







 , Guo-Xing Zhang 4,*
, Guo-Xing Zhang 4,*1 Department of Intensive Care Unit, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Gusu School, Nanjing Medical University, 215008 Suzhou, Jiangsu, China
2 Suzhou Clinical Medical Center of Critical Care Medicine, 215001 Suzhou, Jiangsu, China
3 Department of Nephrology, The First Affiliated Hospital of Soochow University, 215006 Suzhou, Jiangsu, China
4 Department of Physiology and Neurosciences, Medical College of Soochow University, 215123 Suzhou, Jiangsu, China
†These authors contributed equally.
Abstract
Background: Dexmedetomidine (DEX) reportedly protects against
ischemia-reperfusion (I/R) injury and associated damage to the kidneys, but the
underlying mechanisms have yet to be established. Methods: Unilateral
nephrectomy was performed in Wistar rats, and the remaining kidney was clamped
for 1 h prior to reperfusion to establish an experimental model system. These
animals were then randomized into Sham, DEX + Sham, DEX + I/R, ATI (Altepamizole,
Keywords
- acute kidney injury
- renal ischemia and reperfusion
- dexmedetomidine
- autophagy
- α2-AR/AMPK/mTOR pathway
Acute kidney injury (AKI) is a frequently diagnosed disease in the clinic that
can develop for a variety of reasons, resulting in a rapid drop in normal renal
function, rising serum creatinine levels, decreased urine output, and the
accumulation of metabolic waste and toxic compounds that ultimately contribute to
widespread organ failure [1]. According to the KDIGO (Kidney Disease: Improving
Global Outcomes) guidelines, AKI is defined by an elevated serum creatinine level
AKI is a complex, multifactorial condition that is often driven in large part by ischemia-reperfusion injury (IRI) [5]. IRI can result from myocardial infarction, trauma, hypovolemic shock, vascular or cardiac surgery, renal transplantation, and other conditions that cause pronounced hypoxia, increased reactive oxygen species biogenesis, aberrant antioxidant activity, inflammatory cytokine production, and activation of the complement system through the alternative pathway, culminating in renal tubular cell damage and death [6, 7, 8]. The necrotic debris released from these cells in the local environment can serve as a danger-related signal that can provoke further inflammation in a damaging positive feedback loop [9]. At present, no effective treatments for IRI-induced AKI (IRI-AKI) have been established.
The
In total, 30 healthy Wistar rats (males, 180–220 g) were obtained from the Animal Experiment Center of Soochow University and housed in SPF-grade cages with free food and water access under controlled conditions (24–26 °C, 40–60% relative humidity, 12 h light-dark cycle). All animal studies were in accordance with the NIH Guide for the Care of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Soochow University (Suzhou, China).
After intraperitoneal anesthesia using pentobarbital sodium (60 mg/kg), the skin and muscle were cut 1 cm away from the spin and 1 cm away from the lower rib edge such that the bilateral kidneys were visible. After ligating the right kidney at the pedicle with a silk thread, the pedicle was cut and the right kidney was excised. After separating the left renal artery, a vascular clip was applied to arrest blood flow for 1 h. After this ischemic period, the clip was removed to permit reperfusion such that the kidney changed in color from dark to bright red. The wound site was sutured and rats were injected (i.p.) with 0.5 mL of warm normal saline and returned to the incubator until awake, at which time they were transferred into clean cages. At 24 h post-surgery, rates received a pentobarbital sodium (60 mg/kg, i.p.) for anesthesia and were sacrificed, after which samples of blood and renal tissue were harvested.
For this study, rats were randomly assigned to six groups (n = 5/group): (1) A
sham group in which all procedures other than kidney excision and left renal
artery clamping were performed; (2) A DEX + sham group in which sham rats
received an i.p. injection of DEX (30 ug/kg; Yangzijiang Pharmaceutical Company);
(3) An ischemia-reperfusion (I/R) model group that underwent the surgical
procedure detailed above; (4) A DEX + I/R group in which DEX (30 ug/kg, i.p.) was
injected 30 min prior to the clamping of the left renal artery; (5) An ATI + DEX
+ I/R group in which the
Blood samples were rapidly collected via opening the ventricle apex, and were stored for 30 min at room temperature followed by centrifugation (10 min, 3500 cm, 4 °C) to collect the serum samples. Serum creatinine (Cre) and blood urea nitrogen (BUN) levels were analyzed with a fully automatic biochemical analyzer (C8000, ARCHITECH, Kyoto, Japan).
Kidney samples were fixed using 4% paraformaldehyde, paraffin-embedded,
prepared into 5 µm sections, and subjected to hematoxylin and eosin
(H&E) staining followed by imaging under light microscopy (BX-FM; Olympus
Corporation, Tokyo, Japan). The percentages of tubules exhibiting brush border
shedding and dilation were evaluated in 10 renal cortical regions to score the
magnitude of renal injury as follows: 0 (0%), 1 (1–19%), 2 (20–49%), 3
(50–69%), and 4 (
Commercial ELISA kits were used to assess serum IL-1
Serum malondialdehyde (MDA) levels were detected using a lipid oxidation detection kit (S0131S, Beyotime, Shanghai, China). This assay reflects the extent of lipid peroxidation based on the production of a red product from the reaction between MDA and thiobarbituric acid (TBA). Levels of the reduction marker glutathione (GSH) were assessed in serum with GSH and GSSG test kits (S0053, Beyotime).
Beclin1, LC3, and p62 expression in kidney tissue samples were detected via qPCR, Western immunoblotting, and immunohistochemical (IHC) staining.
Trizol (Invitrogen, Waltham, MA, USA) was used to isolate total RNA from renal
tissue samples based on provided directions, after which the RrimeScriptRT kit
(RR036A, Takara, Kyoto, Japan) was used to prepare cDNA. Then, a TB Green Premix
Ex TaqII kit (RR820A, Takara) and a 480 II Detection System (Roche) were used to
assess relative gene expression via qPCR using primers compiled in Table 1. GAPDH served as a normalization control, and relative expression was
quantified via the 2
| Genes | Sequences (5 |
| Beclin1 | (F)TCAAGATCCTGGACCGAGTGACC |
| (R)CTCCTCTCCTGAGTTAGCCTCTTCC | |
| LC3 | (F)GAGCGAGTTGGTCAAGATCATCCG |
| (R)GATGTCAGCGATGGGTGTGGATAC | |
| P62 | (F)CCAGCACAGGCACAGAAGATAAGAG |
| (R)TCCCACCGACTCCAAGGCTATC | |
| GAPDH | (F)ACGGCAAGTTCAACGGCACAG |
| (R)CGACATACTCAGCACCAGCATCAC |
For Western immunoblotting, minced kidney tissue samples were suspended in RIPA
buffer (P0013C, Beyotime, China) containing PMSF (P0100, Solarbio, Beijing,
China), protease inhibitors (05892791001, Roche, Basel, Switzerland), and
phosphatase inhibitors (04906845001, Roche) and homogenized in a tissue grinder
(F6/10 F013200021, JingXin, Xincang, China). After centrifugation (10 min, 3000
rpm, 4 °C) supernatant protein levels were detected via BCA assay (P0012,
Beyotime, China). Equal protein amounts were separated via SDS-PAGE and
transferred onto PVDF blots that were blocked for 2 h at room temperature with
5% skim milk followed by overnight incubation with primary antibodies (diluted
1:1000 unless otherwise noted) specific for the following proteins: Beclin 1
(1:2000; ab207612, abcam, Cambridge, UK); LC3B (2775S, Cell Signaling Technology,
Danvers, MA, USA); p62 (5114S, Cell Signaling Technology); GAPDH (60004-1-Ig,
Proteintech Group, Wuhan, China);
For IHC staining, xylene was used to deparaffinize 3 µm thick
kidney sections that were dehydrated with an ethanol gradient and treated with
3% H
AKI-related ultrastructural changes and autophagosome numbers were evaluated
via transmission electron microscopy (TEM). Briefly, kidney tissue
samples were washed using chilled PBS (pH 7.4), cut into 2 mm
Western immunoblotting was used to detect AMPK and mTOR levels in renal tissue samples as above using the following primary antibodies (diluted 1:1000): anti-AMPK (5831S, Cell Signaling Technology), anti-mTOR (CY5306, Abways, Shanghai, China). Proteins were detected with the same secondary antibodies used above.
Data are reported as means
To explore the impact of DEX on renal function in IRI-AKI model rats, serum
creatinine (Cre) and urea nitrogen (BUN) levels were analyzed as functional
biomarkers. Both Cre and BUN levels in the I/R group were significantly elevated
relative to sham control rats, while they were significantly reduced in the DEX +
I/R group as compared to the I/R model group. This suggests that DEX is capable
of reversing IRI-AKI-associated reductions in kidney function. Strikingly, both
Cre and BUN levels were significantly elevated in the ATI + DEX + I/R and 3-MA +
DEX + I/R groups as compared to the DEX + I/R group, suggesting that
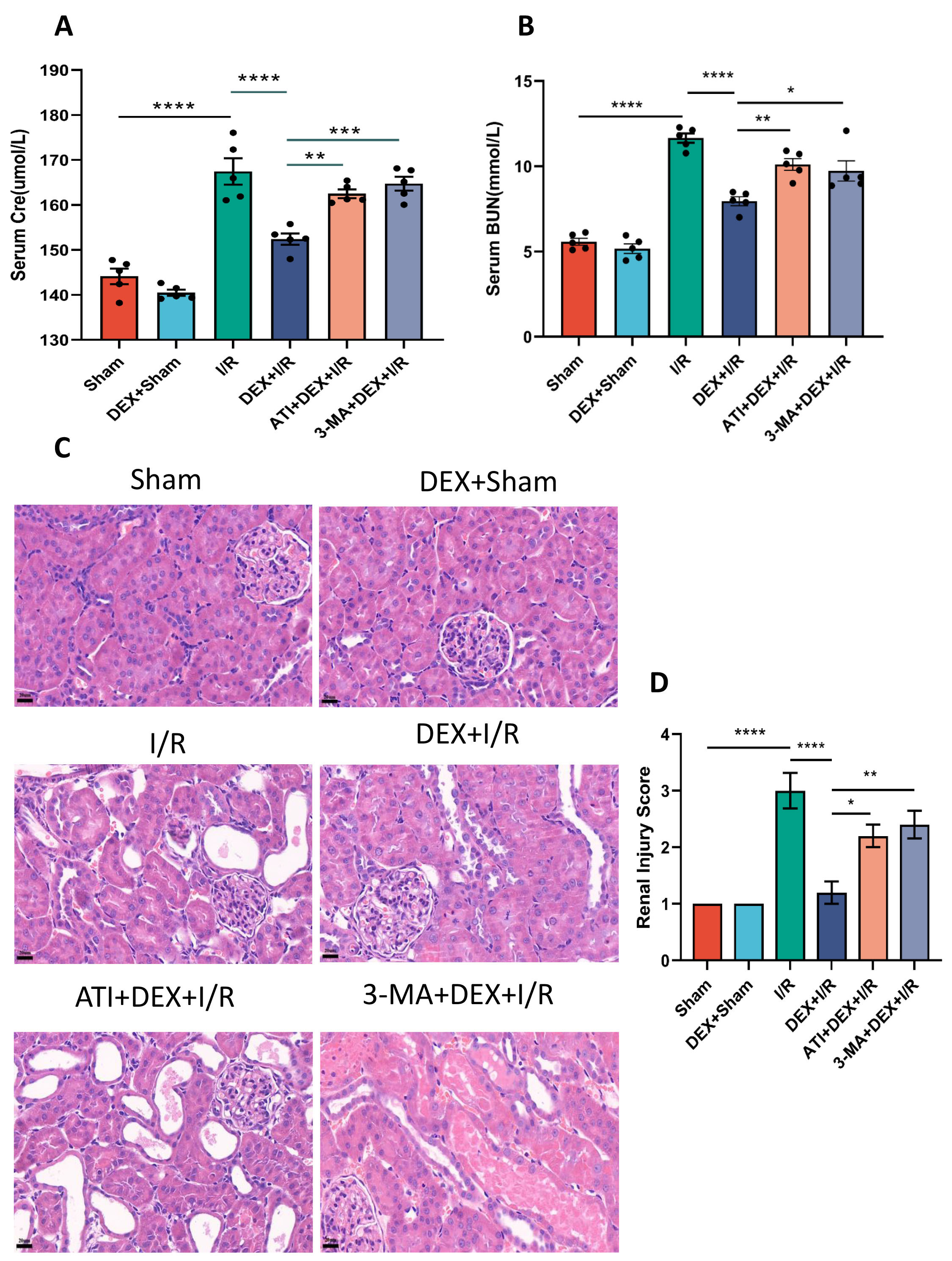 Fig. 1.
Fig. 1.DEX alleviates renal functional impairment in IRI-AKI model
rats. (A,B) Serum Cre (A) and BUN (B) levels. (C) H&E staining of renal
cortical tissues (400
To test the effects of DEX in IRI-AKI model rats, H&E staining and associated scores were used to evaluate the extent of renal pathology. Significantly higher scores were evident in I/R model rats relative to sham controls, while these scores were significantly reduced in the DEX + I/R group, supporting the ability of DEX to reverse I/R-associated kidney damage. Renal scores for ATI + DEX + I/R and 3-MA + DEX + I/R rats were also significantly elevated as compared to the DEX + I/R group, supporting the ability of ATI or 3-MA to eliminate the protective benefits of DEX against IRI-AKI (Fig. 1C,D).
To better understand the effects of DEX on IRI-AKI pathogenesis, inflammatory
cytokine production was next evaluated at serum. Significant increases in serum
IL-6, IL-1
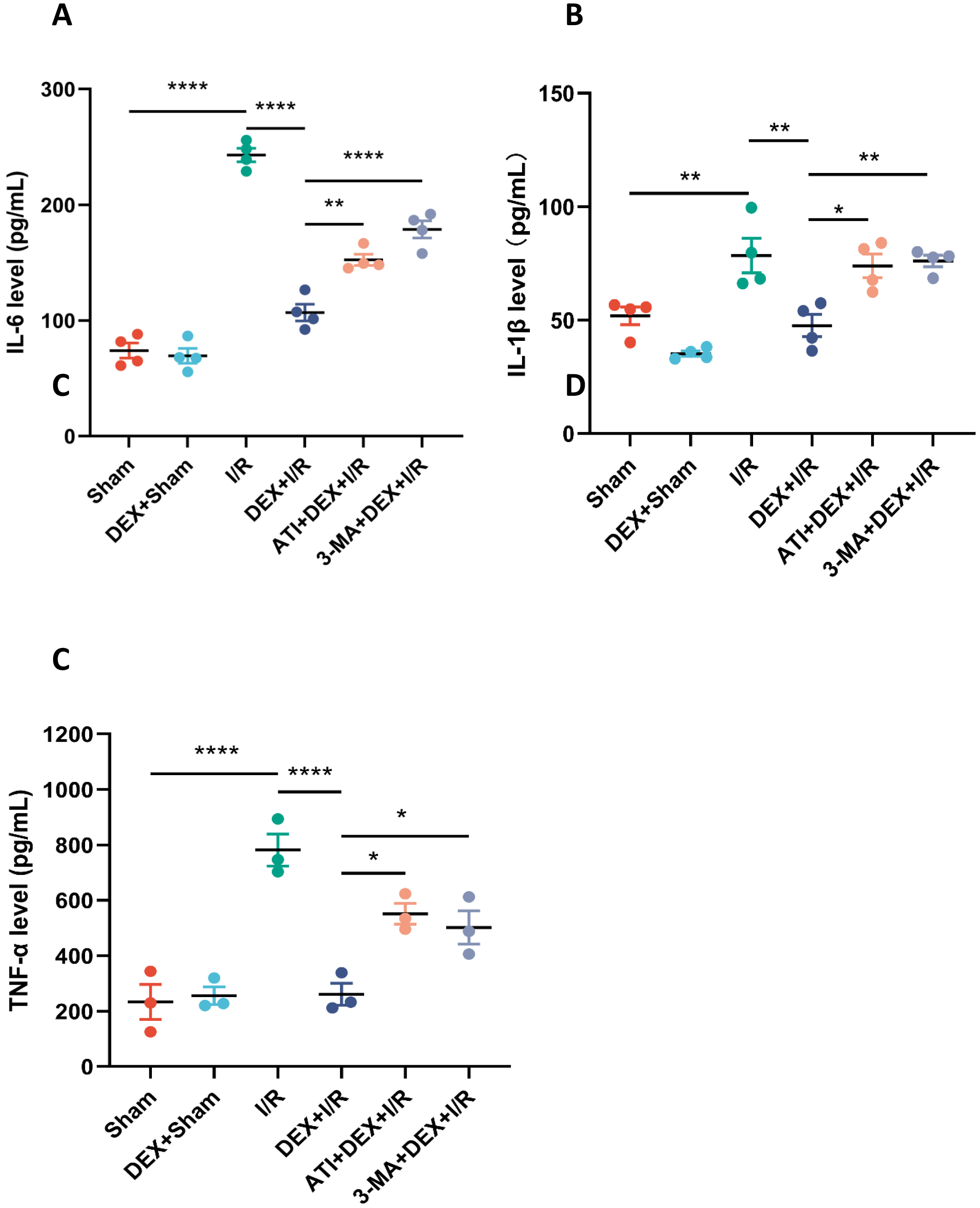 Fig. 2.
Fig. 2.DEX suppresses renal and systemic inflammation in IRI-AKI rats.
Serum levels of IL-6 (A, n = 4), IL-1
The impact of DEX on IRI-AKI-associated oxidative stress was next evaluated.
Serum MDA levels in I/R model rats were significantly elevated over sham
controls, while these levels were significantly reduced in DEX + I/R rats,
demonstrating the ability of DEX to abrogate oxidative stress induced in response
to I/R. However, significantly increased MDA levels were evident in both the ATI
+ DEX + I/R and 3-MA + DEX + I/R groups relative to the DEX + I/R group. Changes
in serum GSH levels were the inverse of those observed for serum MDA in these
rats. Overall, these data are consistent with the ability of DEX to regulate
oxidative stress via
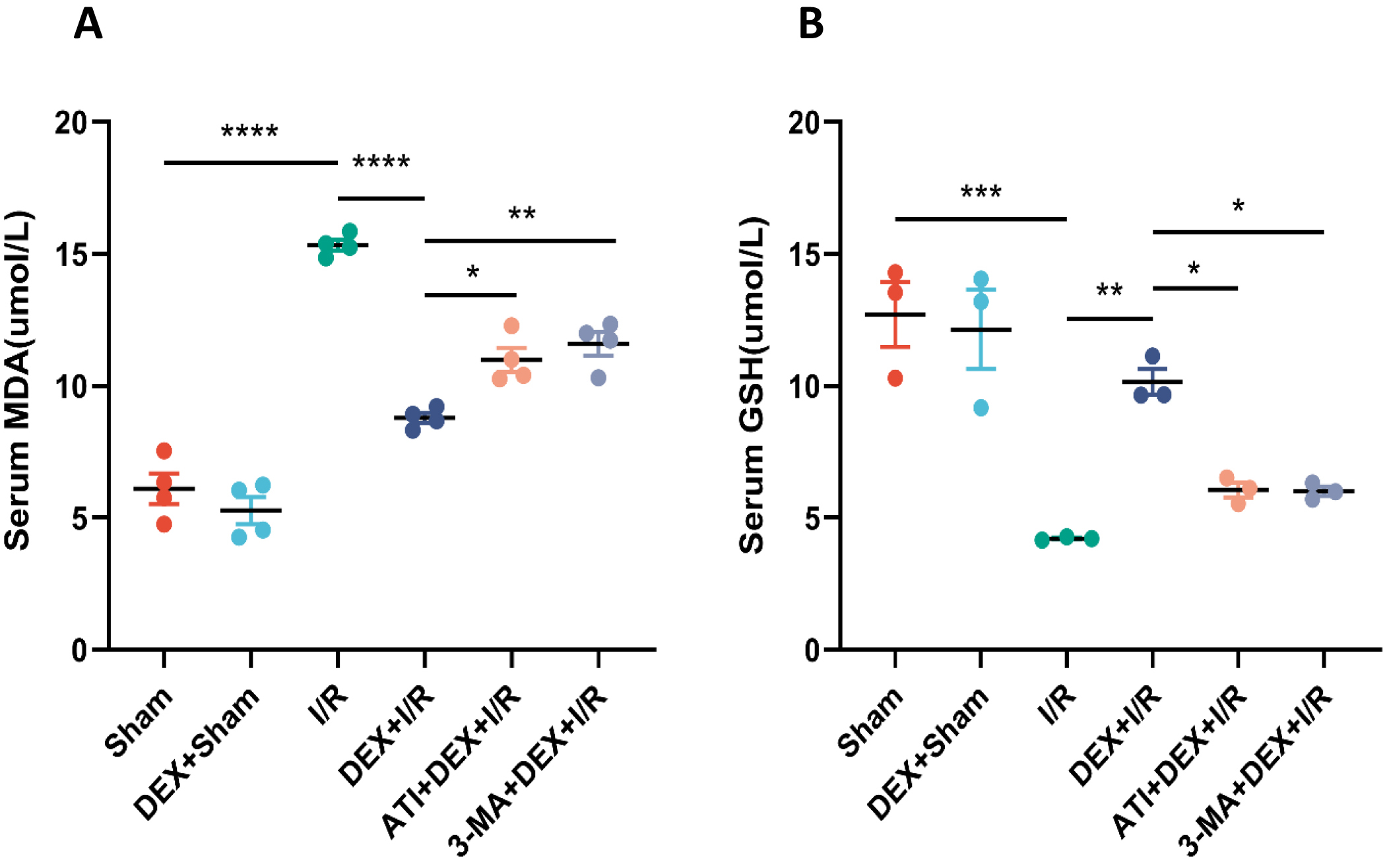 Fig. 3.
Fig. 3.DEX alleviates oxidative stress in IRI-AKI model rats. (A,B)
Serum MDA (A) and GSH (B) levels (n = 4). Data are means
The role that autophagy plays in the protective benefits of DEX in this experimental system was next assessed by evaluating key autophagic biomarkers (Beclin1, LC3, and p62) and quantifying autophagosome numbers in renal tissue samples. Significantly lower Beclin1 levels and LC3-II/LC3-I ratio values were observed in I/R model rats relative to sham controls, with significantly higher levels of expression of these biomarkers in DEX + I/R rats as compared to the I/R model group. These changes were reversed in rats treated with ATI or 3-MA, which ablated the beneficial effects of DEX. Changes in relative p62 expression were the inverse of those for Beclin1 and the LC3-II/LC3-I ratio, suggesting that DEX can reverse the I/R-associated suppression of autophagy in a manner that can be overcome by ATI or 3-MA treatment (Fig. 4).
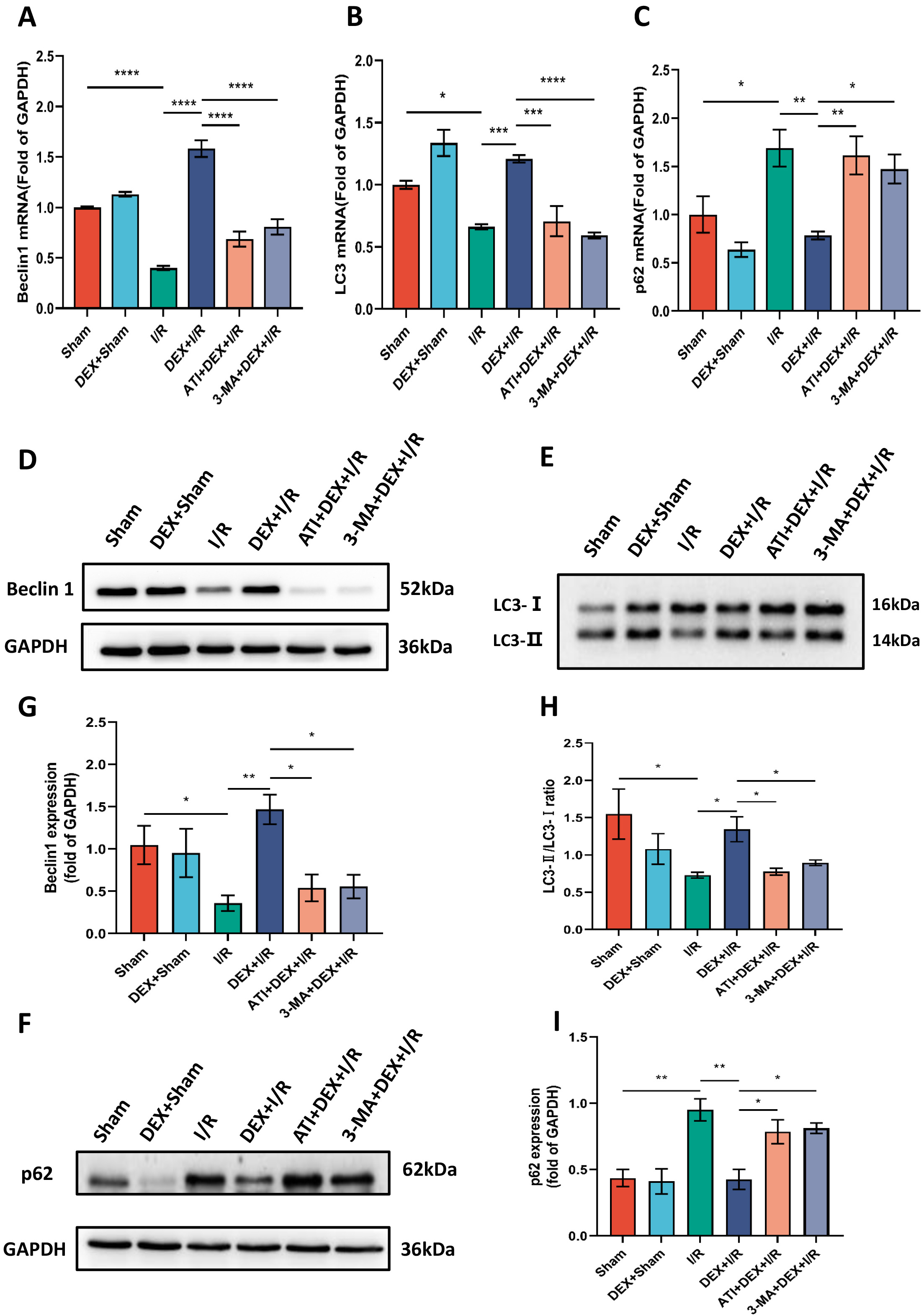 Fig. 4.
Fig. 4.DEX enhances autophagy in a manner reversed by ATI or 3-MA
treatment. (A–C) Beclin1 (n = 4), LC3 (n = 4), and p62 (n = 5) mRNA levels.
(D–F) Beclin1, LC3, and p62 protein levels, with (G–I) corresponding
quantification, using GAPDH as a normalization control (n = 4). Data are means
IHC staining revealed Beclin1, LC3, and p62 expression levels consistent with the qPCR and Western immunoblotting data shown above (Fig. 5).
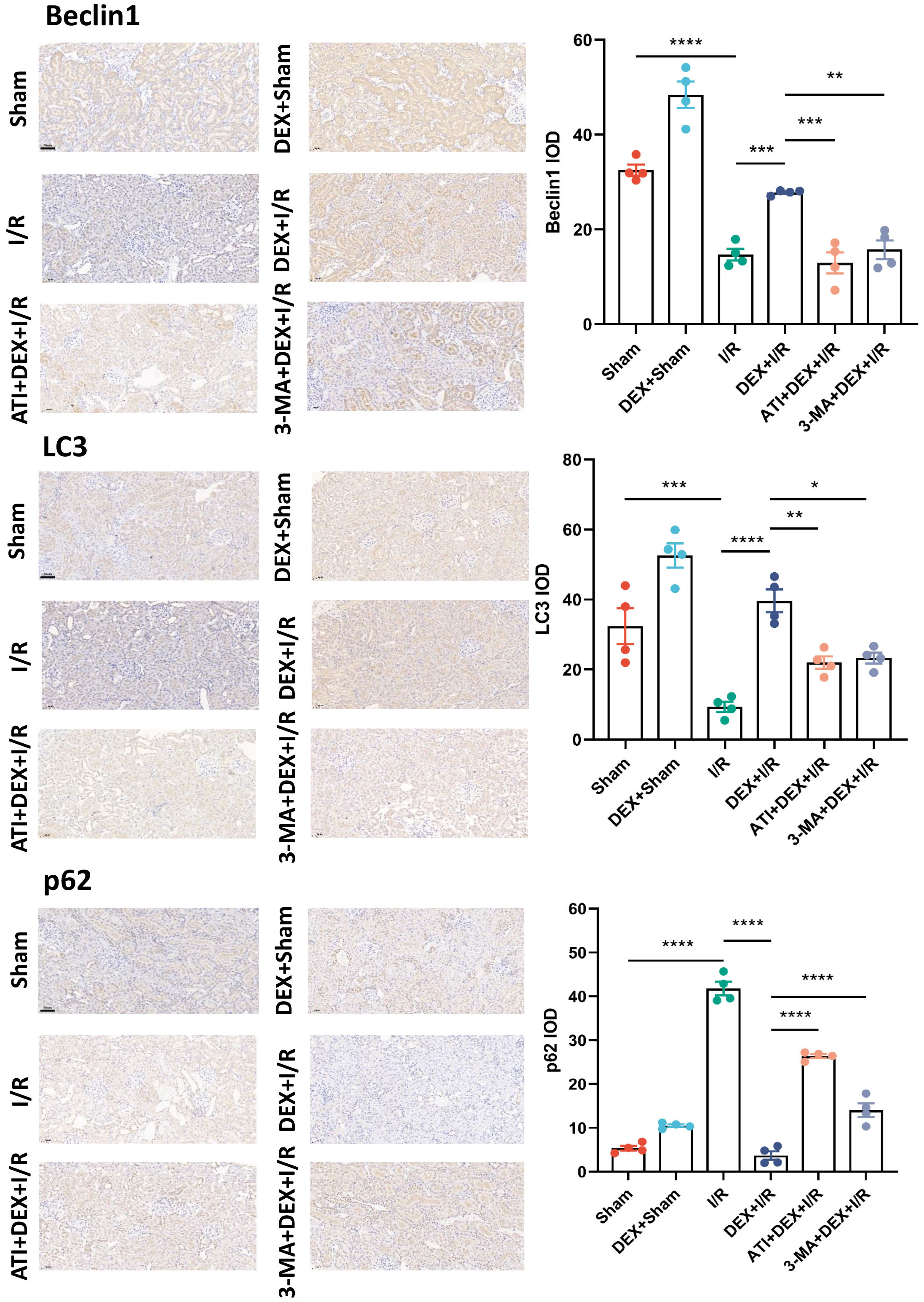 Fig. 5.
Fig. 5.DEX induces enhanced autophagic activity. Beclin1, LC3, and p62
levels were detected via IHC staining with corresponding quantification
(n = 4). Scale bar: 50 µm. Data are means
Next, electron microscopy analyses were conducted that revealed a significant drop in autophagosome numbers in the I/R group as compared to sham control rats, whereas DEX + I/R rats exhibited significantly elevated autophagosome counts as compared to I/R model animals consistent with the ability of DEX to remediate I/R-associated autophagic inhibition. As compared to the DEX + I/R group, both ATI and 3-MA were able to reduce autophagosome numbers, thereby abrogating the protective benefits of DEX administration (Fig. 6).
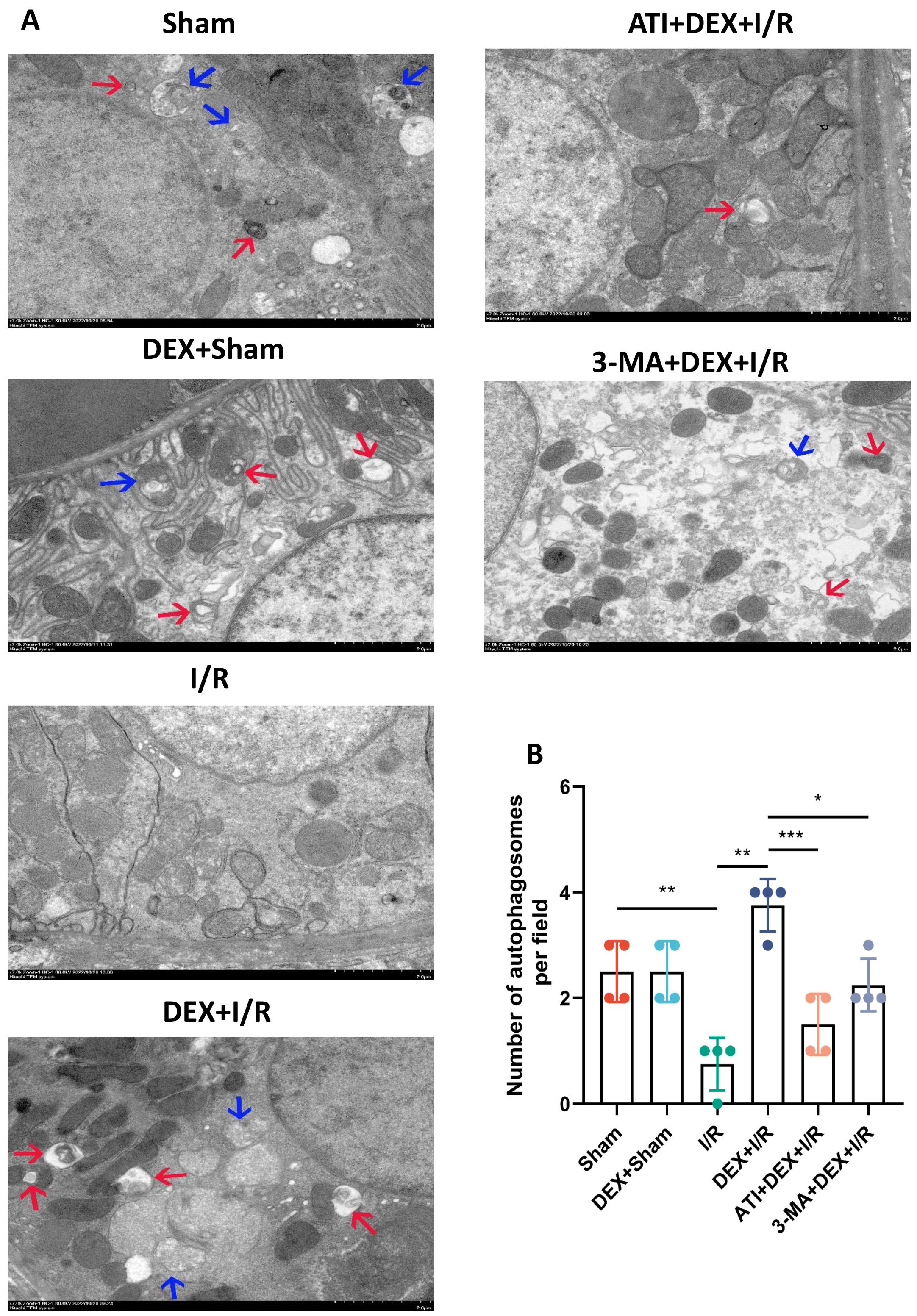 Fig. 6.
Fig. 6.DEX promotes enhanced autophagic activity. (A) Transmission
electron microscopy images of renal tissue samples. Autolysosomes and
autophagosomes are respectively indicated with blue and red arrows. Scale Bar:
2.0 µm. (B) Quantitative analyses of autophagosome numbers from 10 regions
per group (n = 4). Data are means
To determine the extent to which DEX influences AMPK/mTOR pathway activity,
protein levels of both AMPK and mTOR were assessed via Western
immunoblotting. Significant reductions in AMPK levels were detected in I/R model
rats relative to sham controls, while AMPK levels in the DEX + I/R group were
significantly elevated as compared to the I/R group. Compared with the DEX + I/R
group, however, ATI or 3-MA administration resulted in significant decreases in
AMPK expression, suggesting that IRI-AKI is associated with the suppression of
AMPK/mTOR pathway activity, whereas DEX can facilitate the activation of this
pathway in an
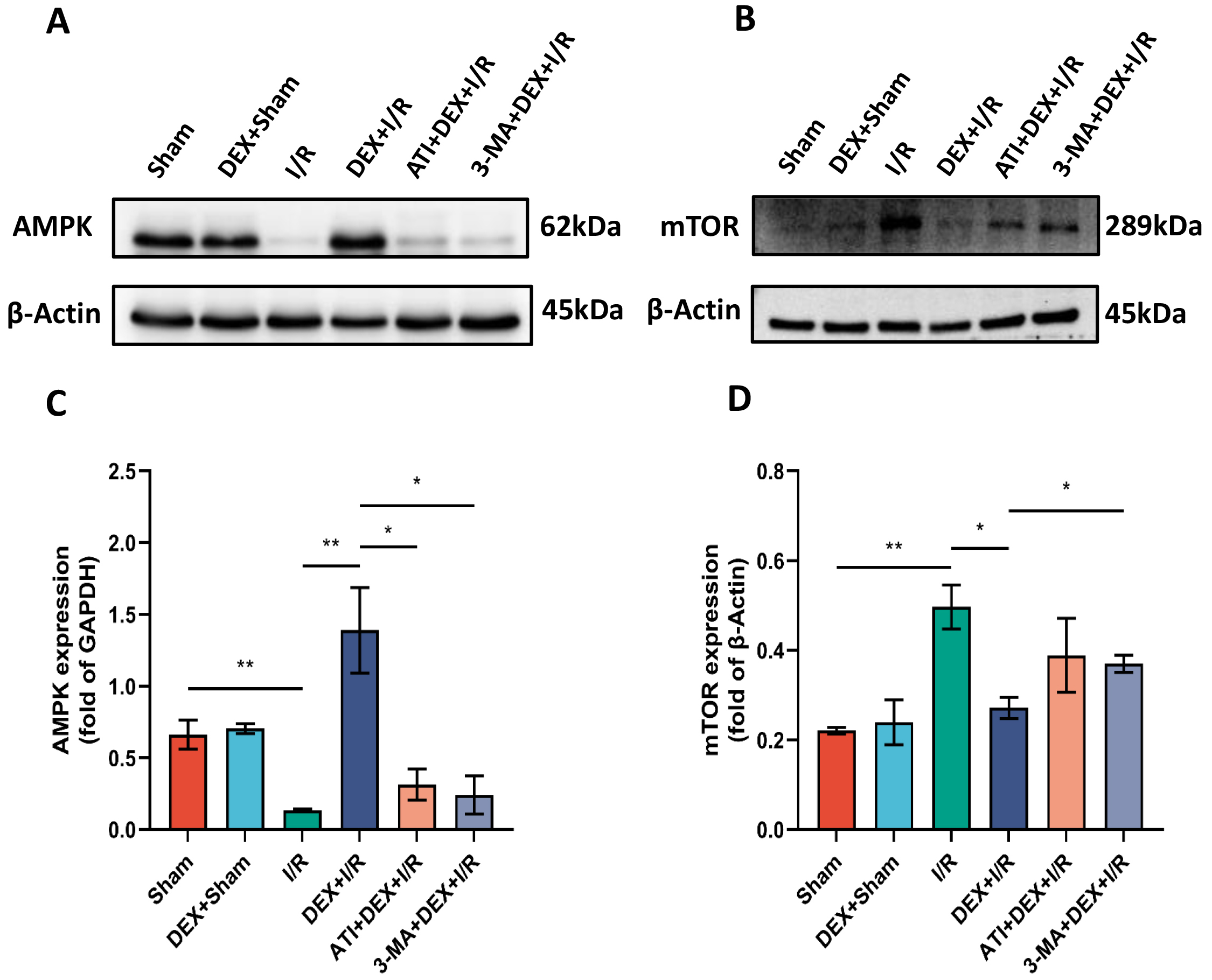 Fig. 7.
Fig. 7.DEX promotes enhanced autophagic activity via the
AMPK/mTOR pathway. (A,B) AMPK (A) and mTOR (B) protein levels. (C,D) The
expression of AMPK (n = 4) (C) and mTOR (n = 3) (D) was normalized to
IRI-AKI is an extremely debilitating clinical syndrome that can cause high rates
of patient morbidity and mortality owing to a lack of effective treatment
options. In an effort to support the design of novel IRI-AKI treatments, a rat
IRI-AKI model system was herein established and leveraged to confirm the
renoprotective effects of DEX administration. In this system, DEX was found to
enhance autophagic activity and to thereby alleviate IRI-AKI severity, decreasing
both oxidative stress and inflammatory cytokine biogenesis. Mechanistically, DEX
was found to promote autophagy through a process dependent on the
After first establishing a rat IRI-AKI model system, pathological changes associated with renal damage were evaluated. Serum biochemical analyses revealed markedly elevated Cre and BUN levels in IRI-AKI model rats, and histological analyses revealed clear tubule dilation and brush border shedding in these animals, thus confirming successful model generation.
These rats were next used to explore the mechanisms whereby DEX is able to exert
its protective benefits. DEX is known to function as an
Lastly, the ability of DEX to enhance autophagic activity via the
In conclusion, the present data provide evidence for the ability of DEX to
enhance autophagic activity and to thereby protect against renal damage in a rat
IRI-AKI model system via the modulation of
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
BZ, JY, and RL designed the work, analyzed the data and interpreted the data, also drafted the work. YS and HZ interpreted the data and participated in the writing. AY interpreted the data, designed, drafted the work and wrote the manuscript. GZ analyzed data, reviewed the work for important intellectual content, made significant revisions and proofread the manuscript, and supervised the program. All authors read and approved the final manuscript. All authors have participated in sufficiently in the work and agreed to be accountable for all aspects of the work.
The experiments were performed in according with the National Institutes of Health Guidelines for the Use of Laboratory Animals (NIH, publication number 85-23, revised 1996.), which were approved by and performed according to guidelines for the care and use of animals established by ethics licensing committee of Soochow University (No. 202201A0303). The present study is reported in accordance with ARRIVE guidelines.
Not applicable.
This work was supported by the National Natural Science Foundation of China (81970422), The research project of Gusu School, Nanjing Medical University (GSKY20210201).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.