensively distributed throughout other connective tissues, excluding hyaline cartilage such as bone, skin, tendon, ligament, sclera, cornea, and blood vessels [24]. This could to some extent explain clinical heterogeneity of OI patients in our reported family. However, the mechanism by which mutations in COL1A1 result in the clinical phenotypes of OI remains a subject of debate. The glycine residue in the Gly-Xaa-Yaa repeat is essential to form a triple helix structure. The alteration p.G242V identified in the OI family is located in the triple helix domain of type I collagen. Recent discoveries suggest that aberrant folding and aggregation of type I collagen could be the main reason for collagenopathy in OI [27, 28, 29], which provide us with a research direction to study the pathogenic mechanisms of OI associated with COL1A1 mutations. To delve into this, a suitable biological model is urgently needed.
iPSCs are generated from somatic cells with reprogramming factors [30] and
possess the following strong points. First, they have unlimited proliferation
capability and the potential to differentiate into cells of three embryonic germ
layers of iPSCs [31]. Second, iPSCs are not targets of immunological rejection or
ethical concerns [32]. Third, various somatic cells can be used for generating
iPSCs such as fibroblasts, keratinocytes, adipose stem cells, cord blood,
peripheral blood, neural cells, and urinary cells [18]. Thus, human iPSCs are a
powerful tool to model human diseases, especially single gene diseases [33].
Several OI-specific iPSC lines have been reported. An OI-iPSC line carrying a
heterozygous COL1A1 (c.3969_3970insT) mutation was obtained by gene
editing an established control iPSC line using CRISPR/Cas9 [34]. Two X-linked OI
iPSC lines harboring the c.1376A
This study reported an OI family. The mutation in the proband and her two
affected children was detected to be heterozygous in COL1A1 exon 10,
c.725G
The data and materials generated during the current study are available from the corresponding author.
DL, conception and design, data collection, data analysis, manuscript writing, review and editing; MO, conception and design, data collection, financial support, review and editing; GD, data analysis, financial support, review and editing; PZ, conception and design, data collection, financial support, review and editing; QL, data analysis, review and editing; JC, data analysis, review and editing; ZS, data collection, review and editing; IMS, conception and design, review and editing; LY, supervision, conception and design, review and editing; GS, financial support, conception and design, review and editing; DT, conception and design, data collection, review and editing; YD, conception and design, data collection, supervision and financial support, review and editing. All authors contributed to editorial changes in the manuscript. All authors have reviewed it critically for important intellectual and approved final manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity.
This study received approval from the Medical Ethics Committee of Shenzhen People’s Hospital (Shenzhen, China), in accordance with the ethical guidelines outlined in the Declaration Helsinki for conducting medical research involving human subjects. All subjects included in this study gave written informed consent to participate. The permission was obtained from the participants to use their photographs. Animal studies were approved by Jinan University Laboratory Animal Ethics Committee (20210301-38).
We thank all participants for their participation in this study.
This work was supported by the Science and Technology Planning Project of Guangdong Province, China (2017B020209001), the National Natural Science Foundation of China (82060393), the Science and Technology Plan of Shenzhen (JCYJ20180305163846927), the Natural Science Foundation of Guangxi (2020GXNSFAA159124), the Natural Science Foundation of Guangdong Province, China (2022A1515010378), the Scientific Research Project of Health System in Pingshan District of Shenzhen (202139, 201801, 202066, 202068) and RSF grant (22-15-20063).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.