
- Academic Editor
†These authors contributed equally.
Background: The mortality rate of colorectal cancer (CRC) ranks second
worldwide. Previous research had indicated that licochalcone A (LA) was a
flavonoid in licorice with diverse anticancer effects. We explored the underlying
mechanisms of LA-triggered anticancer activity in CRC. Methods: Thiazolyl Blue (MTT)
experiment and EdU staining were utilized to evaluate cell proliferation.
Meanwhile, cells were stained by Annexin V/PI to investigate apoptosis through
flow cytometry assay. Moreover, expressions of proteins were detected by
immunoblotting, and the level of related mRNA was investigated using real-time
quantitative PCR. Results: LA selectively suppressed the proliferation
and triggered apoptosis of CRC cells. Strikingly, LA induced cytoprotective
autophagic activities since the suppression of autophagy significantly
strengthened LA-induced cytotoxicity and FLICE inhibitory protein (c-FLIP
According to global cancer data for 2020, colorectal cancer (CRC) had the second-highest mortality rate and accounted for approximately 9% of all cancer-related deaths each year worldwide [1]. It was reported that the incidence of CRC worldwide was expected to rise to about 2.5 million in developing countries until 2035 [2]. Diagnosing CRC is difficult because its symptoms appear only at a late stage, and its mechanisms are elusive. The five-year relative survival rate in low-income countries remained below 50% [3, 4, 5]. The drug resistance of anticancer drugs is becoming gradually serious because of the complexity of the tumorigenesis mechanism. The search for novel anticancer drugs with superior anticancer effects and novel mechanisms has become one of the key targets for researchers.
In recent years, the powerful anticancer effects and low side effects of herbal extracts have attracted the attention of medical researchers worldwide. Our previous studies have demonstrated that Chalcones were a promising class of anticancer agents [6, 7, 8, 9]. Licochalcone A (LA) is a chalcone in licorice with effective antitumor activities [10, 11, 12, 13, 14, 15], and the poor bioavailability of LA was solved by loading on liposome carriers [16]. It was reported that LA induced autophagy by activating mitogen-activated protein kinase (MAPKs), including extracellular signal-regulated protein kinases (ERK), p38, and c-Jun N-terminal kinase (JNK), in lung cancer [9, 17, 18, 19]. However, the effects and mechanisms of MAPKs and autophagy in LA-induced anti-CRC activities remain elusive.
During our research, we found that autophagy could maintain the expression of
FLICE inhibitory protein (c-FLIP
In our research, LA was found to restrain cell viability and trigger cell
apoptosis in CRC cells. However, LA triggered cytoprotective autophagy in CRC
cells. This autophagy enhanced Hsp70 expression, maintaining the expression of
its downstream c-FLIP
Antibodies for phosphor-p38 MAPK (4511), PARP (9542), and LC3 (4108) were from
Cell Signaling Technology (Boston, Massachusetts, USA). Antibody for HSPA4
(381501), JNK1 (201001), P44/42 MAPK (ERK1/2) (250222), p38 (220979), Hsp90 alpha
(209902), GAPDH (200306-7E4),
NCM460 of normal human colonic epithelial and HT29 cell line was from JENNIO
Biological Technology (China). Colon cancer cell lines SW620 and HCT116 were
purchased from Procell Life Science & Technology Co., Ltd. (China). NCM460 grew
in RPIM 1640 mediums. SW620 cells grew in DMEM mediums. HT29 and HCT116 cells
grew in MCCOY’S 5A mediums. These mediums were added with fetal bovine serum
(FBS) (1:10), L-glutamine (2 mmol/L), penicillin (100 U/mL), and streptomycin
(100 µg/mL). All cells were held at 37 °C with 5% CO
Thiazolyl Blue (MTT) experiment was used to investigate the cytotoxic activities
of LA. CRC cells were divided into three parts and plated in 48-well plates (2.5
The proliferation effects of LA on SW620 cells were investigated with a colony
formation experiment. CRC cell line was grown in six-well plates (2
CRC cell line gown in 48-well plates, next, exposed to DMSO (0.1%) or LA for 8 h at 37 °C. Then, discarded the old culture medium from the 48-well plates and added 200 µL medium containing LA and EdU (medium: EdU solution = 1000:1) to each well for 2 h. Cells were treated with 2 mg/mL glycine and 0.5% TritonX-100 after being fixed with 4% paraformaldehyde. Next, DNA syntheses were measured by the EdU Apollo488 Imaging Kit according to recommendations (RiboBio, Guangzhou, China).
Cells were grown in 48-well plates and, next, cultured with CQ, U0126, and LA for 8 h at 37 °C. Then, cells were treated with Methanol precooled at –20 °C and were exposed to Triton X-100 diluted in PBST (1:200) at 20 °C. After 30 min, they were treated with BSA (1:20 diluted in PBST) for 1 h and held at 4 °C for 12 h with an antibody against LC3 (1:200 diluted in PBST). Then, cells were held with an anti-rabbit antibody which was conjugated to Alexa Fluor 594. After 1 h, DAPI was used to stain the nucleus for 5 min, and cells were examined with a fluorescence microscope instrument (Olympus CKX41, Japan) immediately.
CRC cell lines were grown in 6-well plates (6
Apoptotic cell death of CRC was measured by Annexin V-FITC Apoptosis Detection Kit (BD Biosciences). CRC cell line grown in 6-well plates. Then, they were grown with DMSO (0.1%) or 20 µM LA. After 48 h, cells were harvested after being treated with 0.25% Trypsin. Next, the binding buffer with PI (5 µL) and Annexin V (FITC-conjugated) (5 µL) was used to treat cells for 15 min in the dark at room temperature. After that, the Accuri C6 flow cytometry instrument (BD Biosciences) was utilized to detect cell apoptosis. The percentages of AnnexinV-FITC/7AAD stained cells were analyzed by Flowjo 10.0 software (Tree Star, San Carlos, California, USA). Experiments were repeated at least three times, and representative results are shown in each figure.
All RNA was separated from CRC cells by Trizol (Ambion, Waltham, Massachusetts, USA). The process of quantification includes two reactions. The first reaction is reverse transcription which used FastKing RT Kit (gDNase) (TIANGEN, Beijing, China) in a reaction system of 20 µL. Reactions were carried out in a C1000 TouchTM Thermal Cycler (BIO-RAD, Hercules, California, USA) at 42 °C for 15 min, then heat inactivation at 95 °C for 3 min. After that, the reverse transcription reaction system was mixed with water without nuclease and held at –80 °C.
The second reaction is Real-time PCR which was carried out using rotorgene Q
2plex HRM (QIAGEN, Hilden, Germany) with SuperReal PreMix Plus (SYBR Green)
(TIANGEN) with a 20 µL PCR reaction system. Each sample was divided into
three parts to run and analyze. After the PCR cycle reaction, an analysis of the
melting curve was carried out to prove the specificity of PCR products. In
addition, primer sequences of c-FLIP
| Genes name | 5′-3′ primer | 3′-5′ primer |
| c-FLIP |
GCAGTCTCACAGCTCACCAT | GTGCTGCAGCCAGACATAA |
| Hsp70 | GAAGCCTGTAGTTGACTGTGTTGTTTC | AATCGCAAGCAATTAAGACCAGCAATC |
| Hsp90 | AACCCTGACCATTCCATTATTGAGACC | GCAAGATGACCAGATCCTTCACAGAC |
Radio immunoprecipitation assay buffer was used to lyse cells. Then, whole cell lysates were centrifugated, and the supernatant fraction was collected for immunoblotting. Protein concentrations of supernatant fraction were investigated by the bicinchoninic acid (BCA) experiment. Then, proteins were incubated at 100 °C for 10 min. After that, equal proteins were extracted from the lytic sample, then distributed by SDS-PAGE. Afterward, protein molecules were metastasized to polyvinylidene difluoride membrane in an electric field. After that, it was exposed to tris buffered saline with Tween-20 (TBST), including non-fat milk powder (1:20). After 2 h; the membrane was exposed to primary antibodies with specificity at 4 °C for 12 h. The next day, it was exposed to secondary antibodies (horseradish peroxidase-conjugated). Immunoreactivities were investigated by the enhanced chemiluminescence (ECL) system (Millipore). Each experiment was carried out 3 times; then representative consequences were shown.
CRC cell lines were grown in 12-well plates (3
All statistics were shown as mean
To interrogate the anticancer effects of LA in CRC in vitro, SW620, HCT116, HT29, and normal colonic epithelial cell line NCM460 grown with diverse thicknesses of LA for 48 h, and next cell viabilities were investigated by MTT experiment. Then, LA was proved to suppress the viability of CRC cells (Fig. 1A). The 50% inhibition concentrations (IC50) for 48 h of LA treatment were 26.4 µM, 30.6 µM, 41.5 µM, and 89.4 µM for the SW620, HCT116, HT29, and NCM460 cells respectively, indicating that LA selectively inhibited CRC cells. It seems that KRAS-mutated CRC cells (SW620 and HCT116) were more sensitive to LA compared with KRAS wild HT29 cells. Similarly, SW620 cells were exposed to diverse thicknesses of LA, then the cell viabilities were detected by MTT experiment at 24, 48, 72, and 96 h (Fig. 1B). Then, LA was proved attenuated the viabilities of CRC cells dose- and time-dependently. Consistently, colony formation assay demonstrated that LA inhibited single CRC cells proliferation (Fig. 1C), and EdU staining showed that LA inhibited DNA synthesis of CRC cells (Fig. 1D). Moreover, LA-induced cell viability inhibition was significantly enhanced under starvation stimulation, indicating that LA might effectively inhibit CRC cells under stress conditions (Fig. 1E).
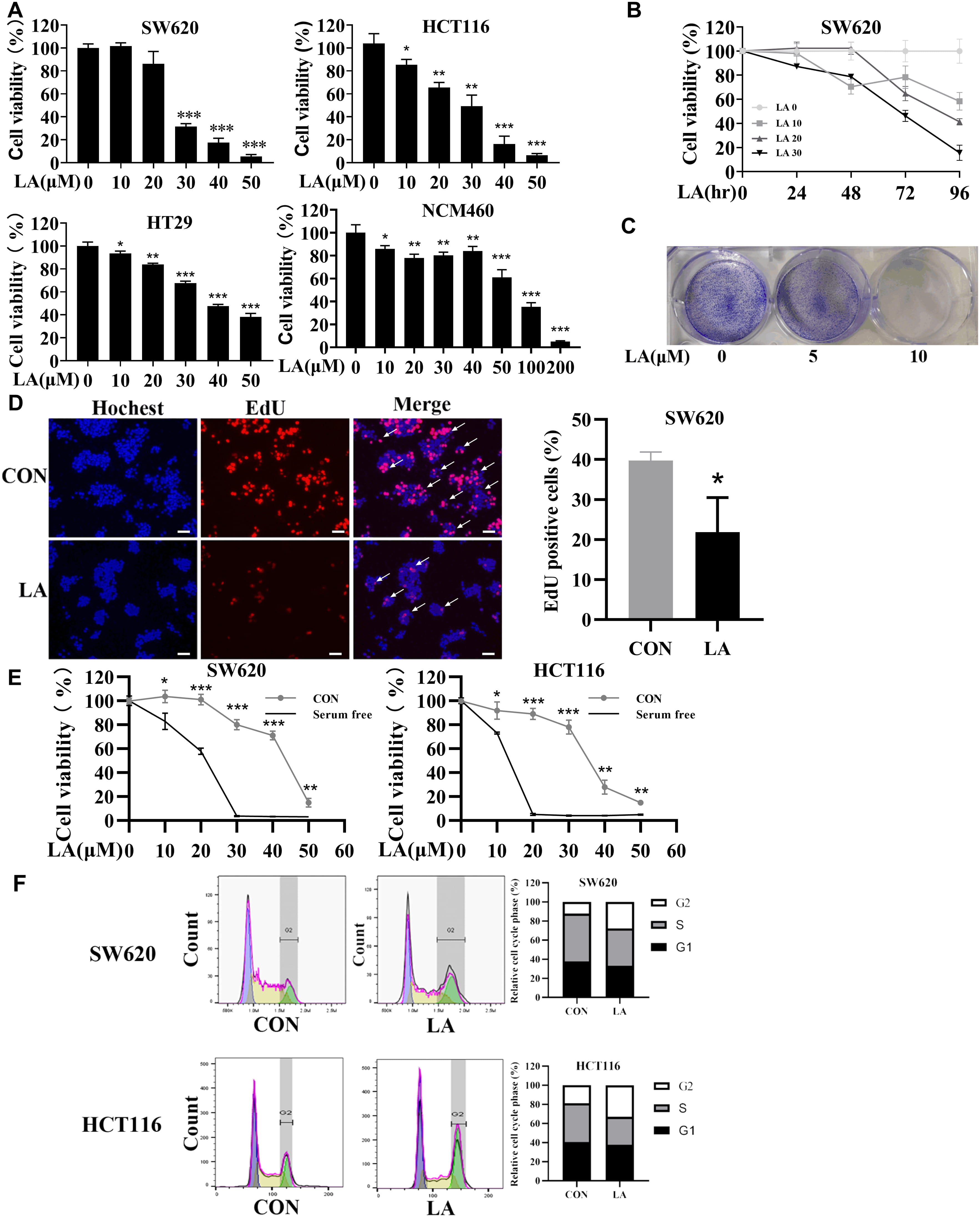 Fig. 1.
Fig. 1.The suppression of licochalcone A (LA) on colorectal cancer (CRC) cell proliferation is significantly
greater than its toxicity to normal cells. (A) different thickness of LA was
used to treat SW620, HCT116, HT29 and NCM460 cells. After 48 h, cell
proliferations were detected by Thiazolyl Blue (MTT) experiment. (B) SW620 cells were cultured
with diverse thickness LA, and then cell viability was tested by MTT experiment
at 24, 48, 72, and 96 h. (C) SW620 cells were cultured with LA (0, 5, and 10
µM) for 7 days; then crystal violet staining was carried out. (D) SW620
cells were cultured with 20 µM LA for 8 h; next, EdU staining was
performed. Scale bar 100 µm. (E) SW620 and HCT116 were cultured with
LA (0, 10, 20, 30, 40, and 50 µM) for 48 h in cultures with serum or not,
and then cell viabilities were measured by MTT experiment. (F) SW620 and HCT116
cells were cultured with LA (20 µM); next, flow cytometric assay was
utilized to examine the cell cycle at 8 h. N = 3. *p
To further investigate the effects of LA in CRC, we detected the cell cycle under LA-induced anticancer activity of SW620 and HCT116 cells. Flow cytometry analysis discovered that significant increase (from 12.1% to 26.5% in SW620 cells and from 18.9% to 33.1% in HCT116 cells) in the ratio of G2 phase in cells after being treated with LA, indicating that the CRC cells cycle was arrest in G2/M phase by LA (Fig. 1F). In summary, LA selectively suppressed CRC cells by inducing arrest in G2/M cell cycle.
Next, we investigated the relationship of apoptosis in LA-triggered cell death by Annexin V/PI experiment. Flow cytometric results indicated that LA enhanced apoptosis of CRC cells (Fig. 2A). Meanwhile, LA greatly augmented the level of hallmark proteins of apoptosis (cleaved caspase-3 and PARP) (Fig. 2B). Furthermore, pan-caspase inhibitor (z-VAD-fmk) greatly compromised LA-triggered cytotoxicity (Fig. 2C), further supporting that LA induced cell apoptosis of CRC.
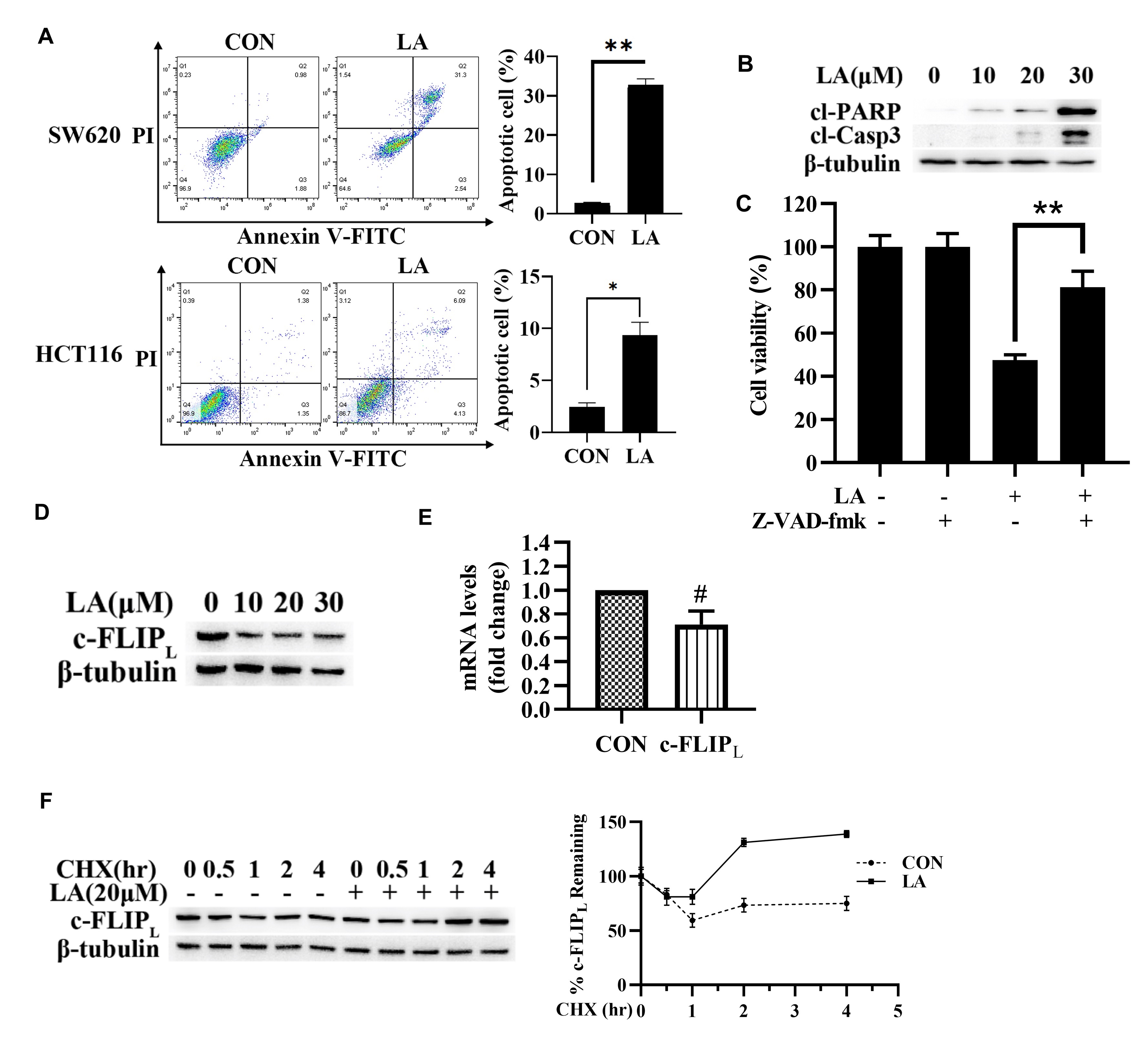 Fig. 2.
Fig. 2.LA triggered CRC cell apoptosis. (A) SW620 and HCT116 cell
lines were cultured in 20 µM LA or DMSO, then flow cytometric assay was
utilized to detect apoptotic cells at 48 h. (B) SW620 cell line grown with
diverse thicknesses of LA. Next, the level of proteins was detected using
immunoblotting at 48 h. (C) SW620 cell line was pretreated with 20 µM
z-VAD-fmk. After 1 h, cells were cultured with 20 µM LA. Next, cell
viabilities were measured using an MTT experiment at 48 h. (D) SW620 cells were
grown with diverse thicknesses of LA; next, the level of c-FLIP
Recently, there was growing evidence that c-FLIP
MAPKs are associated with LA-triggered anticancer effects. However, the roles of
MAPKs are elusive in CRC. Thereby, we explored mechanisms of MAPKs in LA-induced
anticancer activity. Then, LA was found indeed to activate ERK and p38
time-dependently in SW620 and HCT116 cells. Whereas the impact of LA on JNK
activity was not obvious in SW620 and HCT116 cells (Fig. 3A). After that, we
interrogated the role of ERK and p38 in LA-triggered anticancer activity. The
results discovered that ERK inhibitor U0126, not p38 inhibitor SB203580,
remarkably promoted LA-induced inhibition of SW620 cell viability (Fig. 3B),
indicating that ERK activation resisted LA-triggered anticancer activities in
SW620 cells, not HCT116 cells. Therefore, we explored the role of ERK in the
LA-induced apoptosis of SW620 cells. The results showed that U0126 enhanced cell
apoptosis (Fig. 3C), augmented cl-caspase-3 and cl-PARP expression, and further
promoted the downregulation of c-FLIP
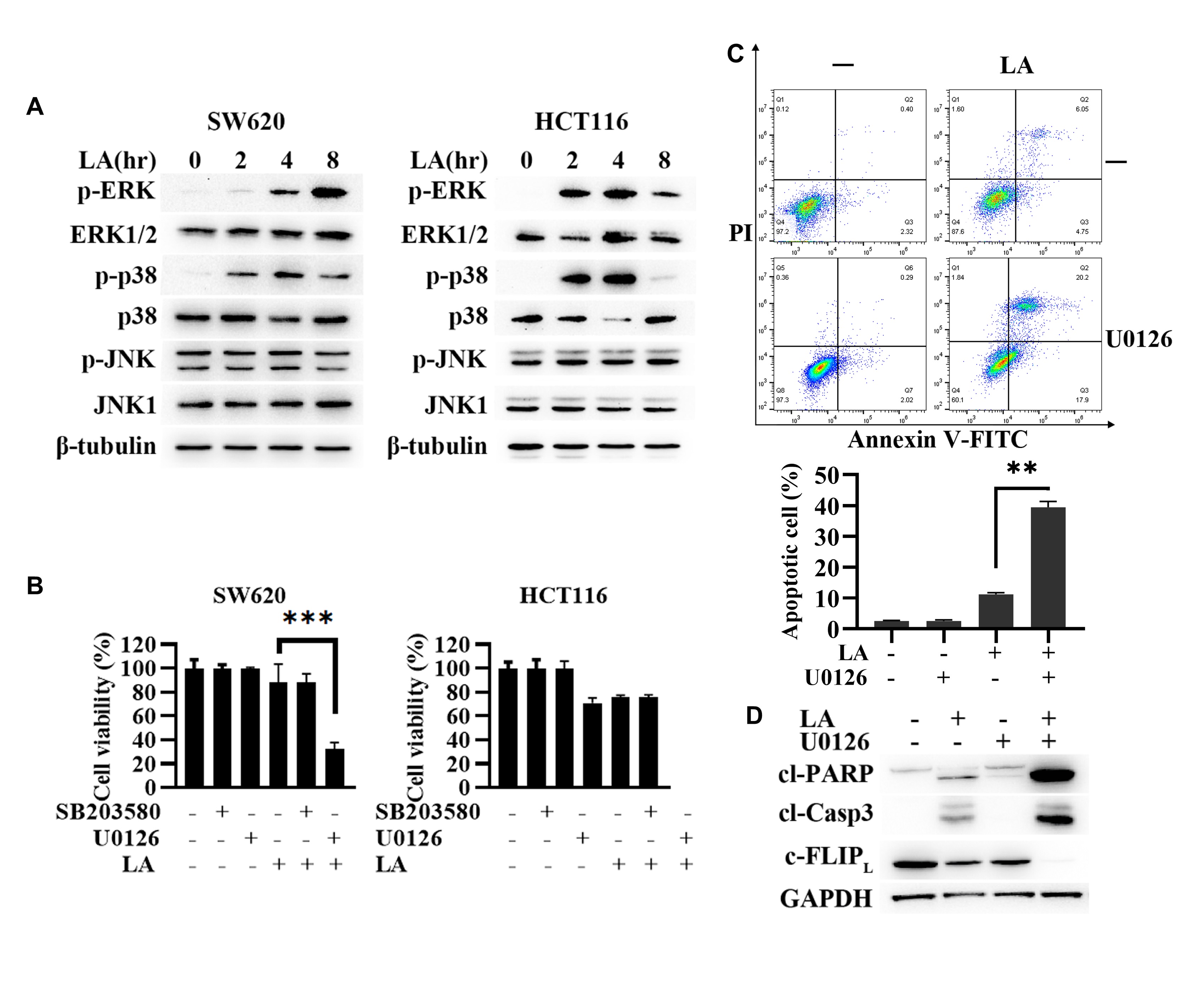 Fig. 3.
Fig. 3.ERK-mediated autophagic induction resisted LA-triggered anti-CRC
activities. (A) SW620 and HCT116 cells were cultured with 20 µM LA; next,
the level of proteins was measured by immunoblotting at different times. (B)
SW620 and HCT116 cell line was pretreated with 20 µM U0126 and 20 µM
SB203580. After 1 h, it was cultured with 20 µM LA for 48 h. After that,
the viability of CRC cells was tested by MTT experiment. (C and D) SW620 cell
line was cultured with 20 µM U0126. After 1 h, it was cultured with 20
µM LA; the apoptotic cell death was tested using flow cytometry (C), and
the level of proteins was measured by immunoblotting (D) at 48 h.
Previous studies have demonstrated LA induced autophagy in diverse types of
cancers [13, 14, 15, 17, 19]. Whereas mechanisms about autophagy in LA-triggered
anticancer activity were controversial. Therefore, we investigated mechanisms of
autophagic induction in LA-triggered anti-CRC effects. Then, LA was discovered to
induce LC3-II accumulation (Fig. 4A), indicating LA might increase autophagic
activities in SW620 cells. These results were further supported by the autophagic
flux assay (Fig. 4B). Consistently, immunofluorescence staining also discovered
that CQ blocked LA-induced autophagic flux (Fig. 4C). To uncover the role of
autophagic induction in LA-triggered anticancer effects, CQ was used to block the
activity of autophagy. As shown in Fig. 4D and 4E, CQ significantly augmented
LA-triggered apoptosis of CRC cells and enhanced the level of cl-caspase-3 and
cl-PARP, meanwhile downregulated the level of c-FLIP
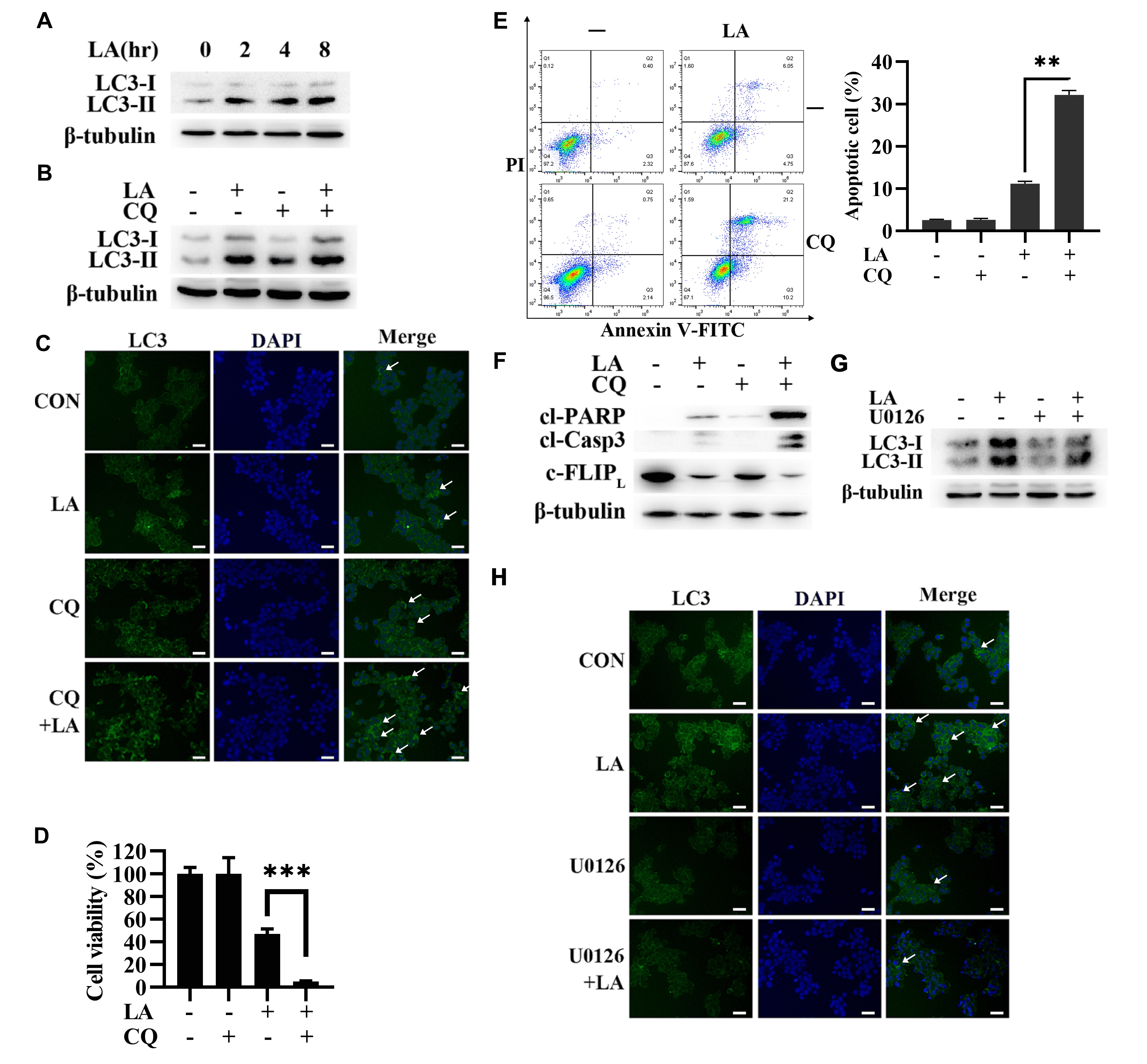 Fig. 4.
Fig. 4.Autophagy played a protective part for CRC cells in LA-triggered
anticancer activities. (A) SW620 cells were cultured with 20 µM LA at
different times; next, LC3 content was detected by immunoblotting. (B) SW620 cell
line grown with 20 µM chloroquine (CQ). After 1 h, cells were cultured with 20 µM
LA, then the level of LC3 was detected by immunoblotting at 8 h. (C) SW620 cell
line grown with 20 µM CQ. After 1 h, cultured with 20 µM LA, then the
level of LC3 was measured by immunofluorescence experiment at 8 h. Scale bar 100
µm. (D, E, and F) SW620 cell line grown with 20 µM CQ. After 1
h, cultured with 20 µM LA, then the viability of CRC cells was tested by
MTT experiment (D), the apoptotic cell death was determined with flow cytometric
assay (E), and the level of protein was determined by immunoblotting (F) at 48 h.
(G and H) SW620 cell line was cultured with 10 µM U0126. After 1 h, it was
cultured with 20 µM LA. After that, the level of LC3 was detected by
immunoblotting (G) and immunofluorescence experiment (H) at 8 h. Scale bar 100
µm.
Thereby, we explored the relationship between ERK and autophagy in LA-triggered anticancer effects. The Western blot (Fig. 4G) and immunofluorescence staining (Fig. 4H) results showed that U0126 suppressed LA-induced LC3-Ⅱ accumulation in SW620 cells, which indicated that LA triggered autophagic induction by ERK activation. Therefore, autophagy activation mediated by ERK played a protective part in LA-triggered anti-CRC effects.
HSPs have been reported to inhibit apoptosis by assisting in the synthesis of
apoptosis-related proteins [23]. Therefore, we detected the expression of Hsp70,
Hsp90, HSPA4 (Hsp70 Family Member), and Hsp90
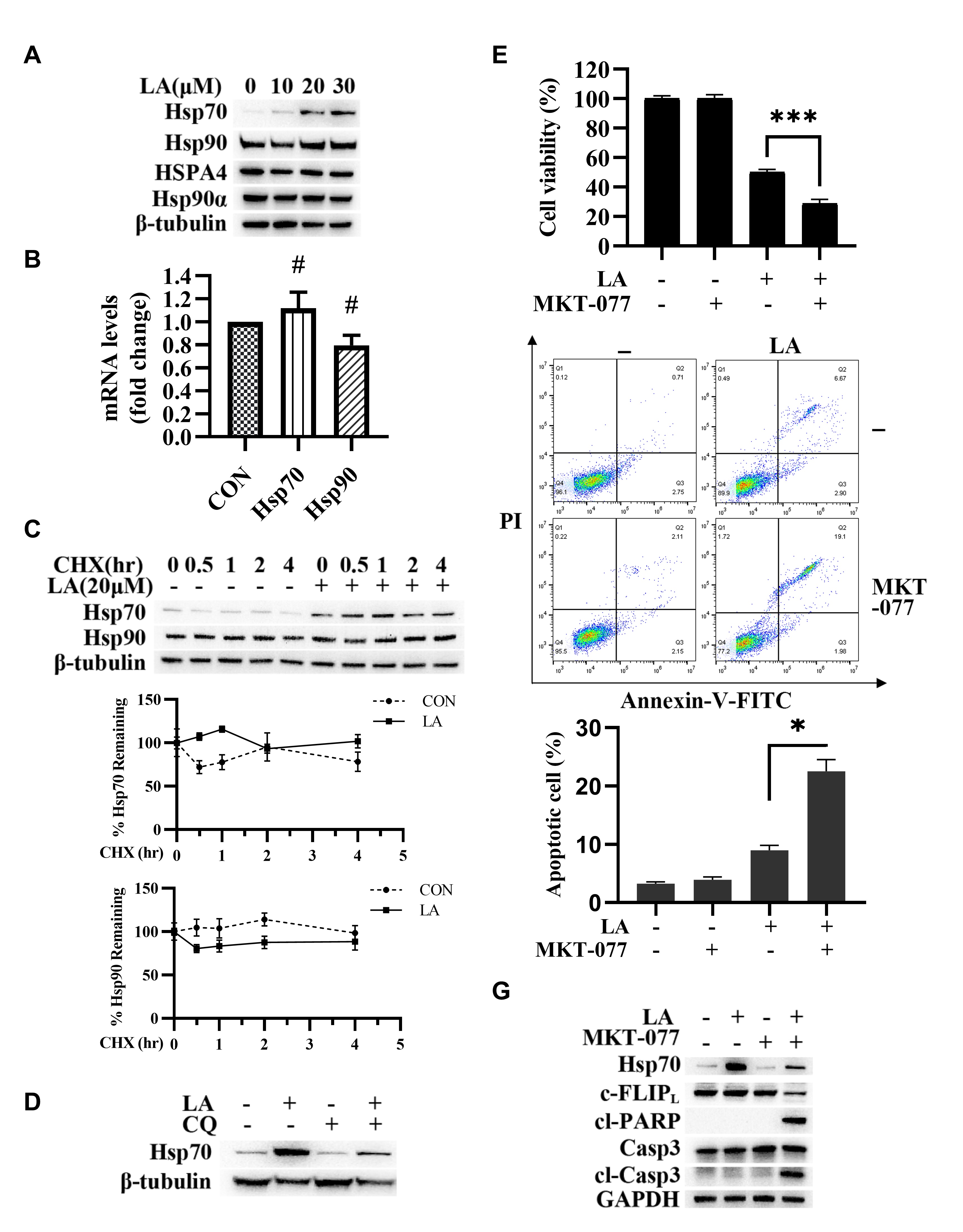 Fig. 5.
Fig. 5.Hsp70 expression maintained c-FLIP
Next, we found that CQ significantly inhibited Hsp70 expression (Fig. 5D),
indicating that LA enhanced Hsp70 expression through autophagy activation. Then,
the results of the MTT experiment and flow cytometry analysis indicated that
MKT-077 (Hsp70 inhibitor) enhanced LA-triggered apoptosis (Fig. 5E and 5F).
Moreover, MKT-077 enhanced cl-caspase-3 and cl-PARP and inhibited c-FLIP
As a tyrosinase inhibitor, LA has a wide range of pharmacological effects [10, 25], and its anticancer effects have received more attention. The anticancer effects of LA were demonstrated in vitro and in vivo for a variety of tumors, including cervical cancer [15], prostate cancer [12, 14], lung cancer [11], and breast cancer [13]. As a flavonoid, LA was affected by its strong hydrophobicity and the first-pass effect of the liver, resulting in its poor bioavailability [26]. However, it was reported that LA significantly increased the bioavailability by loading on liposome carriers or other nanocarriers [16, 27, 28, 29]. In this study, LA was confirmed to have an anti-CRC effect at a dose that had no obvious toxicity to normal colonic epithelial cell line NCM460. In addition, LA had a better inhibitory effect on KRAS mutant cell lines (SW620 and HCT116) than KRAS wild-type HT29 cells. We speculated that this might be due to KRAS promoting tumor growth through activation of the PI3K/Akt pathway [30], while LA inhibited the PI3K/Akt pathway [13, 31, 32]. Colony forming assay demonstrated that LA inhibited clonal proliferation of SW620 cells. Next, we elucidate the DNA synthesis of SW620 cell proliferation using EdU assays. Early studies showed that tumor growth and proliferation rapidly often lead to internal cell starvation without adequate nutrients [33]. Many herbal components, such as Anemarrhena asphodeloides, were reported to have a sensitizing anticancer with Serum starvation [34]. In the present research, LA was found to have a more significant anti-CRC effect with Serum starvation.
MAPK pathway was reported to be involved in LA-induced anti-multiple tumor effects [35, 36, 37]. In particular, activation of the MAPK/ERK plays a promoting role in the carcinogenic activity of human CRC [38]. Our results showed that LA activated ERK and p38 in SW620 and HCT116 cells without significant effect on JNK. Subsequently, we found that inhibitors of ERK enhanced the LA-induced inhibition of SW620 cell viability, while inhibitors of p38 had no significant effect. This result suggested that it was ERK, not p38, involved in the anti-CRC pathway, although LA activated them in SW620 cells.
LA was found induced autophagy of CRC cells in our research. Autophagy is a catabolic process induced under stressful conditions. Lots of studies have confirmed that LA-induced autophagy plays different roles in different types of cancer cells [13, 14, 15, 17, 19]. Shen et al. [39] found that LA triggered autophagy in osteosarcoma cells, thus inhibiting cell proliferation and promoting apoptosis. It was reported that this might be because LA downregulated the autophagy protein p62 (which was described as a cancer-inducing factor [40, 41]), thereby inhibiting tumor progression [42]. However, we found that the LA-induced autophagy was a cytoprotective autophagy and resisted the anticancer effect of LA in CRC. This conclusion was corroborated in studies of the anticancer effects of LA on cervical cancer [15] and lung cancer [9]. A recent study elucidated the mechanism by which some drugs or agents-induced autophagy could reduce the level of FOXO3a (a tumor suppressor protein) and thus downregulate the expression of the Puma/Bbc3 (a pro-apoptotic gene) gene [43]. Then, the decreased Puma/Bbc3 gene expression leads to increased drug resistance in tumor cells [43].
Our results showed that autophagy inhibited apoptosis by maintaining the
continuous expression of the antiapoptotic protein c-FLIP
Conclusively, LA induced cytoprotective autophagy through activation of ERK, and
autophagy maintained c-FLIP
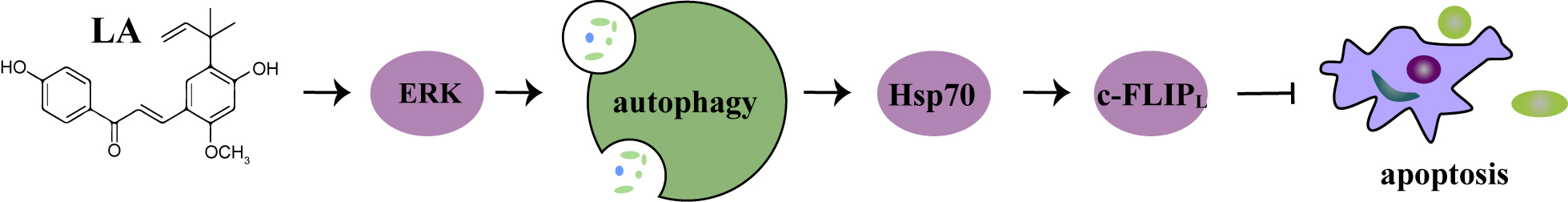 Fig. 6.
Fig. 6.LA-triggered anti-CRC activities. Hsp70 maintained c-FLIP
In conclusion, this study provided evidence that LA inhibited proliferation and
induced apoptotic cell death of colorectal cancer, and Hsp70 sustained
c-FLIP
The datasets created during this research are available from the corresponding author with rational requirement.
TianL—conceptualization, data curation, methodology, project administration, validation, visualization, writing—original draft; TingL—methodology, project administration, validation, visualization, writing—review & editing; HZ—conceptualization, methodology, validation, visualization, writing—original draft; CL—data curation, methodology, validation, visualization, writing—original draft; ML—conceptualization, data curation, formal analysis, validation, visualization, writing—original draft; CW—conceptualization, methodology, data curation, validation, visualization, writing—original draft; YZ—data curation, validation, visualization, writing—original draft; LZ—conceptualization, data curation, validation, visualization, writing—review & editing; XL—data curation, validation, visualization, writing—review & editing; YL—data curation, validation, visualization, writing—review & editing; SS—data curation, validation, visualization, writing—review & editing; WC—data curation, validation, visualization, writing—review & editing. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was funded by National Natural Science Foundation of China (No. 81960156), High Level Reserve Talents in Health Science (H2018002), Medical edible flowers technology innovation team of universities in Yunnan province (2020YGC01), Kunming Medical University Applied basic Research Joint Project (202001AY070001-189, 202201AT070043), the Opening Project of Medical Imaging Key Laboratory of Sichuan Province (MIKLSP202004).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.