
- Academic Editor
Nonalcoholic fatty liver disease (NAFLD) constitutes a commonly diagnosed liver
pathology with perturbed lipid metabolism, which is mainly caused by excessive
accumulation of fat in hepatocytes by various pathogenic factors. Currently,
there are no effective drug treatments for NAFLD. Ferroptosis represents a novel
form of programmed cell death depending on iron, which is driven by large
cellular amounts of reactive oxygen species (ROS) and lipid peroxides.
Ferroptosis plays critical regulatory roles in the pathogenesis of NAFLD, and
overaccumulation of Fe
Nonalcoholic fatty liver disease (NAFLD) constitutes the commonest chronic liver disease, affecting about 25% of humans globally and projected to become the main reason for liver transplantation by 2030, therefore representing a worldwide public health issue [1, 2, 3]. Current evidence shows that NAFLD is a generic term designating a multisystem disease, including a range of liver diseases with metabolic risk factors [4, 5]. NAFL refers to hepatic steatosis, in which fat is present in more than 5% of hepatocytes. Subsequently, nonalcoholic steatohepatitis (NASH) occurs after NAFL aggravates different injuries. NAFL/NASH is associated with hepatocellular damage and usually has four phases: (1) fat overaccumulation in hepatocytes, also termed NAFL; (2) hepatocyte ballooning degeneration and diffuse lobular inflammation; (3) fibrosis, caused by inflammation and hepatocellular damage, which features an excessive extracellular matrix (ECM) accumulation that alters the normal hepatocyte features; (4) cirrhosis that sustainably progresses to the terminal stage of liver disease. Approximately 20% of NAFL cases progress to NASH, and more than 40% of NASH cases develop fibrosis; liver transplantation is considered in case of severe cirrhosis [6, 7].
Recently, ferroptosis has rapidly attracted attention among scientists studying chronic liver disease, as excessive accumulation of iron, which is the main feature of most liver diseases, can cause oxidative damage in the liver. Evidence suggests ferroptosis is tightly associated with lipid peroxidation (LPO) in NAFLD and can therefore be an important target for preventing and treating NAFLD [8]. This article reviews the generation and regulation of ferroptosis and elucidates the association of LPO in ferroptosis with NAFLD.
Ferroptosis, a newly identified iron-dependent cell death, is characterized by iron accumulation and LPO, and has different morphological, biochemical and genetic characteristics from other forms of programmed cell death. Ultrastructural analysis showed the key morphological features distinguishing ferroptosis from apoptosis, necrosis and autophagy include cell membrane rupture and blister, decreased mitochondrial size relative to normal, mitochondrial ridge reduction or disappearance, and increased membrane density [9].
LPO is a key factor inducing ferroptosis, with polyunsaturated fatty acids
(PUFAs) generally serving as reaction substrates. Carbon-carbon double bonds and
carbon-hydrogen bonds in PUFAs are transformed to generate PUFAs-CoA by acyl-CoA
synthetase long chain family member 4 (ACSL4), and PUFAs-coenzyme A (PUFAs-CoA)
are then esterified by lysophosphatidylcholine acyltransferase 3 (LPCAT3) into
substrate PUFAs-PL in membrane phospholipids. Therefore, ACSL4 is considered a
ferroptosis marker [10]. Free radicals interact with the hydrogen of PUFAs-PL to
form the phospholipid radical (PL
LPO occurs in many subcellular structures such as mitochondria, endoplasmic reticulum (ER) and lysosomes [12]. Reactive oxygen species (ROS) generated by the tricarboxylic acid (TCA) cycle and electron transport chain (ETC) in the mitochondria, can target PUFAs-double bonds to produce LPO and induce ferroptosis [13]. Moreover, ER stress has a definite correlation with ferroptosis, and lysosomal ROS may also participate in ferroptosis as ferrostatin was observed in lysosomes [14, 15]. However, the specific mechanism remains undefined. Reactive lipid species generated by LPO include 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), whose contents are the highest and associated with ferroptosis [16].
Currently, three pathways are known to inhibit LPO to prevent ferroptosis,
including the glutathione pathway (GSH-GPX4), the ubiquinone pathway
(NADPH-FSP1-CoQ10) and the tetrahydrobiopterin pathway (GCH1-BH4) [17]. Firstly,
the glutathione peroxidase 4 (GPX4) pathway is one of the commonest defense
systems, which mainly depends on the Xc
Secondly, ferroptosis suppressor protein 1 (FSP1) is a newly reported strong and effective inhibitor of ferroptosis. The FSP1 pathway does not depend on GSH for its inhibitory effect, but instead functions by regulating coenzyme Q10 (CoQ10) [22]. Acetylated FSP1 is targeted to the plasma membrane and CoQ10 stimulates NAD(P)H to shuttle into the lipid bilayer with the help of FSP1; then, free radicals in the droplets are regulated to capture antioxidants, maintaining the quality of lipids and preventing LPO from inducing ferroptosis [23]. Currently, relatively few studies have examined the BH4 pathway. BH4 participates in the antioxidant system and the overexpression of its rate-limiting enzyme GTP cyclohydrolase 1 (GCH1) selectively inhibits the peroxidation of some PUFAs. Therefore, ferroptosis can be inhibited through the GCH1-BH4 pathway [24, 25]. The major regulators of ferroptosis and associated mechanisms are summarized (Fig. 1).
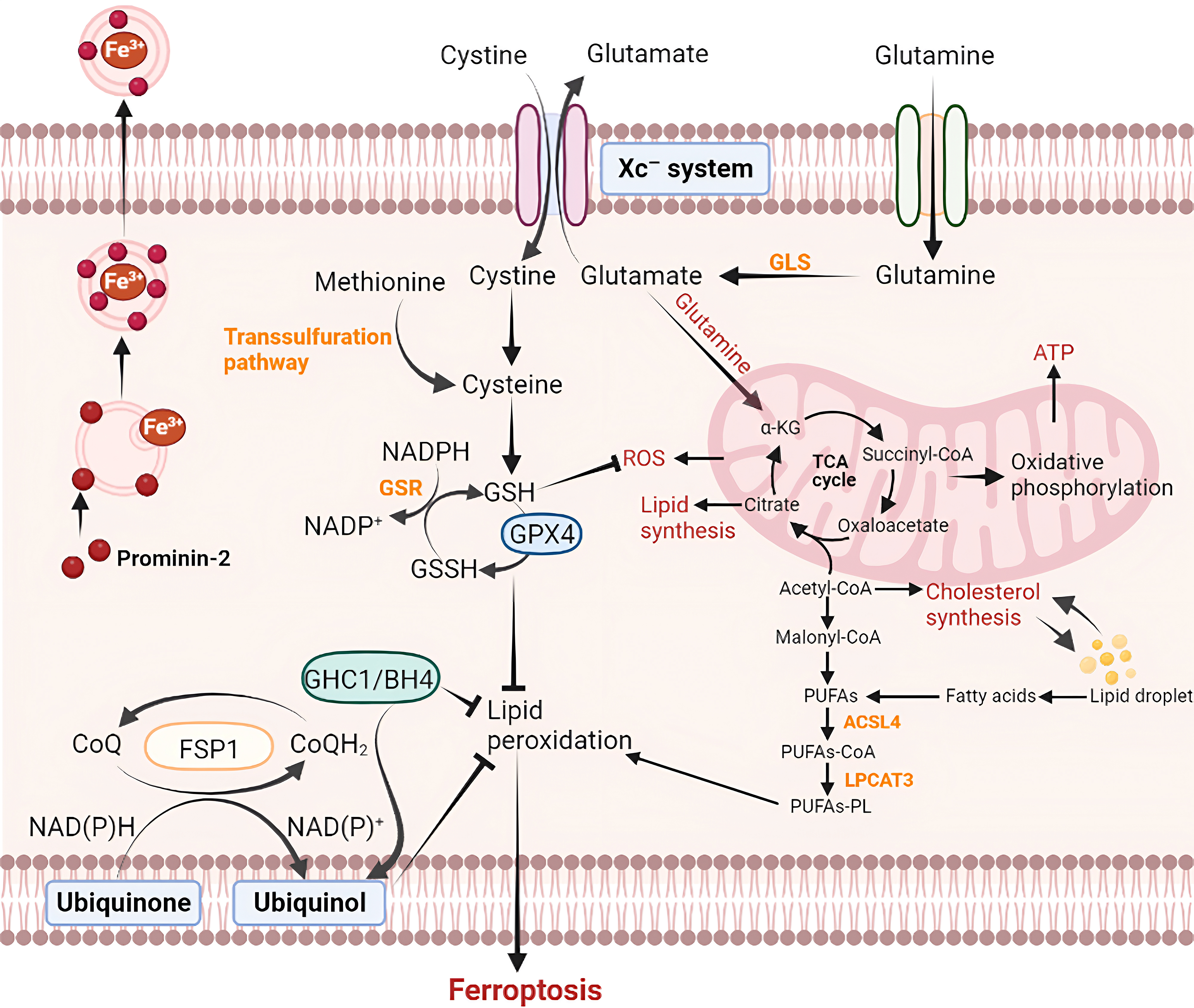 Fig. 1.
Fig. 1.Pathways of ferroptosis. Ferroptosis can be triggered through
inhibiting the system Xc
Necroptosis is a programmed form of cell death that can regulate necrosis with passive and active proinflammatory functions [26]. Numerous studies have consistently shown that ferroptosis is invariably accompanied by necroptosis [27]. The key steps involved in necroptosis include tumor necrosis factor (TNF) signaling, deubiquitination of receptor-interacting protein (RIP) 1, phosphorylation of RIP1 and RIP3, inactivation of caspase-8, and phosphorylation of mixed lineage kinase domain-like protein (MLKL) [28]. Ferroptosis can induce mitochondrial damage, resulting in the opening of the mitochondrial permeability transition pore. Subsequently, the phosphorylation of RIP1/3 was further intensified, ultimately culminating in necroptosis [29]. These findings demonstrate that there is a crosstalk between ferroptosis and necroptosis, but the regulatory relationship between necroptosis and ferroptosis still requires further study.
Autophagy refers to the process where the detached double membrane of the
ribosome-free attachment zone of the rough endoplasmic reticulum envelops
cytoplasm, organelles, proteins, and other cellular components that require
degradation to form autophagosomes [30]. Subsequently, the autophagosomes fuse
with lysosomes to form autophagolysosomes, wherein the enclosed contents are
degraded to fulfill the metabolic requirements and facilitate organelle renewal.
A growing body of evidence suggests that ferroptosis relies on the autophagy
machinery for its execution, making it a form of autophagy-dependent cell death
[31]. Recent studies have discovered that autophagy plays a role in promoting
ferroptosis by degrading the ferroptosis-related protein, ferritin, through the
autophagy-related gene (ATG) 5/ATG7-nuclear receptor coactivator 4 (NCOA4)
autophagic pathway [32]. Moreover, ras-related protein Rab-7a (RAB7A)-mediated
lipophagy, BECN1-mediated system Xc
Apoptosis is a well-established mode of programmed cell death involving both
extracellular and intracellular pathways [34]. The extracellular pathway is
triggered by receptors on the cell membrane, such as TNF-
Pyroptosis, also known as cell inflammatory necrosis, is a form of programmed cell death that relies on inflammatory caspases [37]. Pyroptosis is characterized by nuclear pyknosis, cell swelling, the formation of lipid membrane vacuoles at the plasma membrane, and eventual cell rupture without DNA fragmentation. Both pyroptosis and ferroptosis are associated with damage to the cytoplasmic membrane. Notably, ROS-mediated damage to the cytoplasmic membrane may contribute to reciprocal regulation between pyroptosis and ferroptosis. Furthermore, research has revealed that both types of cell death can be triggered by elevated levels of intracellular iron and ROS [38]. The aforementioned study further demonstrates that a synergistic relationship and crosstalk is existed between pyroptosis and ferroptosis, but the underlying mechanisms still need to be thoroughly investigated.
The liver, an important organ regulating lipid balance, is critical for the metabolism of lipids. Meanwhile, the liver also maintains iron homeostasis in the body and constitutes the main organ that stores excess iron and synthesizes hepcidin. In case of an imbalance between energy intake and consumption or abnormal energy storage function in adipocytes, the liver is most inclined to accumulate fat leading to hepatic steatosis, which subsequently causes systemic metabolic disorders [39, 40]. In this regard, the disruption of iron metabolism is correlated with obesity and insulin resistance (IR) in patients with NAFLD. Furthermore, disturbed iron metabolism has close correlations with the typical features of obesity and pancreatic resistance in NAFLD patients [41]. The “two-hit” and “multiple-hit” hypotheses are classic theoretical explanations for NAFLD pathogenesis [42]. Major risk factors for NAFLD are considered to include IR, altered lipid metabolism, ER stress, mitochondrial dysfunction, lipid toxicity, inflammatory cascade, genetic susceptibility, epigenetic changes and disturbed intestinal flora [43]. Thus, LPO may aggravate ferroptosis. Recently, ferroptosis was suggested to play a critical role by effectively promoting and inhibiting ferroptosis-related factors in NAFLD and NASH mice [44, 45]. In NAFLD, this is mainly associated with the LPO reaction triggered by iron overload and oxidative stress.
Iron participates in multiple cellular processes, including oxygen storage and
transport, mitochondrial respiration, DNA replication, and intercellular
signaling in the body [46]. Intracellular iron balance considers iron absorption,
output, use and storage. Excess iron is mostly stored in ferritin, as a redox
inactive form. Generally, Fe
Hepatocytes are critical for liver iron homeostasis. Iron shortage in hepatocytes and excessive iron accumulation in hepatic stellate cells mediated by Kupffer cells-induced secretion of iron-rich extracellular vesicles (EVs), can lead to NAFLD and NASH. Meanwhile, blocking EV secretion or EV iron transport can restore iron balance in the liver and alleviate NAFLD/NASH-associated hepatic steatosis and fibrosis. A previous study revealed that the lack of hepcidin inhibitor matriptase-2 may alleviate obesity, accelerate lipolysis, improve glucose tolerance and insulin sensitivity, and prevent hepatic steatosis in high-fat diet mice [52]. This suggests that regulation of iron homeostasis in the liver has a significant correlation with anti-steatosis.
Oxidative stress describes an imbalance between oxidative and antioxidant events, where the generation of free radicals exceeds the capacity of antioxidant defense systems. Iron overload promotes the production of ROS, thereby inducing oxidative stress. Furthermore, oxidative stress-related transcription factors downregulate or upregulate ferroptosis-related genes, which regulates ferroptosis [53, 54, 55]. The “multiple-hit” hypothesis considers oxidative stress one of the key mechanisms underlying NAFLD-related liver injury. When ROS are excessively produced through the external environment (e.g., physical and chemical factors) and the internal environment (e.g., mitochondrial dysfunction, ER stress and peroxidation), they target the double bonds of PUFAs, resulting in LPO and the formation of lipid peroxides, which induces ferroptosis and aggravates hepatocyte injury. Recent evidence suggests mitochondrial ROS play a role in NASH-related ferroptosis. Epigallocatechin gallate prevents liver lipotoxicity by suppressing mitochondrial ROS-related liver ferroptosis [41]. Quercetin reduces lipid accumulation, hepatic lipotoxicity and LPO in high-fat diet mice, ultimately inhibiting ferroptosis to alleviate NAFLD [56]. Besides, obeticholic acid reduced the amounts of triglyceride-containing PUFAs, while elevating free PUFAs and phosphatidylethanolamine-containing PUFAs, which are prone to oxidization to generate lipid peroxides, triggering ferroptosis in hepatocytes and causing hepatic stellate cell activation [57]. Taken together, both oxidative stress and iron overload can induce the LPO process, and even synergically worsen the degree of cell damage, resulting in NAFLD progression from simple hepatic steatosis to inflammation and then fibrosis.
GSH is the main antioxidant in mammalian cells, which is composed of glutamate, cysteine and glycine, and represents a cofactor of GPX4 [58]. However, GSH depletion disrupts the dynamic balance of intracellular redox activity, which leads to ROS accumulation and aggravates ferroptosis [59, 60]. Cysteine is the main component and thus the limiting amino acid in GSH biosynthesis; suppressing cysteine import via the cystine/glutamate antiporter SLC7A11 induces ferroptosis via GSH depletion [61]. Therefore, GSH is often used as an evaluation indicator of oxidative stress, especially in studies developing drug therapies for ferroptosis-induced NAFLD.
GPX4 was the first discovered selenium-containing protein inhibiting ferroptosis. It is mainly found in the cytoplasm and mitochondria, and plays a role in scavenging lipid hydrogen peroxide. GPX4 can convert toxic lipid hydroperoxides into corresponding nontoxic lipid alcohols, which forms a defense mechanism against LPO and plays an important regulatory role in ferroptosis [62]. In methionine and choline deficient L-amino acid diet (MCD)-fed NAFLD mice treated with the ferroptosis inducer (1S,3R)-RSL3, liver GPX4 was downregulated while it was upregulated by lipoxygenase and an apoptosis inducer. Subsequently, supplementation of the GPX4 activator sodium nitrite increased GPX4 expression and reduced the severity of NASH. These results indicated that GPX4 plays a significant role in ferroptosis-related changes in hepatocytes that cause NASH [63]. Therefore, GPX4 regulation may control the development of NAFLD inflammation before it progresses to NASH. NAFLD rats were treated with ferroptosis inducer (erastin) and inhibitor (ferrostatin 1 [Fer-1]), and steatosis in human liver cell 7702 (L02) was induced by palmitic acid (PA), respectively. Both in cultured cells and experimental animals, interfered GPX4 expression decreased the protective effects on rat liver and mitochondrial membrane integrity in L02 cells, and also regulated B-cell lymphoma-2 (Bcl-2), Bcl-2 associated x (Bax), Caspase-3 and superoxide dismutase 1 (SOD1), attenuating the treatment effect in NAFLD. This study revealed that suppressing GPX4-related ferroptosis may provide a novel approach for NAFLD treatment [64].
The Xc
Nrf2 interacts with nuclear factors to activate downstream transcription, which
regulates antioxidative events and detoxification [72]. Nrf2 regulates multiple
biological events, e.g., the transcription of the antioxidant system, the
detoxification of endogenous and exogenous substances, NADPH regeneration and
heme metabolism, as well as many cell processes, including autophagy and unfolded
protein response [73]. Mounting evidence suggests a regulatory role for Nrf2 in
ferroptosis. Nrf2 constitutes an upstream effector of some essential
ferroptosis-related genes such as GPX4 and Xc
Besides, the Nrf2 contributes to reducing ROS overproduction to exert
anti-inflammatory effects for NAFLD. Indeed, a small interfering RNA (siRNA) of
Nrf2 or a glutathione peroxidase suppressor could abolish nuclear factor kappa-B
(NF-
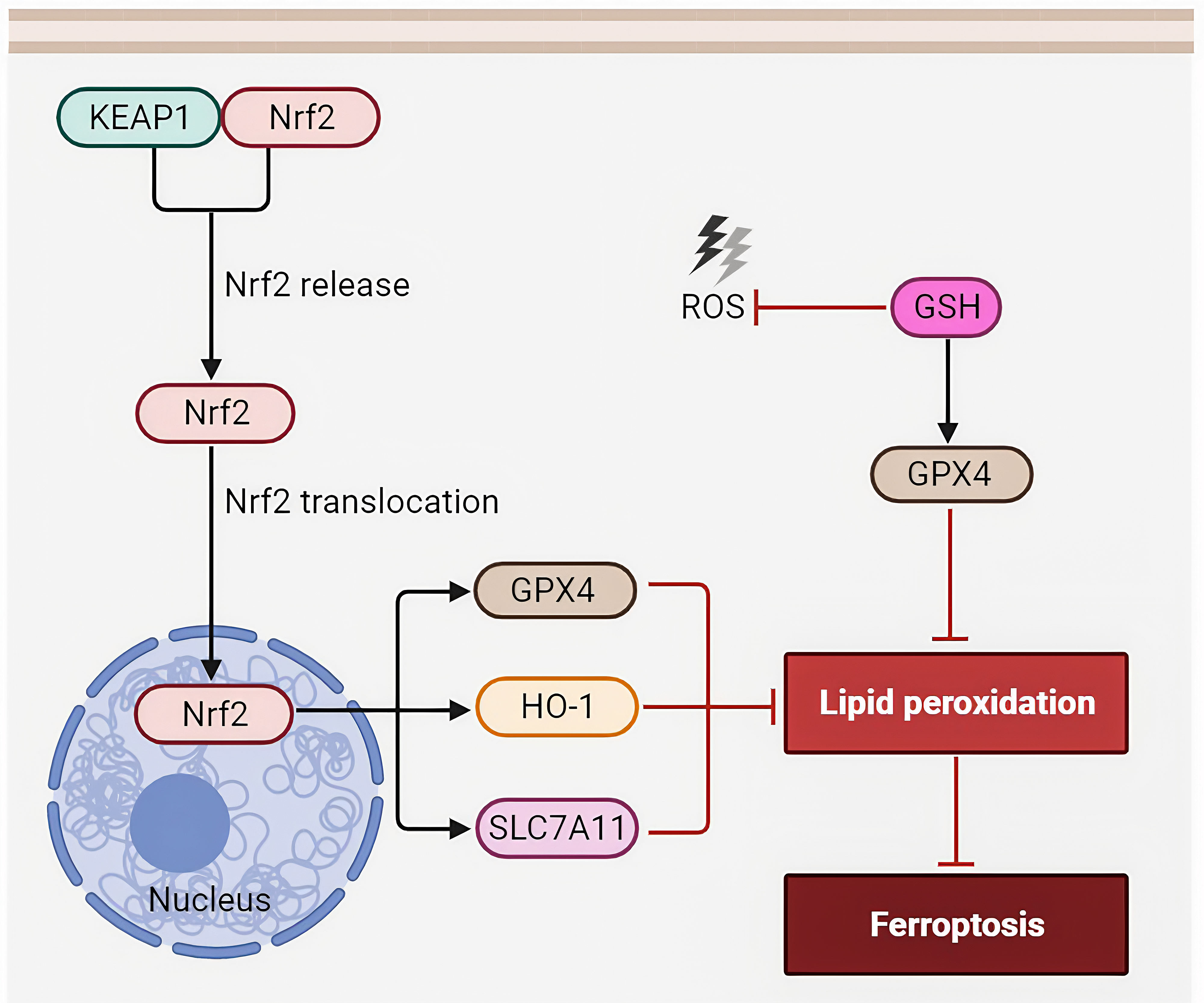 Fig. 2.
Fig. 2.KEAP1-Nrf2 pathway. When cells experience oxidative stress, the KEAP1-Nrf2 interaction is altered, preventing KEAP1 from targeting Nrf2 for degradation. As a result, Nrf2 is stabilized and accumulates in the cell. One of the key mechanisms by which Nrf2 contributes to protecting cells from ferroptosis is through the upregulation of genes involved in iron metabolism. Nrf2 induces the expression of GPX4, HO-1, and SLC7A11, releasing iron in a controlled manner. By regulating iron levels, Nrf2 helps prevent the accumulation of toxic levels of labile iron that can catalyze lipid peroxidation, a hallmark of ferroptosis. Abbreviations: KEAP1, Kelch-like ECH-associated protein 1; GPX4, glutathione peroxidase 4; HO-1, heme oxygenase-1; SLC7A11, subunit solute carrier family 7 member 11; Nrf2, nuclear factor erythroid 2-related factor 2.
In NAFLD, liver inflammation is a critical component of disease progression. FSP1 has been associated with the regulation of inflammatory responses in various cell types. Its involvement in immune cell migration and activation may influence the extent of liver inflammation in NAFLD [18]. FSP1 was initially identified as the homologous regulation factor of the mitochondrial pro-apoptotic protein apoptosis-inducing factor (AIF)/AIF mitochondrion-associated 1 (AIFM1), termed AIFM2. However, it was later confirmed that FSP1 lacks the N-terminal sequence for mitochondrial targeting, is not localized in mitochondria and does not promote apoptosis. It was then renamed by Bersuker et al. [23] as FSP1 and its cellular function was described. FSP1 is present in lipid droplets and cell membranes, and it acts as a suppressor of ferroptosis by reducing lipid peroxidation and protecting cells from oxidative damage. Mechanically, CoQ10 was identified as a major substrate using a ferroptosis activator [80]. FSP1 expression shows a positive correlation with resistance to GPX4 suppressors, and plays a critical role in maintaining cancer cell proliferation in case of no GPX4 expression. Notably, similar to FSP1, GCH1 was recently described as a new protein suppressing ferroptosis based on whole-genome activation screening [81]. GCH1 overexpression occurs in presence of all three ferroptosis activators, including RSL3, erastin and GPX4. High GCH1 amounts enhance BH4 and CoQ10 levels, synergistically acting with the FSP1 pathway to hamper the LPO process. GCH1 expression and resistance to ferroptosis are overtly correlated, suggesting the GCH1-BH4-phospholipid pathway as a potential therapeutic target in iron-related diseases.
Iron metabolism regulatory factors are essential proteins and molecules that play key roles in maintaining the balance of iron levels within cells and tissues. These factors are also known to have a close implications in NAFLD. Understanding its roles in NAFLD, such as Tfr1 and Ferritin, is crucial for elucidating the mechanisms underlying the disease and potentially identifying new therapeutic targets. They are tightly regulated by various iron metabolism regulatory factors that cells have access to adequate iron for their functions while preventing iron overload, which can lead to cellular damage and contribute to the development of iron-related NAFLD.
Transferrin receptor 1 (Tfr1), or cluster of differentiation 71 (CD71), interacts with transferrin and exerts an important effect on iron uptake by hepatocytes in a specific manner [82]. In palmitate-induced IR, palmitate simultaneously upregulates Tfr1 and induces intracellular ferroptosis. Knockdown of Tfr1 prevented palmitate-induced iron uptake and IR, and also translocated Tfr1 via enhancement of calcium influx. However, supplementation of a calcium chelator substantially decreased ferroptosis by suppressing Tfr1 translocation, and ultimately enhanced insulin sensitivity [83]. It was also found that the interaction between complexes was neutralized by antibodies against Tfr1, making Tfr1 a potential target for ferroptosis alleviation in NAFLD [84].
Excess iron in the body is stored in ferritin that plays a critical role in iron
metabolism and storage in cells. It is usually involved in ferritinophagy which
releases Fe
Recently, ferroptosis has attracted increasing attention among investigators studying liver pathologies and was shown to involve the regulation of NCOA4-mediated ferritinophagy [88]. There is a feedback mechanism behind intracellular iron utilization in which NCOA4-mediated ferritinophagy degrades ferritin, and its activation increases the amounts of available iron within the cell (Fig. 3). In terms of mechanism, the selective autophagy receptor NCOA4 interacts with ferritin heavy chain 1 (FTH1) and mediates ferritin transfer to autolysosomes, eventually releasing free iron. Such regulatory effect is not unidirectional, as intracellular iron levels also affect the flux of ferritinophagy [89]. In case of high iron content, HECT and RLD domain containing E3 ubiquitin protein ligase 2 (HERC2), an enzyme 3 (E3) ubiquitin ligase, drives ubiquitin-dependent degradation of NCOA4. This process reduces the ferritinophagy flux, thereby maintaining iron homeostasis [90]. It is noteworthy that NCOA4 level is a central determinant of the ferritinophagy flux. In the non-disease state, ferritinophagy maintains cellular iron balance, but its overactivation can cause iron overload within cells. LPO, which is correlated with iron overload, is an important factor required for ferroptosis [89]. The above findings jointly suggest a close relation between ferritinophagy and ferroptosis. A study confirmed GPX4 suppresses ferroptosis and shows elevated expression in NCOA4-knockout mice, revealing an association of ferroptosis with ferritinophagy [91]. Additionally, several studies have suggested that ferritinophagy promotes ferroptosis, primarily because of iron overload resulting from NCOA4 upregulation. Sorafenib is an iron-dependent inducer of ferroptosis, whose potential mechanism involves increasing NCOA4 expression to accelerate iron overload and ROS generation [92]. Moreover, elevated iron content induced by NCOA4 overexpression increases sensitivity to ferroptosis. Conversely, ferritinophagy protects from ferroptosis. In case of NCOA4 deficiency or obstructed NCOA4 binding to FTH1, ferritinophagy is hindered, which may decrease sensitivity to ferroptosis within cells [32]. Furthermore, NCOA4 knockout blocks sideroflexin 1-mediated mitochondrial iron overload, thereby inhibiting ferroptosis [93]. Taken together, these data indicate iron overload represents the initial step of ferritinophagy-mediated ferroptosis. The major factor in ferritinophagy regulation is to influence its core molecule NCOA4 by controlling its expression or preventing its interaction with ferritin.
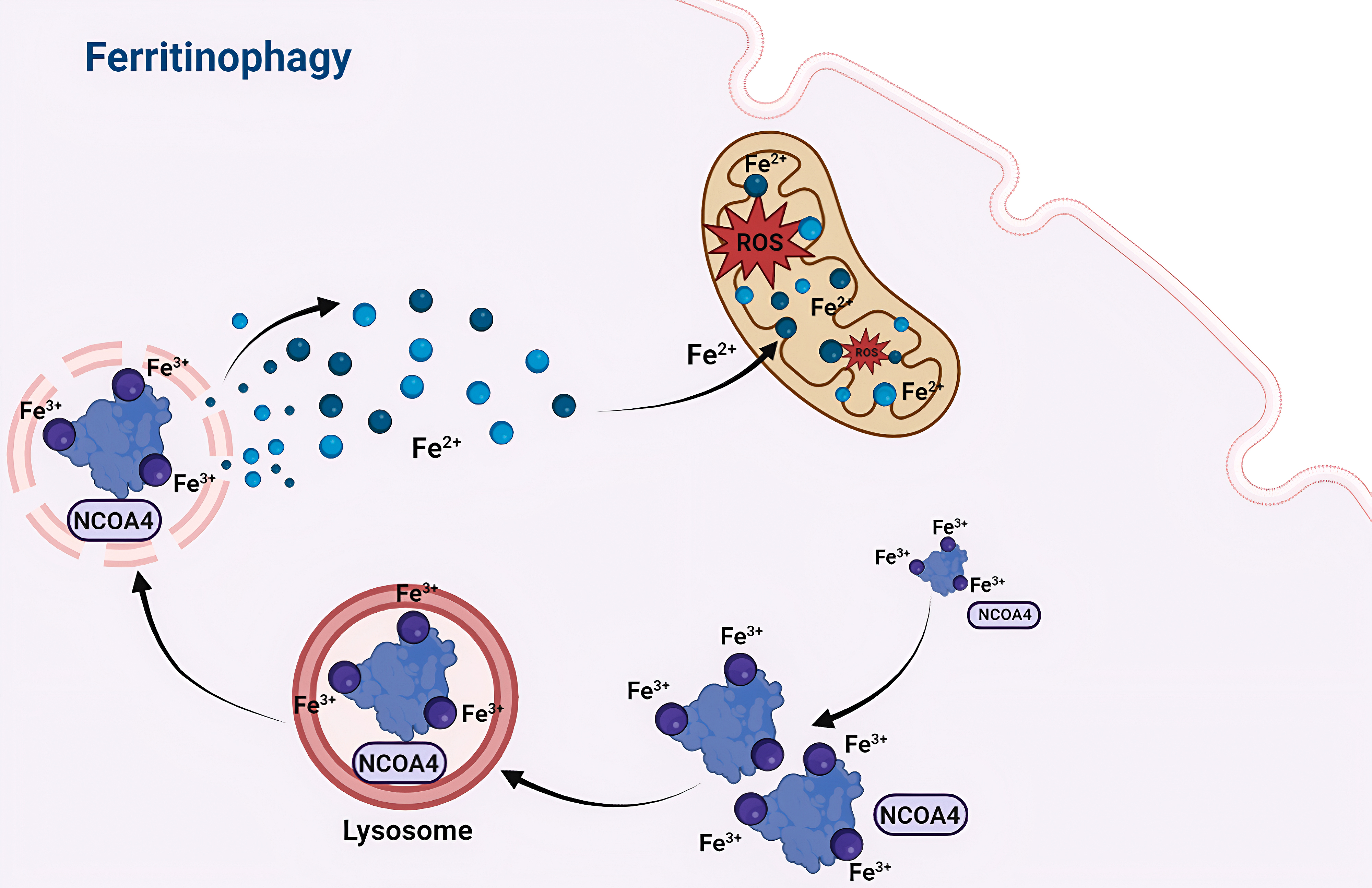 Fig. 3.
Fig. 3.Mechanism of ferritinophagy. NCOA4-mediated ferritinophagy is a selective autophagic process. NCOA4 acts as a selective cargo receptor, specifically recognizing ferritin and targeting it to autophagosomes for degradation in lysosomes. Through ferritinophagy, intracellular ferritin is broken down, releasing free iron, which can contribute to the initiation of ferroptosis. Abbreviations: NCOA4, nuclear receptor coactivator 4.
Overall, NCOA4-dependent ferritinophagy exerts a crucial regulatory effect on ferroptosis by modulating iron homeostasis in cells and controlling ROS biosynthesis. Further research is necessary to investigate the pathways by which ferritinophagy modulates ferroptosis and to develop drugs that may target ferritinophagy as a therapeutic strategy to ameliorate the pathological processes associated with ferroptosis in various diseases.
Agents selectively regulating ferroptosis (inhibitors or inducers) are crucial for the mechanistic studies of ferroptosis-dependent NAFLD and other disorders. Table 1 (Ref. [23, 45, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105]) and Table 2 (Ref. [67, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126]) list the best-studied and most widely-applied inhibitors and inducers of ferroptosis, respectively.
| Compound | Possible mechanisms | Functions and diseases |
| Deferiprone | As an iron chelating agent, has affinity with Fe |
Alleviates NASH and fibrosis [45] |
| Ferrostatin-1 | Prevents lipid ROS accumulation and inhibits LPO, constituting a lipid ROS scavenger | Alleviates NAFLD/NASH [94] |
| Ginkgolide B | Promotes GPX4 and FTH1 expression, and reduces TFR1 expression, ROS levels, and intracellular iron content | Improves fibrosis and diabetic nephropathy [95] |
| Trolox | Damps lipoxygenase and abrogates hydroxyl group radicals, with potent antioxidant properties | Revealed an association between blood redox status and the likelihood of NAFLD [96] |
| Liproxstatin-1 | Functions as a radical-trapping antioxidant to inhibit lipid peroxidation | Ameliorates NAFLD and acute renal failure [45, 97, 98] |
| Selenium | Increases the expression of GPX4 | Affects the progression of NASH [99] |
| CoQ10 | Acts as a lipophilic radical-trapping antioxidant to prevent the generation of lipid peroxides | Exerts hepatoprotective effects and is beneficial in NAFLD; sensitizes malignant cells to ferroptosis-activating chemotherapeutics [23, 100] |
| Baicalein | Targets lipoxygenases and suppresses lipoxygenase-triggered LPO | Exerts pharmacological effects in bladder cancer, vitiligo and ischemia-reperfusion injury [101, 102, 103] |
| XJB-5-131 | Exerts antioxidant effects and suppresses LPO | Improves ischemia-reperfusion injury and acute kidney injury [104, 105] |
NASH, nonalcoholic steatohepatitis; NAFLD, nonalcoholic fatty liver disease; LPO, lipid peroxidation; CoQ10, coenzyme Q10; GPX4, glutathione peroxidase 4; FTH1, ferritin heavy chain 1.
| Compound | Possible mechanisms | Functions and diseases |
| RSL-3 | Suppresses GPX4 and induces the accumulation of lipid hydroperoxides | Aggravates gastric cancer, hepatocellular carcinoma, triple-negative breast cancer, and colorectal cancer [106, 107, 108, 109] |
| High-fat diet | Promotes iron overload and oxidative stress, and inhibits the expression of GPX4 and SLC7A11 | Aggravates NASH [110] |
| Erastin | Targets the Xc |
Weakens therapeutic effects in NAFLD and enhances the growth of tumors, e.g., melanoma and triple negative breast cancer [111, 112] |
| Sorafenib | Suppresses cystine uptake through the Xc |
Ameliorates hepatocellular carcinoma and murine liver fibrosis [67, 113] |
| Sulfasalazine | Suppresses cystine uptake through the Xc |
Improves mesenchymal traits and disabled antioxidant program in head and neck cancer, hepatocellular carcinoma and pancreatic cancer [114, 115] |
| Glutamate | High concentrations of glutamate inhibit cystine uptake by the Xc |
Improves non-small cell lung cancer [116] |
| ML-162 | Suppresses GPX4 and causes disorders of lipid hydroperoxides | Improves head and neck cancer [117] |
| FIN 56 | Decreases GPX4 expression, activates squalene synthase and suppresses CoQ10 | Exerts anticancer effects in human fibrosarcoma and head and neck cancer [117, 118] |
| Dihydro-artemisinin | Decreases GPX4 expression | Increases sensitivity to ferroptosis in cancer cells [119] |
| Ferric ammonium citrate | Causes iron overload and LPO | Protects against iron overload-associated knee osteoarthritis and osteoblast differentiation [120, 121] |
| Trigonelline | Inhibits Nrf2 | Reduces blood glucose and kidney injury in diabetes, and renders cancer cells more susceptible to apoptosis [122, 123] |
| FINO(2) | Suppresses GPX4-related enzymatic activity and oxidizes iron, and causes LPO | Exerts anticancer effects in human fibrosarcoma [124] |
| Brequinar | Inhibits DHODH and thus decreases ubiquinone reduction to ubiquinol | Suppresses renal, cervical and nasopharyngeal cancer growth [125, 126] |
RSL-3, (1S,3R)-RSL3; SLC7A11, subunit solute carrier family 7 member 11; DHODH, dihydroorotate dehydrogenase.
Ferroptosis plays a crucial role in liver pathophysiology and is also a key factor in initiating the progression from NAFLD to NASH. Therefore, it holds great promise as a potential therapeutic target for preventing the development of NAFLD. While the existing data strongly suggest that targeted ferroptosis could be an excellent approach for treating NAFLD, it is essential to acknowledge the current gaps in research on this topic. To facilitate its understanding and potential applications, certain issues should be addressed in the future. These mainly include: (1) elucidating the mechanisms: further researches are needed to elucidate deeper and detailed molecular mechanism that are aiming to pave the way for more effective and targeted therapeutic interventions; (2) identifying biomarkers: in the context of NAFLD, the identification of reliable biomarkers associated with ferroptosis in NAFLD is essential for early detection and diagnosis; (3) addressing specific subtypes: investigating how ferroptosis contributes to the progression of different NAFLD subtypes will aid in tailoring treatments to specific patient populations; (4) investigating combination therapies: considering wide spectrum in NAFLD, exploring the combinations involving ferroptosis-targeting agents along with other existing treatments may offer a more comprehensive approach to management. By addressing the aforementioned gaps, we may have the opportunity to fully unleash the potential of targeted ferroptosis as a powerful strategy for effectively preventing and treating NAFLD.
Ferroptosis represents a newly discovered form of cell death associated with
iron-dependent LPO. Currently, studies exploring ferroptosis mechanistically in
human health are mainly focused on cancer, and based on reported mechanisms, new
targets for developing anti-cancer drugs have been discovered. Summarizing recent
studies on the pathways and defense mechanisms associated with ferroptosis,
accumulating evidence indicates a certain correlation among the pathological
processes of NAFLD such as lipid accumulation, hepatic steatosis, inflammatory
cell infiltration, fibrosis, mitochondrial damage and ferroptosis. Iron overload
and oxidative stress-induced LPO may mediate the progression of NAFLD, and the
main antioxidant mechanism and regulatory factors controlling iron metabolism may
become potential targets for NAFLD-related research. Among them, targeting Nrf2
activation can cause SLC7A11 to accelerate the transport of cysteine and
glutamate through the Xc
With multiple potential targets and overarching approaches for regulating ferroptosis emerging from recent reports, ferritinophagy intervention could pave a novel way for ferroptosis regulation. Ferritinophagy-mediated ferroptosis is a potential target for preventing the onset of NAFLD. Therefore, it is necessary to also take ferritinophagy into account in strategies for NAFLD therapy. The concept of ferroptosis has enormous research potential and clinical value in the field of NAFLD. This review provides new insights into ferroptosis relevant to the pathogenesis of NAFLD. However, more detailed mechanism of action for ferroptosis in NAFLD still needs further investigation.
4-HNE, 4-hydroxynonenal; ACSL4, acyl-CoA synthetase long chain family member 4;
Bmp6, bone morphogenetic protein 6; CD71, cluster of differentiation 71; CoQ10,
coenzyme Q10; ECM, extracellular matrix; ER, endoplasmic reticulum; ETC, electron
transport chain; EVs, extracellular vesicles; Fer-1, ferrostatin 1; FSP1,
ferroptosis suppressor protein 1; FTH1, ferritin heavy chain 1; GCH1, GTP
cyclohydrolase 1; HO-1, heme oxygenase-1; IR, insulin resistance; GSH,
glutathione; KEAP1, Kelch-like ECH-associated protein 1; LPCAT3,
lysophosphatidylcholine acyltransferase 3; LPO, lipid peroxidation; MDA,
malondialdehyde; MVBs, multivesicular bodies; NAFLD, nonalcoholic fatty liver
disease; NASH, nonalcoholic steatohepatitis; NCOA4, nuclear receptor coactivator
4; Nrf2, nuclear factor erythroid 2-related factor 2; PA, palmitic acid;
PL
SNZ and XZY designed the study. SNZ, YG and XZY collected and analyzed the literatures. SNZ and XZY wrote the manuscript. YG and XZY discussed and critically revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the Science and Technology Development Plan of Jilin Province (20210204029YY and YDZJ202201ZYTS151).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.