


 , Tao Li 4,*, Jiefeng He 1,2,*, Haoliang Zhao 1,2,*
, Tao Li 4,*, Jiefeng He 1,2,*, Haoliang Zhao 1,2,*1 Third Hospital of Shanxi Medical University, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, 030032 Taiyuan, Shanxi, China
2 Department of Hepatobiliary Surgery, Shanxi Bethune Hospital, Shanxi Academy of Medical Sciences, Tongji Shanxi Hospital, 030032 Taiyuan, Shanxi, China
3 Hepatic Surgery Center, Institute of Hepato-Pancreato-Biliary Surgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430030 Wuhan, Hubei, China
4 Department of Surgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, 430022 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
Toll-like receptor 3 (TLR3) is a prominent member of the Toll-like receptor (TLR) family and has the ability to recognize and bind intracellular double-stranded RNA (dsRNA). Once triggered by a viral infection or other pathological condition, TLR3 activates immune cells and induces the production of interferons and other immune response molecules. Additionally, TLR3 is considered an important immune modulator, as it can regulate cell apoptosis and promote anticancer immunity. The investigation and application of TLR3 agonists in digestive system tumors have attracted widespread attention and are regarded as a promising cancer treatment strategy with potential clinical applications. TLR3 expression levels are generally elevated in most digestive system tumors, and higher TLR3 expression is associated with a better prognosis. Therefore, TLR3 has emerged as a novel therapeutic target for digestive system tumors. It has been used in combination with chemotherapy, radiotherapy, and targeted therapy and demonstrated excellent efficacy and tolerability. This has provided new ideas and hopes for the treatment of digestive system tumors. This review discusses the mechanisms of TLR3 and its frontier research in digestive system tumors.
Keywords
- Toll-like receptor 3
- digestive system tumors
- dsRNA
- TLR3 agonist
Cancer represents a significant public health concern globally, as there were approximately 10 million reported cancer related-deaths in 2019 [1]. Among the leading causes of cancer-related mortality, colorectal cancer, liver cancer, and stomach cancer rank as the second, third, and fourth most prevalent types, respectively [2]. Consequently, digestive tract tumors have emerged as significant contributors to human mortality, with current treatment options still lacking efficacy. Chronic inflammation serves as a common risk factor for digestive system tumors. For instance, digestive tract cancers are closely associated with inflammatory bowel disease, viral hepatitis, alcohol-related pancreatitis, and gastroesophageal reflux disease [3]. These chronic inflammatory conditions can create a microenvironment that promotes tumor growth [4]. In low- and middle-income countries, chronic infectious inflammation caused by human papillomavirus and hepatitis accounts for approximately 30% of cancer cases [2]. Additionally, a substantial presence of inflammatory cells, chemokines, and inflammatory mediators is observed within the tumor microenvironment; these factors play indispensable roles in the process of tumor formation [4, 5]. Hence, it is imperative to investigate the relationship between inflammation and cancer, aiming to gain a better understanding of the mechanisms underlying cancer development and to improve diagnostic and therapeutic strategies.
Toll-like receptors (TLRs) are a class of crucial proteins that recognize
pathogenic microorganisms and elicit inflammatory immune responses [6, 7]. TLRs
are widely expressed in human immune cells and play a crucial regulatory role in
human immunity [8]. TLRs belong to the family of pattern recognition receptors
(PRRs) and can specifically recognize structure-conserved molecules produced by
pathogens and damaged cells, thus triggering the production of the inflammatory
response and proinflammatory factors [9]. TLRs were first discovered in
Drosophila, and later, TLR4 was identified in humans [10]. Currently, 10 TLR
subtypes (TLR1–TLR10) are known to exist in the human body [11]. Among them,
TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are extracellular receptors located on
the cell membrane. These receptors are positioned on the surface of the cell
membrane, allowing them to sense microbial molecules in contact with them
[12, 13, 14, 15, 16]. TLR3, TLR7, TLR8, and TLR9 are located inside the cell membrane in the
endoplasmic reticulum, lysosomes, and the endoplasmic reticulum-Golgi
intermediate compartment [17]. TLRs are type I transmembrane proteins composed of
a transmembrane domain, an amino-terminal extracellular domain responsible for
pathogen associated molecular patterns (PAMP) recognition, and a cytoplasmic
carboxy-terminal Toll/interleukin-1 receptor domain (TIR) responsible for
downstream signal transduction [18, 19, 20]. The extracellular domain of TLRs exhibits
a “horseshoe-shaped” structure composed of several protein domains, mainly
consisting of 19–25 leucine-rich repeats (LRRs) that are rich in leucine
residues [21, 22]. Through the LRR domain, different TLR subtypes can recognize
specific pathogen components. For example, TLR1 and TLR6 form a complex with TLR2
to recognize microbial lipopeptides, TLR3 recognizes viral double-stranded RNA
(dsRNA), TLR4 recognizes LPS, TLR5 is a receptor for flagellin, and TLR7/9 are
receptors for unmethylated Cytosine-phosphate-Guanine (CpG)DNA and
single-stranded RNA (ssRNA) [23]. The TIR domain is responsible for activating
downstream signaling pathways, which include four adapters: MyD88, TIR
domain-containing adaptor protein (TIRAP), TIR domain-containing adapter inducing
interferon-
TLRs not only play a role in human innate immunity but have also been involved
in cancer immunotherapy, cognitive function, autoimmune diseases, cardiovascular
diseases, and other areas in the past decade of research [32, 33, 34, 35, 36]. The study of
TLR function has expanded beyond the classic NF-
TLR3 is a crucial member of the TLR family. It is located on the long arm of
chromosome 4 (4q35.1) and encodes a 904-amino acid residue protein with a
molecular weight of 103.8 kDa [42]. Unlike other TLRs, TLR3 has 5 exons in its
DNA; the coding sequence for the structural protein located on exons 2, 3, 4, and
5. However, most other TLRs are encoded by only one or two exons [20]. The
overall structure of TLR3-extracellular domain (ECD) is characterized by a large
curved helical tube containing multiple subdomains in the membrane region. The
most important subdomains are the N-terminal domain consisting of 23 leucine-rich
repeats (LRR) and the C-terminal domain (LRR-CT), which together form a globular
LRR domain. The LRR domain is composed of a
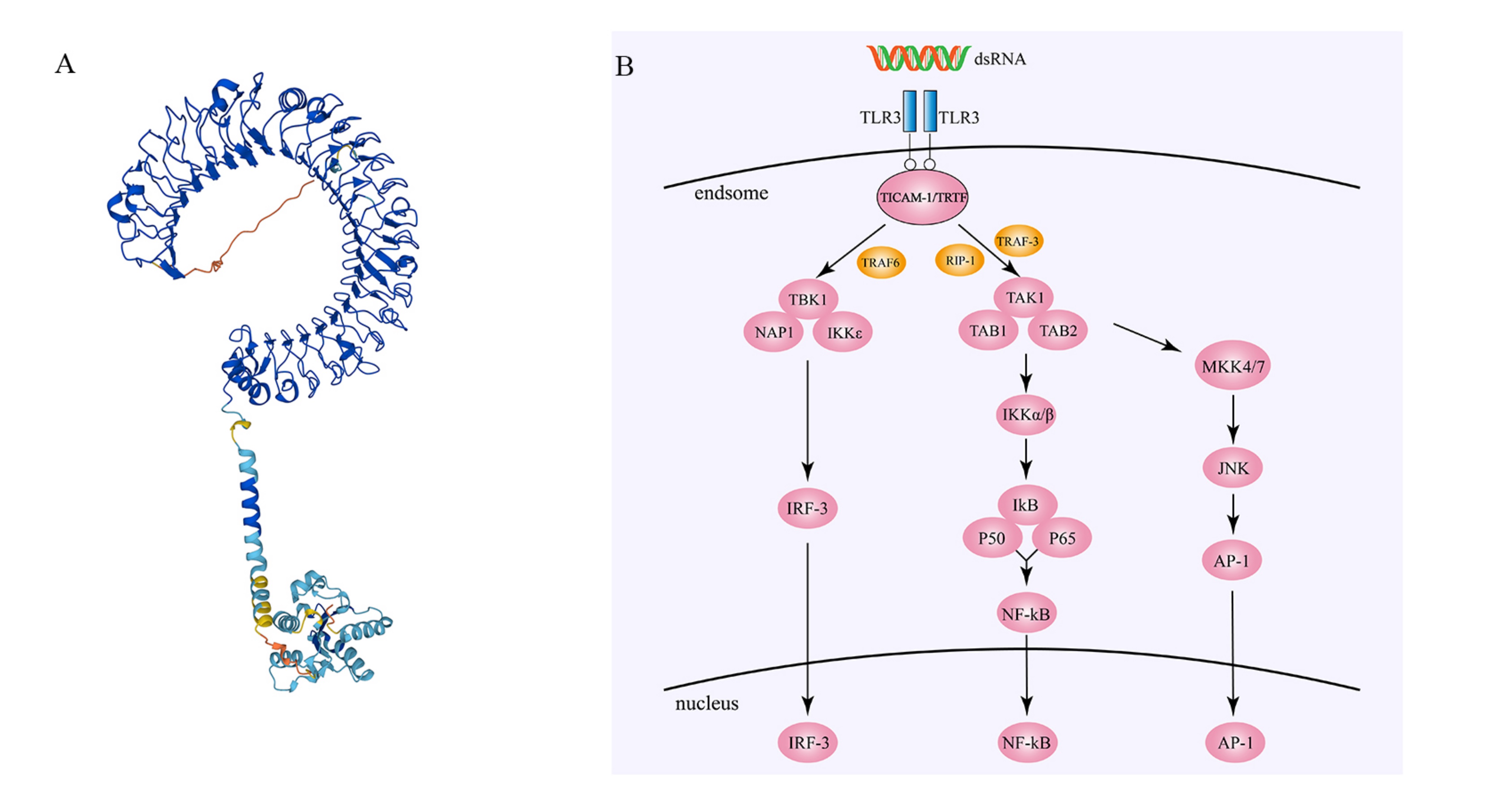 Fig. 1.
Fig. 1.Structure and regulatory mechanisms of Toll-like receptor 3
(TLR3). (A) The three-dimensional structure of Toll-like receptor 3 (source:
https://alphafold.ebi.ac.uk/entry/O15455, Copyright [2023], permission granted).
(B) After double-stranded RNA (dsRNA) binds to two TLR3 molecules, the TIR domain
of TLR3 binds to the TIR domain of TICAM-1/TRIF, and TRAF3 links TBK1 to
TICAM-1/TRIF. TBK1, NAP1, and IKK
TLR3 is expressed in various cell types, including dendritic cells, macrophages, epithelial cells, endothelial cells, and fibroblasts. In macrophages, fibroblasts, and certain epithelial cells, TRL3 is found both on the cell surface and within the cytoplasm. However, in dendritic cells, TLR3 is predominantly located in the endoplasmic reticulum (ER) and lysosomal membranes [45, 46]. In the ER, TLR3 forms a complex with other proteins, such as Unc-93 homolog B1 (UNC93B1), a transmembrane protein that helps TLR3-fold properly and localize correctly in the ER [47, 48, 49]. In lysosomes, TLR3 is involved in the recognition of viral RNA and regulates the immune response along with other proteins, such as TRIF and IRF3 [50]. Additionally, TLR3 can modulate its function by translocating to the cell membrane or nucleus, and upregulating UNC93B1 can promote TLR3 translocation to the cell membrane and activation by external dsRNA [51]. Some studies have also suggested that TLR3 can translocate from the ER to the nucleus after cellular stimulation, although the mechanism of this translocation is not yet clear [52]. In addition to the aforementioned subcellular locations, TLR3 also exists on the cell surface of cells such as in human fibroblasts and lung epithelial BEAS-2B cells [46, 53]. Despite some exploration of the subcellular localization of TLR3, further research is still needed.
The ligands of TLR3 are primarily double-stranded RNA (dsRNA), which is a double-stranded structure formed by complementary base pairing of two RNA strands. dsRNA is widely present both inside and outside of cells and is a structural component of the genomic RNA of many RNA viruses. dsRNA can enter cells through various pathways, such as viral infection, autophagy, and endocytosis. In addition to viral infection, dsRNA also has an endogenous source, as cellular RNA produced by tissue damage or cell death can serve as a potential source of dsRNA [54].
TLR3 exists as a monomer and is membrane-bound in resting cells. Upon binding of the extracellular domain (ECD) of TLR3 to dsRNA or other TLR3 agonists, dimerization occurs [55, 56]. The extracellular domain (ECD) of TLR3 undergoes dimerization upon binding to these dsRNAs [57]. Notably, the binding of TLR3-ECD to dsRNA requires an acidic environment [58]. TLR3-ECD binds to dsRNA at two sites located at the two ends of its “horseshoe-shaped” structure (N- and C-termini), forming a coordinated and stable dsRNA-TLR3 signaling complex composed of one dsRNA and two TLR3 molecules [59]. Subsequently, the oligomerization structure of TLR3 undergoes a change, causing TLR3 to be internalized with dsRNA into endoplasmic reticulum (ER) vesicles [60]. Inside ER vesicles, TLR3 binds to the TRIF adaptor protein. This thereby activates a cascade of signaling molecules, induces the expression of various cytokines and antiviral proteins, and promotes the activation of innate immune cells and immune responses.
TLR3 is fully dependent on TICAM-1/TRIF for downstream signaling, and the
TICAM-1/TRIF pathway ultimately activates AP-1, NF-
The signaling pathway mediated by Toll-like receptor 3 is critical for host
defense against pathogen invasion. However, excessive responses can also cause
harmful damage to the host. It has been found that the E3 ubiquitin ligase TRIM32
can affect the recruitment of TBK1 to TRIF, thereby preventing further signal
transduction by TRIF [63]. In addition, suppressor of cytokine signaling 1
(SOCS1) has been found to inhibit the cytokine receptor-associated signal
transduction (JAK-STAT) pathway in the TLR3 signaling pathway [64], thus
suppressing immune responses. It is noteworthy that SOCS1 can also be induced by
cytokines (such as IFN-
In addition to its own regulatory mechanisms, TLR3 can also cross-regulate other signaling pathways and participate in various immune responses [66]. Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) can also recognize dsRNA and trigger interferon responses to inhibit viral replication and spread. Studies have shown that there is cross-regulation between the TLR3 and RIG-I pathways. TLR3 and RIG-I can mutually activate each other, thereby enhancing interferon production and synergistically acting against viral infections [67]. In addition, TLR3 and TLR4 act together on the TIR domain. It has been found that the TIR domains of TLR3 and TLR4 can form heterodimers and coactivate downstream signaling pathways, thereby enhancing host immune responses against pathogens [68]. However, the TLR3 and TLR4 signaling pathways can activate both the TRIF pathway and the MyD88 pathway, playing important roles in host immune responses [69].
TLR3 expression in tumors is not limited to the endoplasmic reticulum membrane.
Upon activation in tumors, TLR3 triggers the activation of IRF3. This leads to
the production of IFN-
Studying the regulatory mechanisms of TLR3 is of great significance for a deeper understanding of how the immune system fights against pathogens and for providing a scientific basis for the development of more effective vaccines and antitumor drugs. Furthermore, by investigating the regulatory mechanisms of the TLR3 signaling pathway, we can also better understand the pathogenesis of autoimmune diseases, cancer immunotherapy, and other fields and provide a scientific basis for the development of new treatment strategies, which has important clinical implications.
The tumor immune microenvironment (TME) refers to a complex system composed of
various cells and molecules adjacent to cancer cells, including immune cells,
vascular endothelial cells, fibroblasts, matrix molecules, cytokines, and
chemokines, among others [78]. These components interact with each other within
the tumor microenvironment, influencing cancer occurrence, development, and
treatment outcomes. TLR3 is expressed on various immune cells, including
epithelial cells, T cells, B cells, NK cells, and dendritic cells (DCs) [79, 80, 81].
Several studies have shown that TLR3 induces the production of IL-12 and IFN via
the TICAM-1 (TRIF) signaling pathway. Thus, it promotes the maturation of
CD8
In summary, TLR3 plays a critical role in the tumor microenvironment (TME), but research on the immune microenvironment of digestive system tumors involving TLR3 is currently limited. Using The Cancer Genome Atlas Program (TCGA) database and ssGSEA method, we evaluated the expression levels of TLR3 in 22 immune cell subtypes and presented its distribution in digestive system-related tumors in Fig. 2. Additionally, we applied the ESTIMATE algorithm to estimate the stromal score and immune score of TLR3 in digestive system tumors and demonstrated the correlation between TLR3 and the tumor microenvironment in Fig. 2. We found that TLR3 is correlated with the immune microenvironment of most digestive system tumors. These findings offer insight into the potential therapeutic applications of TLR3 modulation in digestive system tumors.
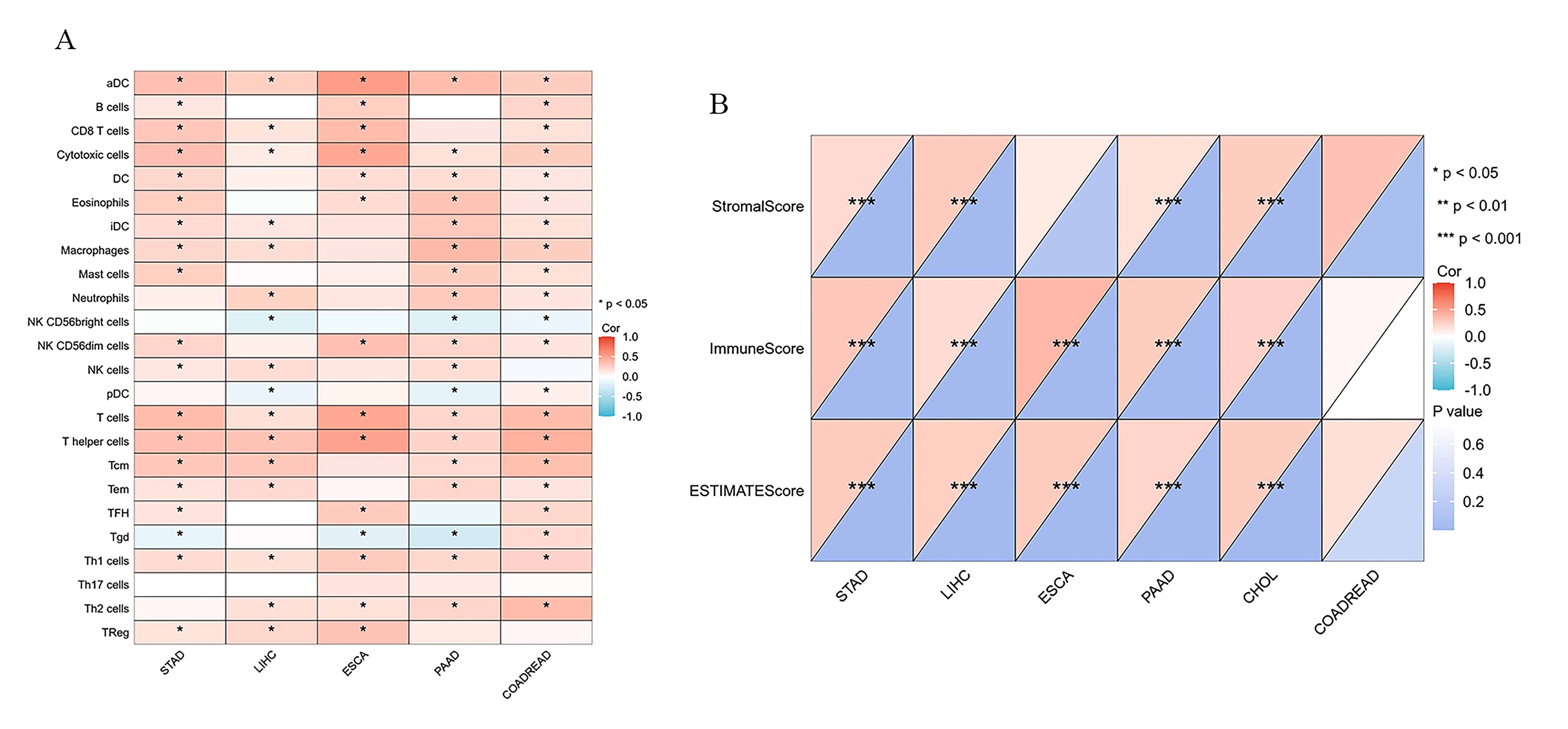 Fig. 2.
Fig. 2.Correlation of TLR3 with immune infiltration in digestive system
tumors. (A) The relationship between TLR3 and immune cell subtype infiltration
in digestive system tumors was analyzed using the ssGSEA algorithm based on The Cancer Genome Atlas Program
(TCGA) database. (B) The correlation between TLR3 and tumor immune score, stromal
score, and ESTIMATE score was analyzed based on the ESTIMATE algorithm.
Correlations were determined using the Pearson method, with *p
Therefore, we infer that the activation of the TLR3 signaling pathway may promote the infiltration and activation of immune cells to enhance the expression and presentation of tumor antigens, thus enhancing the antitumor immune response in most digestive system tumors. These findings provide a potential therapeutic target for improving the immune microenvironment of digestive system tumors.
Esophageal cancer (EC) is divided into two major types: esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC). Among all ECs, 87% are ESCC, but EAC is spreading in developed countries in Europe and America [97, 98]. Although the survival rate of esophageal cancer has significantly increased over the past fifty years, the five-year survival rate remains below 20% [98]. Early detection difficulties and limitations in late-stage treatments are obstacles to improving survival for patients with esophageal cancer [99]. Chronic inflammatory stimuli, such as smoking, alcohol consumption, spicy foods, Barrett’s esophagus, and gastroesophageal reflux disease (GERD), may contribute to the development of esophageal cancer [100].
The TLR family is involved in the inflammatory response in esophageal cancer.
The expression of TLR3 gradually increases as the disease progresses, from
esophageal simple hyperplasia (ESSH) and intraepithelial neoplasia (IEN) to
esophageal squamous cell carcinoma, and TLR3 expression gradually increases in
tumor tissue. Cases with high TLR3 expression also have a higher rate of lymph
node metastasis [101, 102, 103]. Patients with high expression of TLR3 in esophageal
cancer tissues demonstrate a better 5-year survival rate than those with low
expression, and they also exhibit increased sensitivity to chemotherapy. This
outcome is associated with the correlation between high TLR3 expression and
infiltration and activation of immune effector cells, as well as the involvement
of apoptotic pathways. Therefore, for esophageal cancer patients with high TLR3
expression, adjuvant chemotherapy following surgery may further improve the
prognosis of advanced-stage patients [100, 101]. The activation of the
TLR3-mediated inflammatory response by necrotic esophageal epithelial cells
upregulates the expression of interleukin-8 and NF-
Gastric cancer is the fifth most common cancer and the third leading cause of cancer-related deaths worldwide [106]. Among the various pathological types, gastric adenocarcinoma (GAC) is the most prevalent and is typically treated with a multidisciplinary approach involving surgery and chemotherapy. Ning Wang and Dingsheng Liu [107] found that TLR3 mRNA expression in GAC tissues is significantly lower than that in normal gastric tissues. Furthermore, copy number variation (CNV) analysis indicated a significant deletion of TLR3 DNA copies [107]. In contrast, Belen Fernandez-Garcia et al. [108] indicated that the expression of TLR3 in gastric cancer cells, as detected by immunohistochemistry (IHC), is relatively higher than that in stromal cells. Furthermore, high expression levels of TLR3 in gastric cancer cells are significantly associated with unfavorable pathological types and lower overall survival rates in patients [108]. A retrospective study involving 564 patients with gastric adenocarcinoma revealed that patients with high nuclear expression of TLR3 had a lower 5-year survival rate. However, there was no significant difference in the expression of TLR3 in the cytoplasm [109]. Zhihao Huang et al. [110] also noted a positive correlation between TLR3 expression levels and various immune biomarkers, including CD8+ T cells, CD4+ T cells, macrophages, neutrophils, dendritic cells, and others. Additionally, they found that high expression of TLR3, compared to that in normal tissues, was associated with increased resistance to treatment in patients [110]. In addition, another study found that the TLR3 agonist poly(I:C) can induce apoptosis of human gastric adenocarcinoma cells through endogenous delivery and inhibit the growth of human gastric adenocarcinoma in a nude mouse model [111]. Therefore, further investigation of the role played by TLR3 in the progression of gastric cancer is expected to provide a promising prognostic marker and a potential viable target for immunotherapy in the treatment of gastric cancer.
Hepatocellular carcinoma (HCC) accounts for approximately 90% of primary liver cancers. Risk factors associated with HCC include viral, metabolic, and immune-related diseases [112, 113]. The incidence of HCC in the middle-aged population (30–59 years old) has significantly decreased worldwide, mainly due to the successful implementation of the hepatitis B virus (HBV) vaccination program. Liver transplantation and surgical resection are early treatment options for HCC. However, most patients are diagnosed at an advanced stage [114]. Therefore, exploring new treatment directions is necessary. Researchers have found substantial expression levels of TLR3 in the cell membrane and cytoplasm of HCC cells, suggesting the need to investigate the role of TLR3 in HCC [115, 116].
Marc Bonnin et al. [115] found through multimodal experiments that in primary liver cancer tissues, the mRNA and protein expression levels of TLR3 were lower than those in normal tissues. Chronic infection with HBV and HCV can also lead to decreased expression of TLR3 in liver cells, which may be attributed to the suppression of the hepatic pro-inflammatory system. However, there are differing views on the expression changes of TLR3 during the stages of chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma, which may be influenced by the use of antiviral medications [117]. In addition, Cheng Zhou et al. [118] also discovered that HCC patients with high TLR3 expression have a more complete capsule. High expression of TLR3 can promote the activation of natural killer (NK) cells and increase their cytotoxicity. This finding suggests that HCC patients with high TLR3 expression may have a longer overall survival [119]. Weiyun Li et al. [120] found that serine/threonine protein kinase 4 (STK4) could regulate the TLR3-mediated inflammatory response in macrophages, thereby inhibiting the progression of chronic inflammation to hepatocellular carcinoma. Additionally, in a study conducted by Cheng Zhou et al. [118], lenvatinib enhanced tumor apoptosis by upregulating TLR3 expression in Huh 7 and MHCC-97H cells. In a transgenic mouse study by Marc Bonnin et al. [115], the absence of TLR3 led to a reduction in apoptosis rates in precancerous liver cells and accelerated liver cancer development without affecting tumor malignancy and tumor size. In addition, it did not affect tumor immune infiltration. These findings suggest a potential beneficial role of TLR3 in HCC [115]. However, not all studies support this notion. One study suggested that low TLR3 expression may be associated with smaller tumor size. Moreover, TLR3 mutations can influence the progression from precancerous lesions to liver cancer, indicating that the TLR3+1234CT and TLR3+1234TT genotypes may be risk factors for the development of HCC in HBV- and HCV-related liver cirrhosis [121, 122].
Most HCC occurs in patients with chronic liver diseases, especially viral hepatitis (i.e., HBV and HCV) and liver cirrhosis caused by various etiologies [123]. Studies have shown that using TLR3 agonists can significantly inhibit HBV replication [116, 124, 125], and the upregulation of TLR3 can promote apoptosis of HBV-positive liver cancer cells [116]. We speculate that TLR3 may enhance the control of HBV by the hepatic intrinsic immune system by recognizing HBV virus RNA, thereby increasing the resistance of liver cells to HBV. Similarly, regarding HCV, spontaneous clearance of HCV infection was found in treated patients with elevated TLR3 expression, indicating a role for TLR3 in the existing antiviral state against HCV infection [117, 126].
dsRNA, as an important ligand of TLR3, plays a crucial role in host defense and disease prevention. BM-06 and poly(I:C) are two analogs of dsRNA, and their stimulation of liver cancer cells can significantly inhibit apoptosis and migration while inducing autophagy [96, 127, 128, 129]. However, the inhibitory effects of TLR3 agonists alone are limited. Peng Shen et al. [130] found that poly(I:C) can inhibit HCC proliferation and induce apoptosis through TLR3-dependent mechanisms. Furthermore, when used in combination with arsenic trioxide (ATO), poly(I:C) can enhance ATO-induced apoptosis in SMMC7721 cells by promoting ROS-dependent mitochondrial membrane potential loss (Dym loss) Nevertheless, some studies have shown that low concentrations of poly(I:C) not only fail to significantly inhibit the proliferation of liver cancer cells and induce cell apoptosis but also increase their migration and invasion ability.
Pancreatic cancer (PC) is a highly malignant tumor and the seventh leading cause
of cancer-related deaths [131]. Current treatment options, such as the modified
FOLFIRINOX regimen, have limited efficacy in improving patient survival rates. In
recent years, immunotherapy has emerged as a new treatment option for pancreatic
cancer [132]. Spas Dimitrov Markov et al. [133] utilized a vaccine
targeting MUC1 with IgE in combination with a TLR3 agonist and immune checkpoint
inhibitors to induce NK cell and CD8+ T-cell-dependent antitumor responses in
mice. Studies have shown that poly(I:C) can enhance the cytotoxic effects of
tyrosine kinase inhibitors (TKIs) on human pancreatic cancer cell lines (hPDA)
and can induce hPDA cell line lysis in vitro by enhancing the cytotoxic
activity of
However, some studies have shown that the expression of TLR3 in PC cells is significantly higher than that in normal pancreatic ductal epithelial cells, and the overexpression of TLR3 promotes malignant behaviors, such as proliferation, migration, and invasion of PC cells [139]. Additionally, it has been reported that poly(I:C) can promote the occurrence of PC in KRAS mutant mice [140]. Benzimidazole (C10) is a TLR3 inhibitor that does not rely on Wnt5a, and it can significantly inhibit the ability of hPDA cells to proliferate, migrate, and invade in vitro [141]. Therefore, despite the varying roles of TLR3 observed in PC in different studies, the potential of TLR3 agonists in the treatment of pancreatic cancer is evident. Consequently, TLR3 agonists hold promise as a future therapeutic option for pancreatic cancer.
Colorectal cancer (CRC) is the second leading cause of cancer-related deaths
worldwide, and the inflammatory pathway promotes its development [142]. Patients
with inflammatory bowel diseases, including Crohn’s disease and ulcerative
colitis, have a significantly increased risk of developing CRC [143]. In CRC
research, the efficacy of TLR3 agonists is diminished when facing tumors with
KRAS mutations. However, Shiqi Long et al. [144] proposed a solution to
this problem by combining respiratory syncytial virus and cetuximab. Both
respiratory syncytial virus and poly(I:C) serve as TLR3 agonists and can enhance
antibody-dependent cell-mediated cytotoxicity (ADCC) against CRC cells by
activating TLR3 and affecting the IKK
Cholangiocarcinoma (CCA) is a malignant tumor that originates from the epithelium of the bile duct, accounting for 3% of gastrointestinal tumors. In recent years, the incidence of CCA has increased, but the specific etiology has not been determined [151]. Chronic inflammation (such as cirrhosis, viral hepatitis, primary sclerosing cholangitis, biliary infections, and parasitic infections) and an immunosuppressive tumor microenvironment are generally considered risk factors for CCA [152]. Research on TLR3 in CCA is not yet fully developed, and only Thanpisit Lomphithak et al. [153] mentioned TLR3 and its activators in a new CCA treatment direction in 2020. Using in vitro experiments, Thanpisit Lomphithak et al. [153] confirmed that the combination of poly(I:C) with an SMAC mimetic can induce apoptosis and necroptosis in cholangiocarcinoma cells. While poly(I:C) induces apoptosis and enhances invasion, the SMAC mimetic inhibits cIAP to release RIPK1-dependent cell death and reduce invasion. Although the exact mechanism of reduced invasion is unknown, in vitro experiments support this finding. Therefore, this study presents a novel treatment approach of combining poly(I:C) and SMAC mimetics in patients with tumors overexpressing RIPK1 to inhibit tumor invasion and increase cancer cell apoptosis and necroptosis. This idea provides a new option for CCA treatment, although it may only be effective in patients with high RIPK1 expression in tumor tissue [153].
TLR3 is generally expressed in tumors, but its role and mechanism in different
cancers are very diverse. In digestive tumors, the expression of TLR3 in tumor
cells is often higher than that in normal tissues (Table 1, Ref. [97, 100, 105, 108, 111, 115, 116, 118, 121, 127, 129, 130, 134, 135, 139, 149, 153]). High
expression of TLR3 can serve as a prognostic indicator in cancer patients, as it
may increase the invasive ability of tumors. However, it is also associated with
a favorable response to chemotherapy. High expression of TLR3 may be associated
with a longer overall survival, which could be attributed to the intrinsic
antitumor response of TLR3. In liver cancer tissues, the expression of TLR3
gradually increases during the progression of hepatitis and liver cirrhosis but
decreases in liver cancer tissues. However, it remains higher than in normal
tissues. This phenomenon may be attributed to the immune evasion mechanisms of
liver cancer [115]. In the tumor microenvironment, dendritic cells (DCs) act as
the most effective antigen-presenting cells (APCs) to initiate and modulate
innate and adaptive immunity. However, the tumor microenvironment suppresses the
maturation of DCs. Therefore, using TLR3 agonists to enhance the anticancer
effect to treat tumors seems to be a feasible direction for conventional DCs
(cDCs). In a study conducted by Huang et al. [87], an in situ
dendritic cell (DC) vaccine, HELA-Exos, was constructed by loading the human
neutrophil elastase protein (ELANE) and the TLR3 agonist Hiltonol into exosomes.
It can specifically induce the immunogenic cell death (ICD) of breast cancer
cells, thereby activating the antigen presentation function of cDC1s and
cross-activating tumor-reactive CD8+ T cells. Thus it plays an anticancer role
[87]. The use of vaccines or drugs that transport TLR3 agonists and other
anticancer drugs through specific carriers has been studied and even applied in
many types of cancers. TLR3 agonists have been combined with 5-fluorouracil or
IFN-
| Cancer type | Sample | Alteration | Related clinical and pathological features | Significance | Reference |
| Esophageal cancer | Esophageal cancer tissue | / | / | Potential immunologic adjuvant | [105] |
| Esophageal cancer tissue | / | High expression has better overall and disease specific survival at 5 years; high expression has stronger sensitivity to chemotherapy | Independent prognostic factors | [100] | |
| KYSE140 KYSE150 KYSE180 KYSE220 KYSE270 | Upregulation | / | / | [100] | |
| Gastric cancer | Gastric cancer tissue | Upregulation | Patients with high TLR3 expression have poor prognosis | Negative regulator | [108] |
| BGC-823 | / | / | Antitumor activity | [111] | |
| Hepatocellular carcinoma | Primary liver cancer tissue | Downregulation | High expression has better recurrence free survival | Antitumor activity | [115] |
| Primary liver cancer tissue | / | High expression has better prognosis and smaller tumors | Antitumor activity | [118] | |
| SK-HEP-1 Hep3B PLC/PRF/5 HepG2 Focus HUh7 | Downregulation | / | Antitumor activity | [115] | |
| Primary liver cancer tissue | No difference | The positive rate of TLR3 is negatively correlated with serum AFP levels, while HBsAg infection is positively correlated | Antitumor activity | [116] | |
| Huh 7 | / | / | Antitumor activity | [97, 127, 129, 130] | |
| MHCC-97H SMMC7721 | |||||
| HepG2 HepG2.2.15 | |||||
| Primary liver cancer tissue | / | Positive correlation with tumor size | Negative regulator | [121] | |
| Pancreatic cancer | BXPC-3 PAC1 | Upregulation | / | Negative regulator | [139] |
| HPDA | / | / | Antitumor activity | [134] | |
| Panc 89 PancTul Colo 357 | / | / | Antitumor activity | [135] | |
| Colorectal cancer | Colorectal adenocarcinoma tissue | Downregulation | / | / | [149] |
| Cholangiocarcinoma | Cholangiocarcinoma tissue | Upregulation | / | / | [153] |
HPDA, human pancreatic cancer cell lines.
Furthermore, the mechanisms of TLR3 agonist therapy for digestive system tumors are not yet fully understood. Their interactions with other signaling pathways, differential expression and functions in different types and stages of tumors and their regulatory role in the tumor microenvironment require further exploration. Additionally, the toxicity of TLR3 agonists should be carefully considered. TLR3 has emerged as a new breakthrough in the treatment of digestive system tumors, and in the future, TLR3 agonists may extend their remarkable potential beyond digestive system tumors.
TLR3 is widely expressed in digestive system tumors, and its activation generally promotes tumor cell apoptosis and inhibits tumor progression. However, the effects of TLR3 may vary depending on its subcellular localization. This highlights that TLR3 agonists are not suitable for standalone therapy in digestive system tumors but rather as adjuvants in combination with other chemotherapy drugs, targeted agents, immune checkpoint inhibitors, etc. This combination approach allows for maximizing the impact on tumors and human tolerance. In the case of precancerous lesions in the digestive system, TLR3 expression undergoes a dynamic process, generally showing an increase compared to normal tissues. This may be attributed to the occurrence of chronic inflammation, leading to prolonged activation of the TLR3 pathway. However, research on TLR3 agonists in the context of precancerous lesions is still limited. Overall, TLR3 agonists have promising prospects in the treatment of digestive system tumors, as several are already in clinical trial stages and showing positive outcomes. Currently, TLR3 agonists have been used in human cancer treatment, but challenges remain in terms of their appropriate use and the development of combination treatment strategies.
AP-1, activating protein-1; CNV, copy number variation; dsRNA, double-stranded
RNA; GSEA, gene set enrichment analysis; IFN-
The data used in this study were obtained from publicly available databases, specifically, TCGA datasets.
BH, LZ, TL, JH, and HZ conceptualized the study idea for the review. BH and CZ performed the article’s editing. BH and XW created all figures. CZ and HS analyzed and interpreted the data from TCGA. BH and CZ wrote the manuscript. LZ, TL, JH, and HZ made significant revisions to the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final version. All authors participated sufficiently in the work and agreed to be accountable for all aspects of the study.
Not applicable.
The author thanks current members of the laboratory for their valuable contributions.
This article was supported by the National Natural Science Foundation of China (Grant No. 82073090), Human Resources and Social Security Department System of Shanxi Province (Grant No. 20210001), Research Project Supported by Shanxi Scholarship Council of China (Grant No. 2021-116), Shanxi ‘136’ Leading Clinical Key Specialty (Grant No. 2019XY002), and Key Laboratory of Hepatobiliary and Pancreatic Diseases of Shanxi Province (Preparatory).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.