








1 Institute for Physiological Chemistry, Medical Faculty, Martin-Luther-University Halle-Wittenberg, 06114 Halle, Germany
†These authors contributed equally.
Abstract
Background: A key mechanism in the neuromuscular disease GNE myopathy (GNEM) is believed to be that point mutations in the GNE gene impair sialic acid synthesis – maybe due to UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) activity restrictions – and resulting in muscle tissue loss. N-acetylmannosamine (ManNAc) is the first product of the bifunctional GNE enzyme and can therefore be regarded as a precursor of sialic acids. This study investigates whether this is also a suitable substance for restoring the sialic acid content in GNE-deficient cells. Methods: A HEK-293 GNE-knockout cell line was generated using CRISPR-Cas9 and analyzed for its ability to synthesize sialic acids. The cells were then supplemented with ManNAc to compensate for possible GNE inactivity and thereby restore sialic acid synthesis. Sialic acid levels were monitored by immunoblot and high performance liquid chromatography (HPLC). Results: The HEK-293 GNE-knockout cells showed almost no polysialylation signal (immunoblot) and a reduced overall (–71%) N-acetylneuraminic acid (Neu5Ac) level (HPLC) relative to total protein and normalized to wild type level. Supplementation of GNE-deficient HEK-293 cells with 2 mM ManNAc can restore polysialylation and free intracellular sialic acid levels to wild type levels. The addition of 1 mM ManNAc is sufficient to restore the membrane-bound sialic acid level. Conclusions: Although the mechanism behind this needs further investigation and although it remains unclear why adding ManNAc to GNE-deficient cells is sufficient to elevate polysialylation back to wild type levels – since this substance is also converted by the GNE, all of this might yet prove helpful in the development of an appropriate therapy for GNEM.
Keywords
- GNE myopathy
- ManNAc
- GNE
- sialic acids
- glycobiology
- glycosylation
- posttranslational modification
First described in 1981 [1], GNE myopathy is a rare neuromuscular disease (GNEM; OMIM: 605820) with an estimated prevalence of one to nine per million [2]. The cellular phenotype of this disease is characterized by the formation of rimmed vacuoles and protein aggregates in muscle cells, followed by loss of affected muscle cells [2, 3]. The disease first affects the distal muscle tissue of the feet and lower legs. In later stages, proximal body regions are also affected. About 10 years after the onset of the disease, patients need walking aids and external help with their daily activities [4]. GNEM is a genetic disease, resulting from point mutations in the gene encoding the bifunctional enzyme UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) [5]. This enzyme is known for its key role in sialic acid synthesis [6].
Sialic acids are a family of negatively charged 9-carbon sugars with a prominent role in protein glycosylation [7, 8]. Sialic acids occur in two forms on glycoproteins: in terminal sialylation, where the sialic acid forms the last sugar on the glycoconjugate, or in polysialic acids (PolySia), where up to 400 sialic acid monomers are conjugated into a large, negatively charged structure [9]. Both sialic acid forms occur on membrane proteins and decorate the cell surface extensively. Surface sialylation is an important regulator of cell-cell interaction. While sialylation creates a negative charge, it reduces adhesion, which, for example, prevents blood cells from clotting [8]. Furthermore, the expression of PolySia plays an important role in maturation and regeneration of muscle cells [10, 11].
Most steps in sialic acid synthesis take place in the cytosol. The activation of N-acetylneuraminic acid (Neu5Ac) to cytidine 5′-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac) takes place in the nucleus. Afterwards it is transferred to the Golgi apparatus, where it serves as a substrate for terminal protein sialylation or for polysialylation (Fig. 1) [12]. The synthesis is highly dependent on the bifunctional enzyme GNE, which catalyzes the first two steps of the synthesis and epimerizes UDP-N-acetylglucosamine (UDP-GlcNAc) to N-acetylmannosamine (ManNAc), which is then phosphorylated by GNE to ManNAc-6-phosphate. GNE itself can then be feedback inhibited by the end product of the sialic acid biosynthesis pathway, CMP-Neu5Ac, making GNE one of the major regulators of sialic acid synthesis [6].
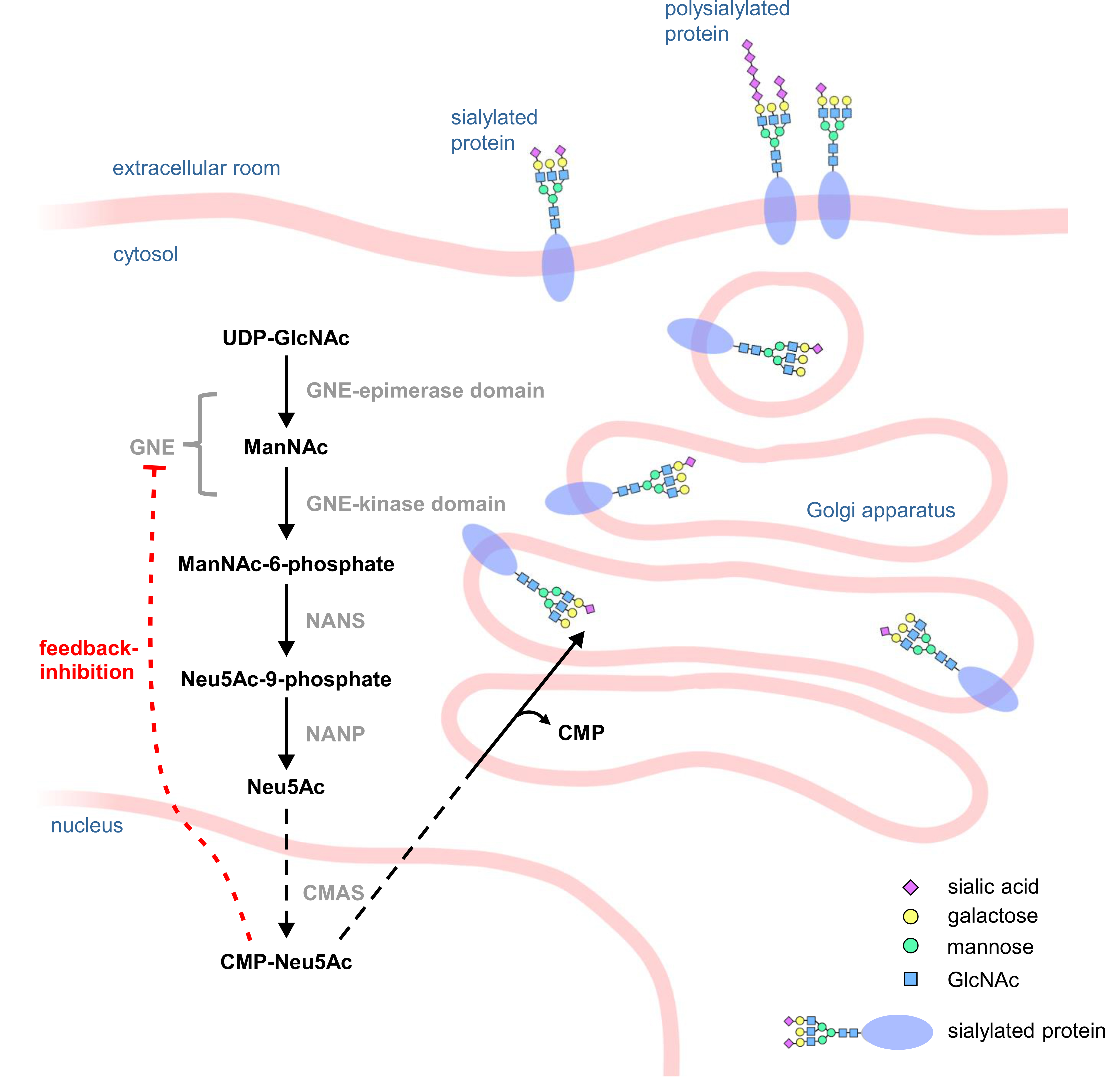 Fig. 1.
Fig. 1.Scheme of the sialic acid biosynthesis pathway. In the cytosol of a cell, UDP-N-acetylglucosamine (UDP-GlcNAc) is epimerized by the epimerase domain of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) to N-acetylmannosamine (ManNAc) which is then phosphorylated by the kinase domain of GNE to ManNAc-6-phosphate. Subsequently, sialic acid synthase (NANS) converts ManNAc-6-phosphate into Neu5Ac-9-phosphate, which is then dephosphorylated by N-acetylneuraminic acid phosphatase (NANP) to Neu5Ac. In the cell nucleus, N-acetylneuraminic acid (Neu5Ac) is then activated by CMP-N-acetylneuraminic acid synthase (CMAS) to cytidine 5′-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac), which can then act as a substrate for sialyltransferases in the Golgi apparatus.
Although the genetic cause for GNE myopathy has been identified [5], the mechanism leading to the disease needs to be clarified. Interestingly the expression of the GNE protein does not seem to be impaired/altered in GNEM patients [13], raising the likelihood that GNE activity is the – or at least a – critical factor for the pathophysiology of the disease. The question of whether the introduction of a point mutation also leads to changes in the intracellular localization of the GNE protein is controversial due to contradictory results [13, 14].
Changes in GNE activity can be attributed to pathogenic point mutations in the GNE gene [15, 16], which can lead to changes in the three-dimensional or quaternary structure of the protein [16, 17]. In addition, posttranslational modifications such as glycation [18], O-GlcNAcylation [19], phosphorylation [20], or a combination of these factors can also have an impact on GNE activity. If limitations in the activity of the enzyme are indeed present in vivo and have a significant impact on the disease, it is likely that this will also affect the “outcome” – Neu5Ac/sialic acid synthesis.
Various studies indicate hyposialylation in the patient’s muscle [21, 22]. However, this could not be proven in all studies, leaving room for other possible disease mechanisms/GNE intervention pathways. It has been shown that GNE also seems to play a role in the DNA damage/repair pathway [23], in the muscle filamentous apparatus [24], in mitochondria-dependent cell apoptosis [25], and in impaired apoptotic signaling [26]. On the other hand, the studies by Noguchi et al. [21], Sparks et al. [15], and Penner et al. [16] showed quite clearly that each variant must be considered individually, as each do not have the same effect on the activity of the GNE/of the domains of the GNE. It is therefore also possible that a mutation – e.g., the M712T variant – has an influence on the kinase activity (–30%; recombinant GNE-protein), but this does not necessarily have to be sufficient to also influence the sialic acid biosynthesis so that hyposialylation becomes detectable [6, 7].
Based on the hypothesis that sialic acid deficiency is largely responsible for the symptoms and manifestations of GNEM, several treatments to counteract this deficiency have been proposed, including supplementation with sialic acids or related compounds like sialyllactose [27, 28]. Many studies focus on ManNAc, the intermediate product of the GNE enzyme. ManNAc is taken up in the gastrointestinal tract, diffuses through the cell membrane and distributes to various tissues including the muscles [28, 29]. ManNAc enters the sialic acid synthesis pathway after being phosphorylated either by residual GNE kinase activity or by alternative kinases like GlcNAc kinase [17, 30].
In cell culture and mouse models, ManNAc proved to be a suitable supplement to increase sialylation [31, 32]. A phase 2 clinical study was built on these positive preliminary tests, in which either 3 g or 6 g ManNAc were administered orally twice a day. ManNAc was found to be ‘safe’ for long-term use. Increased sarcolemmal sialylation as well as increased plasma and intracellular CMP-Neu5Ac levels were reported. Decline in muscle strength of the upper and lower limbs showed to be reduced compared to a control group. Performance in other tests for physical function and endurance were however found not to be improved. A phase 3 clinical trial involving a randomized double-blind placebo-controlled study is to be conducted [30].
In addition to ManNAc, ace-neuraminic acid, an extended-release sialic acid product that lasts longer in the body, was evaluated in a phase 3 clinical trial. Patients received doses of 6 g per day taken orally for 48 weeks. While the study measured an increase in serum sialic acids, no positive effect on muscle strength and function was observed [33].
An orally administered peracetylated ManNAc derivate was tested in a mouse model for 54 weeks. Sialic acid content of various tissues, muscle mass, fitness as well as survival rates were reported to be increased [28]. These initial positive results laid the foundation for the current search for a peracetylated prodrug suitable for the treatment of GNEM-patients. Peracetylated ManNAc has the advantage that it can pass through the cell membrane more easily. A derivate of a series of ManNAc-6-phosphate prodrugs was shown to increase sialic acid content in GNEM patient-derived myoblasts by more than 50-fold at a concentration of 1 mM [34]. Research on peracetylated prodrugs represents a promising approach for the development of a drug for the treatment of GNEM.
Overall, however, there is a major problem with studies on the pathophysiology or treatment options for this disease: there is often a lack of comparability. The first question is which variant should be examined. With more than 200 known point mutations in the GNE gene, all leading to GNEM [2, 4], this is quite a challenge – especially given that this can already have an impact on enzyme activity [15, 16, 21]. Then a decision must be made between studying the protein in vitro (protein purification; whole protein vs. single GNE-domains; cell culture; appropriate cell line), or in vivo (animal model). In turn, they all have their advantages and disadvantages and it depends very much on the question to be examined, which approach is the right one.
In our study, we aimed to investigate how much ManNAc is required to restore sialylation to wild type levels in hyposialylated cells, and whether this is even possible. Other groups have used human embryonal kidney (HEK-293) cells with GNE-knockdown as model to gain insights into GNEM [14, 35], since knockdown cells always contain some residual expression (about 30% of the GNE expression remains [14]) and activity. With this in mind, we decided to perform our study with a GNE-knockout model with HEK-293 cells. Of course, a GNE-deficient model is different from a more disease-oriented model in which the GNE is still present, but for simplicity and to better focus on the impact of sialylation and its recovery, such a model should suffice. Once the model is proven to be effective, this would ensure that future tasks in this direction could be carried out easily, quickly, and, if necessary, with a high throughput.
The CRISPR/Cas9-technique was used to introduce GNE-knockout into
HEK-293 cells – later in the article these cells will be referred to as HEK-293
GNE
The cells of the human embryonal kidney line (HEK-293) were provided by the DSMZ (ACC 305; Heidelberg, Germany). The HEK-293 cell line as well as the GNE-knockout HEK-293 cell line was cultured in DMEM (Dulbecco’s Modified Eagle Medium; 11960044; Gibco/Thermo Fisher Scientific; Waltham, MA, USA) supplemented with 10% FCS (fetal calf serum) and 1% penicillin/streptomycin (10,000 units/mL (penicillin) and 10,000 µg/mL (streptomycin)) and passaged every two to three days. Cells were grown to 50–70% confluency, then supplemented with ManNAc (New Zealand Pharmaceuticals, Palmerston North, New Zealand) for 24 h and harvested. The ManNAc concentrations used are given in the corresponding results.
The cell lines were maintained at 37 °C in a humidified atmosphere with
5% CO
HEK-293 GNE-knockout cells (HEK-293 GNE
The cells were resuspended in solubilization buffer (150 mM NaCl, 50 mM Tris,
1% Triton, 100 mM PMSF (phenylmethylsulfinyl fluoride), 1:500 PIC (protein
inhibitor complex)) and opened by squeezing several times through a syringe. The
lysate was then incubated on a rocker at 4 °C for approximately 1 h and
then centrifuged at 13000 rpm for 10 min at 4 °C. The supernatant was
collected and the protein content was determined using the Pierce
For SDS-PAGE, 20 µg protein was loaded onto a 10% gel. The proteins were then blotted onto a nitrocellulose membrane for 1 h. The membrane was stained with Ponceau S and then blocked with 5% milk in Tris-buffered saline and 0.1% Tween 20 (TBS-T) for 1 h. The membrane was then washed three times with TBS-T, with each washing step lasting 10 min. Afterwards the primary antibody was added and incubated overnight. The washing procedure was repeated and afterwards the secondary antibody was added and incubated for 1 h. Again, the washing procedure was repeated. Subsequently, bands were detected with ECL immunoblot detection reagent (Merck, Darmstadt, Germany) and the ChemiDoc MP imaging system (Bio-Rad Laboratories, Hercules, CA, USA).
Cells were resuspended in 2 M propionic acid and lysed by squeezing through a
syringe four to six times. The lysate was then incubated at 80 °C for
4 h. After acidic hydrolysis, samples were centrifuged for 20 min at 13,000 rpm
and the supernatant was collected. Samples were lyophilized overnight and
resuspended in ddH
Cells were resuspended in ddH
100 µL sample and reference solutions were labelled (2.5 h, 50 °C, shaking) by 20 µL DMB-labelling reagent (1,2-diamino-4,5-methylenedioxybenzene.2HCl (DMB), 1.6 mg/mL, DMB Sialic Acid Labelling Kit, QA Bio, Palm Desert, CA, USA) as mainly described in the protocol of Ludger Ltd. [36]. Samples were diluted in 1:3 ratio with water and measured as duplicates.
To measure the sialic acid concentration, 20 µL of labelled sample and reference were injected into the device. A LiChroCART®250-4 LiChrospher®100 RP-18e (5 µm) column (Merck KGaA, Darmstadt, Germany) was used. Reference panel (1.25 nmol, AdvanceBio Sialic Acid reference panel, Agilent, Santa Clara, CA, USA) were used as reference for every measurement. Solvent A (acetonitrile: methanol: water 9:7:84) and solvent B (acetonitrile) were used as flux with a flow of 0.5 mL/min. The concentrations were determined using a diluted standard of N-acetylneuraminic acid (50 ng/mL, 500 ng/mL, 1 µg/mL, 20 µg/mL, Sigma Aldrich, St. Louis, MO, USA). The standard was prepared using N-acetylneuraminic acid powder diluted in aqua dest. Output of data were realized by Clarity Software (DataApex, Prague, Czech Republic).
Statistical analysis were performed using GraphPad Prism 9 2021 software (GraphPad Software, LLC, Dotmatics, Boston, MA, USA). Student’s t-test was used for statistical analysis. All bar charts show the mean and, as an error bar, the standard error of the mean (SEM).
A HEK-293 GNE
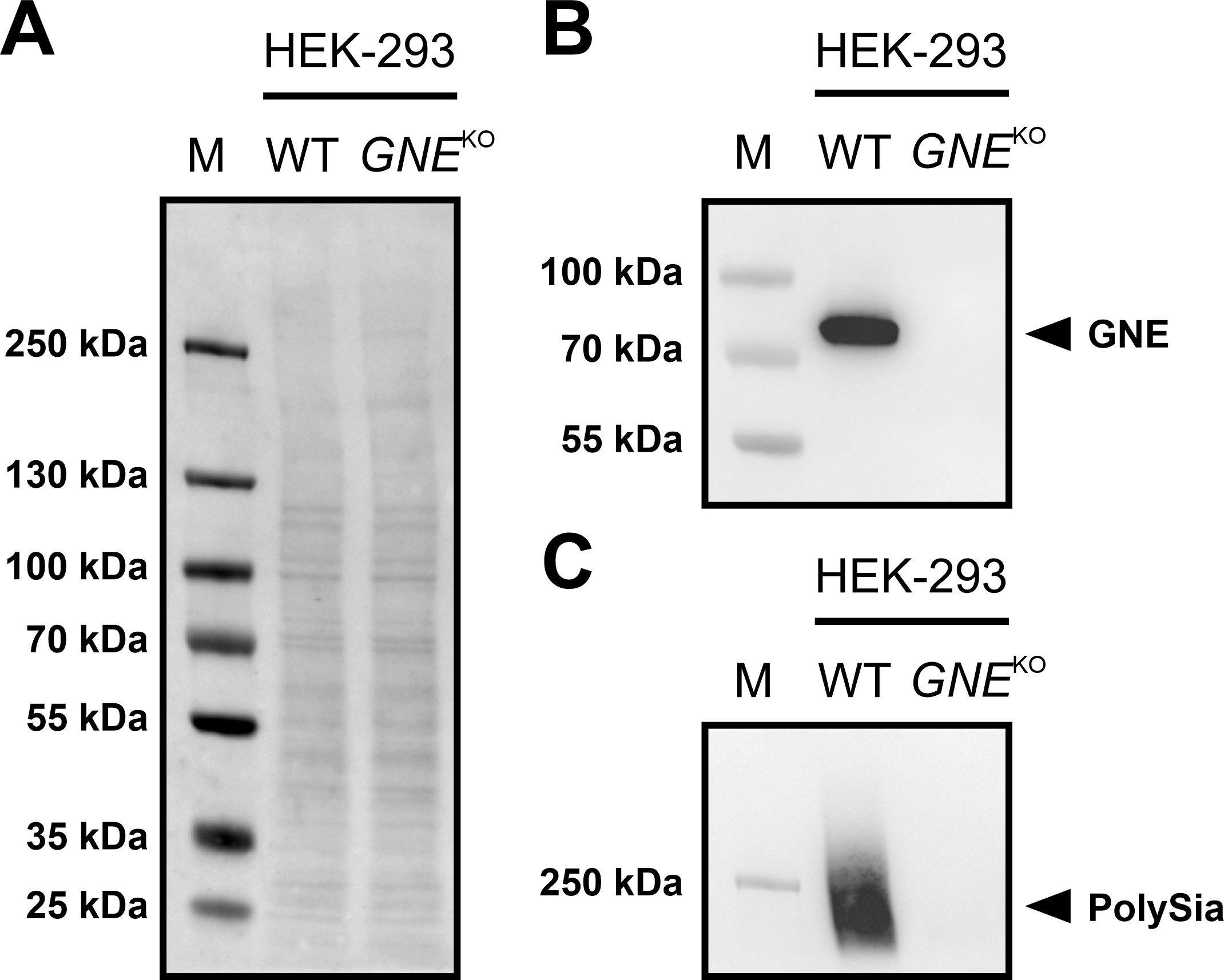 Fig. 2.
Fig. 2.Immunoblots of GNE and PolySia expression in HEK-293 WT and
HEK-293 GNE
If the protein is present, a band near the 70 kDa marker band can be expected
using a specific antibody against the protein. The corresponding band could be
detected in the HEK-293 WT cells, but not in the HEK-293 GNE
| Name | Target protein/modification | Dilution | Company and ordering number | |
| Primary antibody | ||||
| anti-GNE antibody (EPR15059) C-terminal; rabbit; monoclonal | GNE | 1:2000 | Abcam; ab189927 | |
| anti-PolySia (735); mouse; monoclonal | Polysialylation | 1:2000 | Kind gift from Rita Gerardy-Schahn (Hannover, Germany) | |
| Secondary antibody | ||||
| goat anti-rabbit | IgG-H&L (HRP) | 1:20,000 | Abcam; ab6721 | |
| goat anti-mouse | IgG-H&L (HRP) | 1:10,000 | Abcam; ab6789 | |
We next examined whether the polysialylation level of HEK-293
GNE
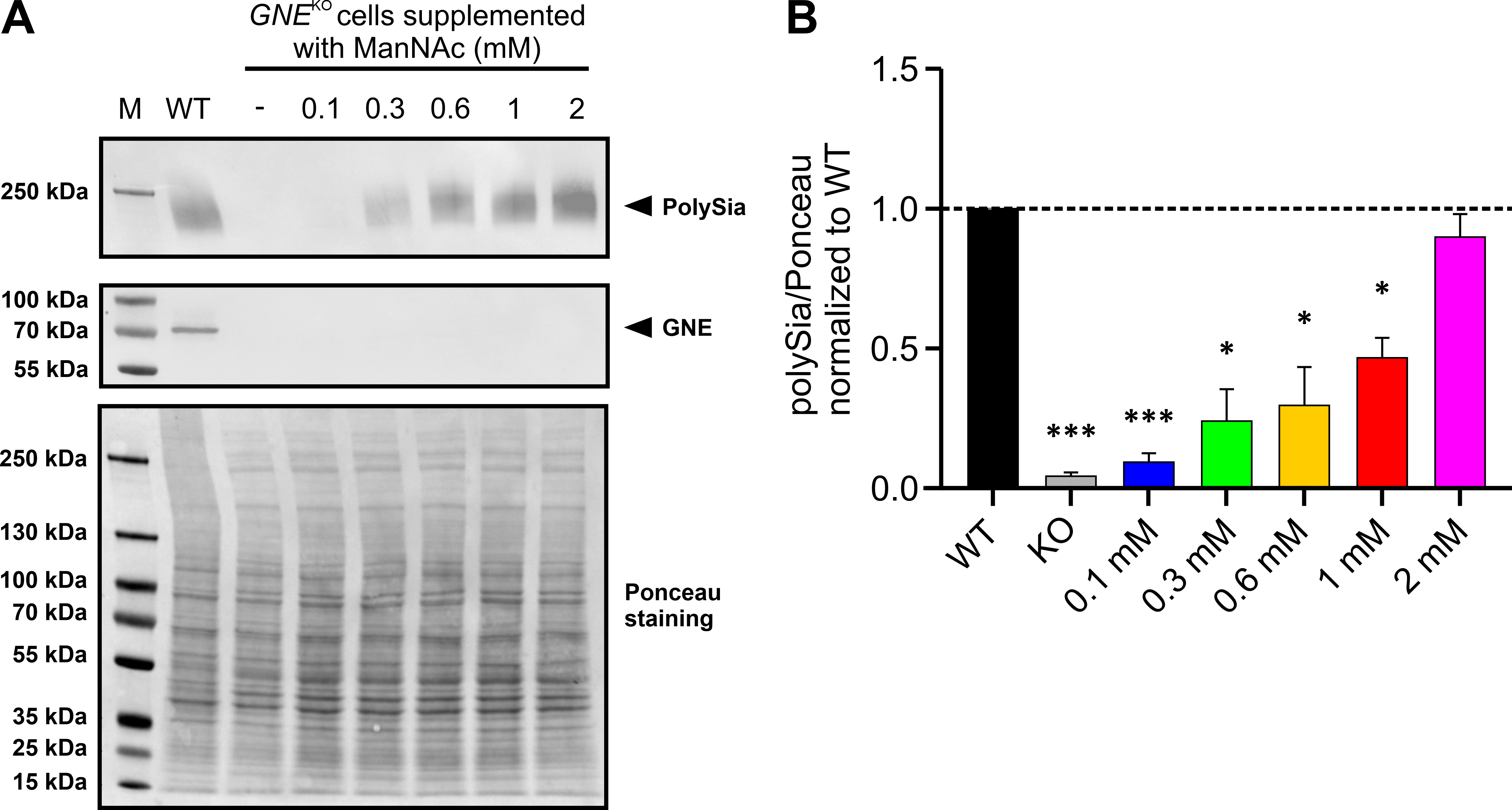 Fig. 3.
Fig. 3.Supplementation of HEK-293 WT and HEK-293 GNE
Afterwards the immunoblot results were quantified by normalizing the PolySia
band intensity to whole Ponceau stain (Fig. 3A bottom). This showed the same
trend as described above (Fig. 3B). Lower amounts of ManNAc resulted in a
relative normalized polysialylation signal of 0.0931
Polysialic acids are the product of one form of sialylation. To understand whether the PolySia detected by immunoblot corresponds to the total cellular sialic acid content, Neu5Ac – the most abundant sialic acid – was measured by HPLC and related to the amount of protein per sample and normalized to the HEK-293 WT samples (Fig. 4A). It can be seen that the total sialic acid level also showed an increasing trend, which was associated with increasing ManNAc supplementation levels – as was the case with the PolySia signal in the immunoblot (see previous section).
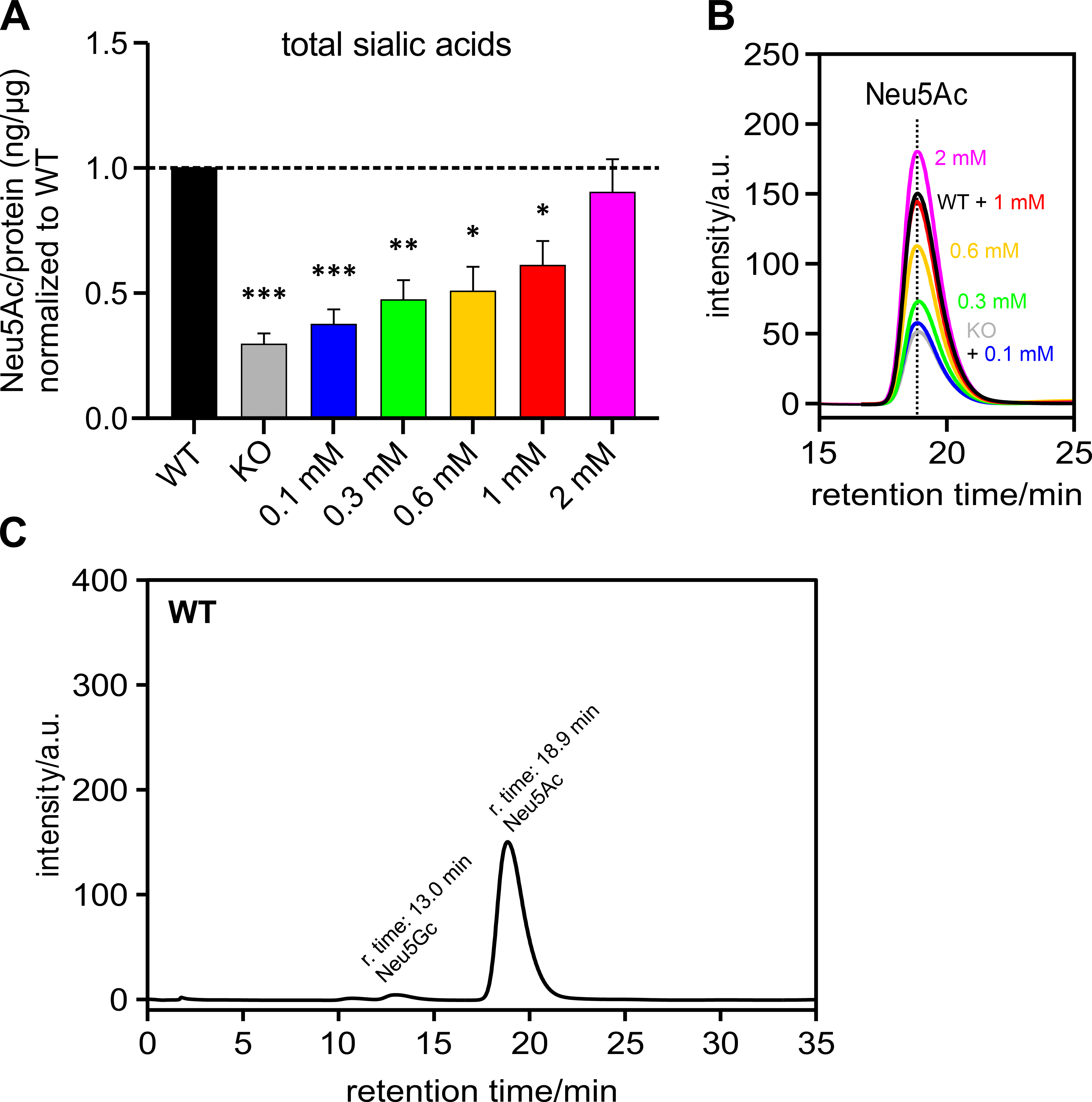 Fig. 4.
Fig. 4.Sialic acid (Neu5Ac) per protein (ng/µg) normalized
to HEK-293 WT cells. HEK-293 GNE
Fig. 4B shows an overlay of several representative chromatograms of Neu5Ac –
HEK-293 WT, HEK-293 GNE
Fig. 4C shows a representative chromatogram of a HEK-293 WT sample over the retention time range of 0 to 35 min. The peak with a retention time of 13 min corresponds to Neu5Gc. The peak corresponding to Neu5Ac has a retention time of 18.9 min.
In addition, we determined the Pearson correlation coefficient of the means for polysialylation and total sialic acid content. It was 0.9980, showing a very good correlation between these two.
In addition, to test whether the level of polysialylation is also representative of free and bound sialic acids, the insoluble membrane protein-bound sialic acids were first separated from the free cellular sialic acids. The content of free cellular sialic acids per protein increases in proportion to the likewise increasing ManNAc concentrations (Fig. 5A).
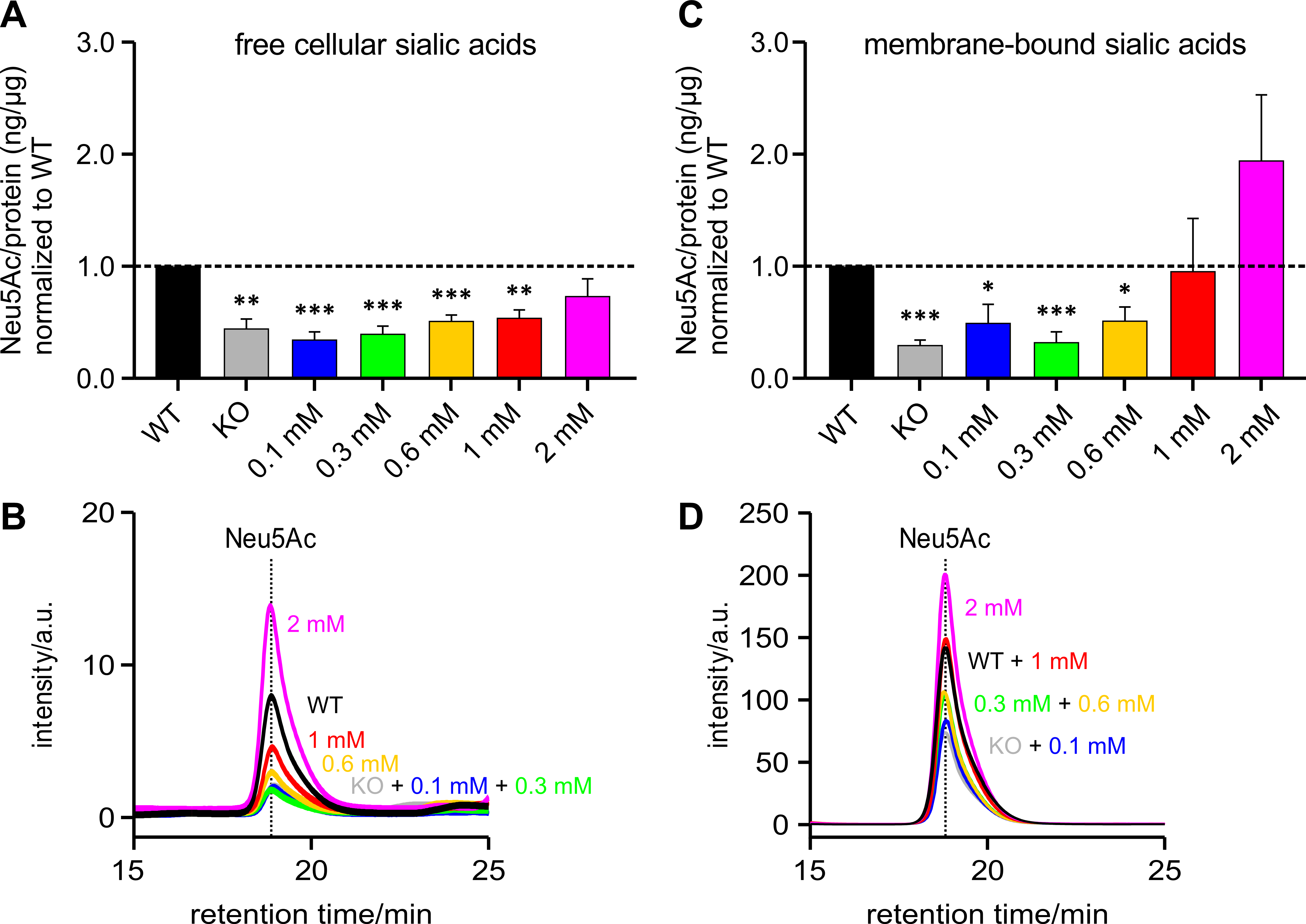 Fig. 5.
Fig. 5.Free and membrane-bound sialic acid (Neu5Ac) per protein
(ng/µg) normalized to WT. HEK-293 GNE
Fig. 5B shows an overlay of several representative chromatograms of free
cellular Neu5Ac – HEK-293 WT, HEK-293 GNE
The Pearson correlation coefficient of the means for polysialylation and free sialic acid content was 0.9345 and for polysialylation and membrane-bound sialic acid content was 0.8200. Here too, both values showed a good correlation with the degree of polysialylation.
In this study, we generated a GNE-knockout in HEK-293 cells and characterized it with and without ManNAc supplementation in terms of the degree of polysialylation and the amount of free and membrane-bound sialic acid. To detect polysialylation, we performed a western blot using an antibody specific against it (anti-PolySia (735)). We decided against actin or other “classic” cytoskeletal proteins to control gel loading, as it is already known that GNE/clinically relevant GNE-variants appear to have an impact on actin dynamics/actin turnover [37, 38]. Therefore, an influence on the actin signal strength in the immunoblot after GNE-knockout cannot be completely ruled out. To avoid this, we used Ponceau staining as gel loading control. The amount of free and membrane-bound sialic acid (Neu5Ac) was quantified using HPLC.
In HEK-293 WT cells, both a PolySia and a GNE signal could be detected in the
western blot. However, none of the signals could be detected in the HEK-293
GNE
We have also shown that supplementation with 2 mM ManNAc, the product of the
GNE-epimerase domain, restores the level of polysialylation in HEK-293
GNE
In addition, HPLC measurements of the Neu5Ac levels showed that the higher the
supplemented ManNAc concentration, the higher the Neu5Ac levels as well. These
results correlate with previous studies [40, 41, 42]. The different concentrations
of ManNAc also have a different impact on the concentrations of free sialic acid
in the cytosol compared to membrane-bound Neu5Ac of HEK-293 GNE
When looking at the results, however, it must also be taken into account that
the cells are always supplied with a small proportion of sialic acids directly via the medium and the serum it contains. However, this proportion is the same under all conditions and should therefore be negligible. Furthermore, it is clear that the content of sialic acids in the medium is not sufficient to increase the sialic acid content/level of polysialylation in HEK-293 GNE
Nevertheless, the question now arises as to why the sialylation level of the
HEK-293 GNE
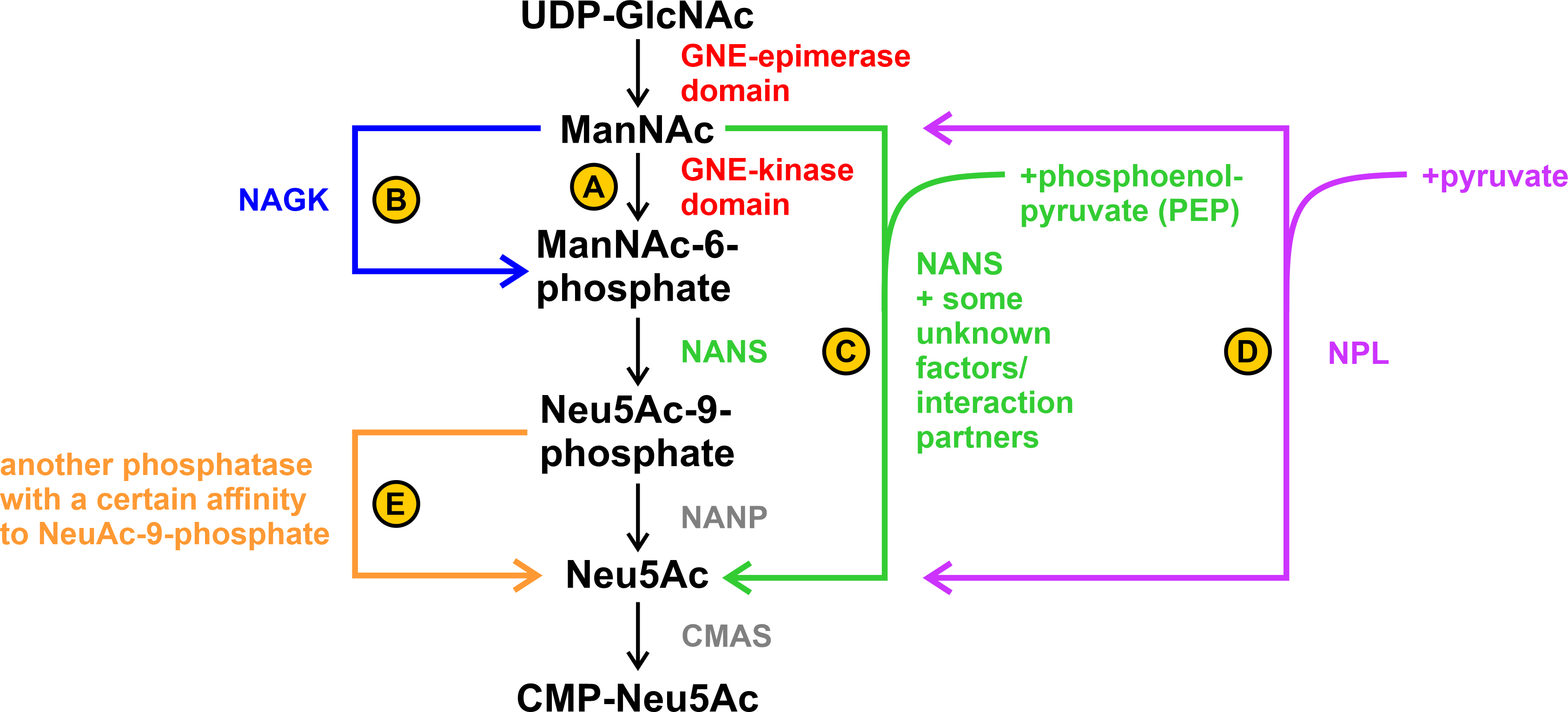 Fig. 6.
Fig. 6.Possible alternative steps in sialic acid biosynthesis. (A) ManNAc is phosphorylated by the GNE-kinase domain. (B) ManNAc is phosphorylated by the N-acetylglucosamine kinase (NAGK). (C) ManNAc is directly converted to Neu5Ac by sialic acid synthase (NANS) with the addition of phosphoenolpyruvate (PEP). Unknown tissue-specific factors/interaction partners may play a role in this. (D) ManNAc is directly converted to Neu5Ac by N-acetylneuraminate pyruvate lyase (NPL) with the addition of pyruvate. This reaction is reversible. (E) Dephosphorylation of Neu5Ac-9-phosphate to Neu5Ac occurs by an unknown phosphatase with a specific side-affinity to Neu5Ac-9-phosphate.
A possible explanation could be that the phosphorylation step from ManNAc to ManNAc-6-phosphate was alternatively performed by the N-acetylglucosamine kinase (NAGK) ([46], see B in Fig. 6). Another alternative explanation can be found by looking at the enzymes and its associated diseases further down the sialic acid biosynthesis pathway: Genevieve type spondyloepimetaphyseal dysplasia (SEMDG; OMIM 610442) is a disease caused by mutations in sialic acid synthase (NANS) that results in developmental delays such as reduced growth rate and mental impairment. However, the sialylation of plasma proteins and the biosynthesis of N- or O-linked glycans did not appear to be affected in the disease. In addition, ManNAc and not ManNAc-6-phoshate, the, so to speak, “classic” substrate of NANS in the biosynthesis pathway, was found in the plasma and urine of an affected patient [47].
This leads to the question whether NANS – as known from bacterial pathways [48]
– can also use ManNAc under certain circumstances directly as a substrate (see C
in Fig. 6; see also UniProt.org: Q9NR45 and Rhea-db.org: 19273) and whether the
GNE-kinase domain is not always the main ManNAc processing enzyme. That could
explain the accumulation of ManNAc in the patient’s body fluids. Furthermore,
they spoke directly of an “accumulation of substrates of the missing enzyme”
([47], page 782, lines 8 and 9 of discussion), suggesting that ManNAc may indeed
be a substrate of NANS. It could therefore be possible that under “normal”
conditions/in certain types of tissues NANS has a significantly higher affinity
for ManNAc-6-phosphate than for ManNAc (shown e.g., in [49, 50] where the enzyme
did not show any activity with ManNAc). However, it is also possible that in
other tissues a cofactor or substance is present that reverses this substrate
affinity, resulting in ManNAc becoming an acceptable, possibly even more
favorable, substrate for NANS. Perhaps the GNE-epimerase and the NANS interact in
certain tissues in a kind of complex, which in turn cannot be formed in other
tissues. Consistent with the assumption of tissue-specific effects, it has
already been shown that there may indeed be such tissue-specific effects on
sialic acid biosynthesis. For example, almost no Neu5Ac-9-phosphate or Neu5Ac
were detected in kidney cells after incubation with
N-acetyl-[
Furthermore, it is of course also possible that a phosphatase dephosphorylates ManNAc-6-phosphate, so that then only ManNAc can be detected in the body fluids of the SEMDG-patients. This would be consistent with the elevated levels of ManNAc-6-phosphate found in patient-generated fibroblasts [47]. Of course, this can also indicate a tissue effect in fibroblasts, in which NANS cannot process ManNAc but only ManNAc-6-phosphate as a substrate.
In addition to NAGK and (perhaps also) NANS, there appears to be a third enzyme – the N-acetylneuraminate pyruvate lyase (NPL) that can cleave sialic acids into ManNAc and pyruvate, but interestingly can also act in reverse ([53], see D in Fig. 6). It has been found in kidney extracts of various species, at least suggesting that it might also be present in HEK-293 cells.
In a study of Willems et al. [39] they worked on CHO (chinese hamster ovary) and HAP1 cells and knocked out each of the so far known enzymes that might be involved in sialic acid biosynthesis. They were able to show that a knockout of NANP (N-acetylneuraminic acid phosphatase) did not result in altered levels of CMP-sialic acids, whereas knockout of GNE, NANS, and CMAS (CMP-N-acetylneuraminic acid synthase) each resulted in drastic effects. Furthermore, they suggest the existence of an alternative pathway capable of bypassing the NANP step in sialic acid biosynthesis. However, in their experimental setup, this does not happen either by NANS (see C in Fig. 6) – the accumulation of Neu5Ac-9-phosphate suggests that Neu5Ac-9-phosphate is an important intermediate in NANP knockouts – nor by NPL, as the results from the double knockout show. Rather, they suggest the presence of another phosphatase, which does not necessarily have to use Neu5Ac-9-phosphate as the main substrate, but nevertheless has a certain affinity for it (see E in Fig. 6).
All of this needs to be further clarified in future experiments, ranging from a confirmation of NAGK, NANS, and NPL expression in HEK-293 cells to a full investigation of the sialic acid biosynthesis pathway in this cell line, e.g., by knockout of each of these enzymes as done in the study of Willems et al. [39]. With other cell lines, unfortunately, all of this has to be done again – due to the tissue-related effects that are becoming apparent.
In other future experiments that also address the question of comparability, it may be of interest to repeat some of the previously performed experiments on Gne-knockout Sol8 cells [23] – a murine muscle cell line – to further see whether there are species and/or tissue-specific differences. Thinking back to the overarching goal of better understanding GNEM, its phenotypes and therapeutic options, this is particularly interesting since Sol8 cells represent a muscle cell line and muscles are the most affected cells in GNEM.
Overall, there appears to be sufficient evidence to suggest that sialylation problems due to reduced GNE activity may not be the sole cause for the disease phenotype, but rather that it involves a variety of complex relationships/interactions – perhaps hyposialylation represents only part of the picture. This too needs further investigation.
The methods used here prove to be a handy tool for assessing the ability of a substance to restore sialylation and determining the amount that must be available to the cell to do so. PolySia detected by immunoblot is easily quantified and, as shown by HPLC measurement, representative of total, free and membrane-bound sialic acid content. The method described here can then also be used to test the ability of ManNAc-6-phosphate prodrugs and other sialic acid precursors.
HPLC, high performance liquid chromatography; CMAS, CMP-N-acetylneuraminic acid synthase; GNE,
UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase;
GNEM, GNE myopathy; HEK-293 GNE
All data will be available in the paper and its supplement. Potential missing information can be obtained from the authors upon reasonable request.
EP, AG, KB and RH designed the research study. EP and PS performed the research and analyzed the data. EP, KB, PS and AG wrote the manuscript. All authors contributed to editorial changes in the manuscript and read and approved the final version. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We acknowledge the contribution of the 735 polySia antibody, kindly provided by Prof. Gerardy-Schahn, Hannover Medical School, Germany. We thank James Morewood, Cardiff University, United Kingdom, for critically proofreading our article.
This research was funded by the Deutsche Forschungsgemeinschaft (460683975) as part of the European Joint Programme Rare Diseases (ProDGNE). We acknowledge the financial support of the Open Access Publication Fund of the Martin-Luther-University Halle-Wittenberg.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.