
 , Rui Wang 2,*
, Rui Wang 2,*1 Department of Medical Oncology, Affiliated Tumor Hospital, Nantong University, 226300 Nantong, Jiangsu, China
2 Department of Medical Oncology, Affiliated Jinling Hospital, Medical School of Nanjing University, 210016 Nanjing, Jiangsu, China
†These authors contributed equally.
Abstract
Oxidative stress (OS) is linked to hepatocellular carcinoma (HCC) progression.
HCC may develop as a result of genetic changes, including oxidative injury to
both nuclear and mitochondrial DNA. Signaling pathways regulated by OS, such as
Wnt/
Keywords
- oxidative stress
- HCC
- genetic changes
- signaling pathways
- transcription factors
- tumor microenvironment
- treatment targets
Hepatocellular carcinoma (HCC) represents the predominant type of hepatic
cancer, and is currently the sixth most frequently diagnosed cancer and the third
leading contributor to cancer-associated mortalities, globally [1]. The
progression of HCC is a complicated and multifaceted phenomenon, with its
underlying mechanism still not fully understood [2]. Prominent risk factors for
HCC include chronic infection with hepatitis B virus (HBV) and/or hepatitis C
virus (HCV), exposure to aflatoxin, type 2 diabetes, liver cirrhosis due to
excessive alcohol use, nonalcoholic fatty liver disease (NAFLD), which is linked
to obesity, and smoking [3, 4, 5]. HCC progression involves multiple stages and
several intricate pathways, such as phosphatidylinositol 3-kinase (PI3K)/protein
kinase B (AKT)/mammalian target of rapamycin (mTOR), Wnt/
OS occurs when an imbalance develops between ROS production and accumulation
following stimulation by harmful endogenous or exogenous factors. Free radicals,
such as ROS and reactive nitrogen species (RNS), commonly function as metabolites
in various redox reactions during normal cellular metabolism, and are increased
upon OS initiation [11]. Both ROS and RNS include organic or inorganic molecules
with an odd number of electrons and such molecules are produced during redox
reactions within the body, and each exhibit high reactivity. Despite oxygen’s
crucial role as a substrate in oxidative metabolism, its partial reduction can
lead to the formation of ROS [12]. ROS species generally include hydroxyl radical
(OH
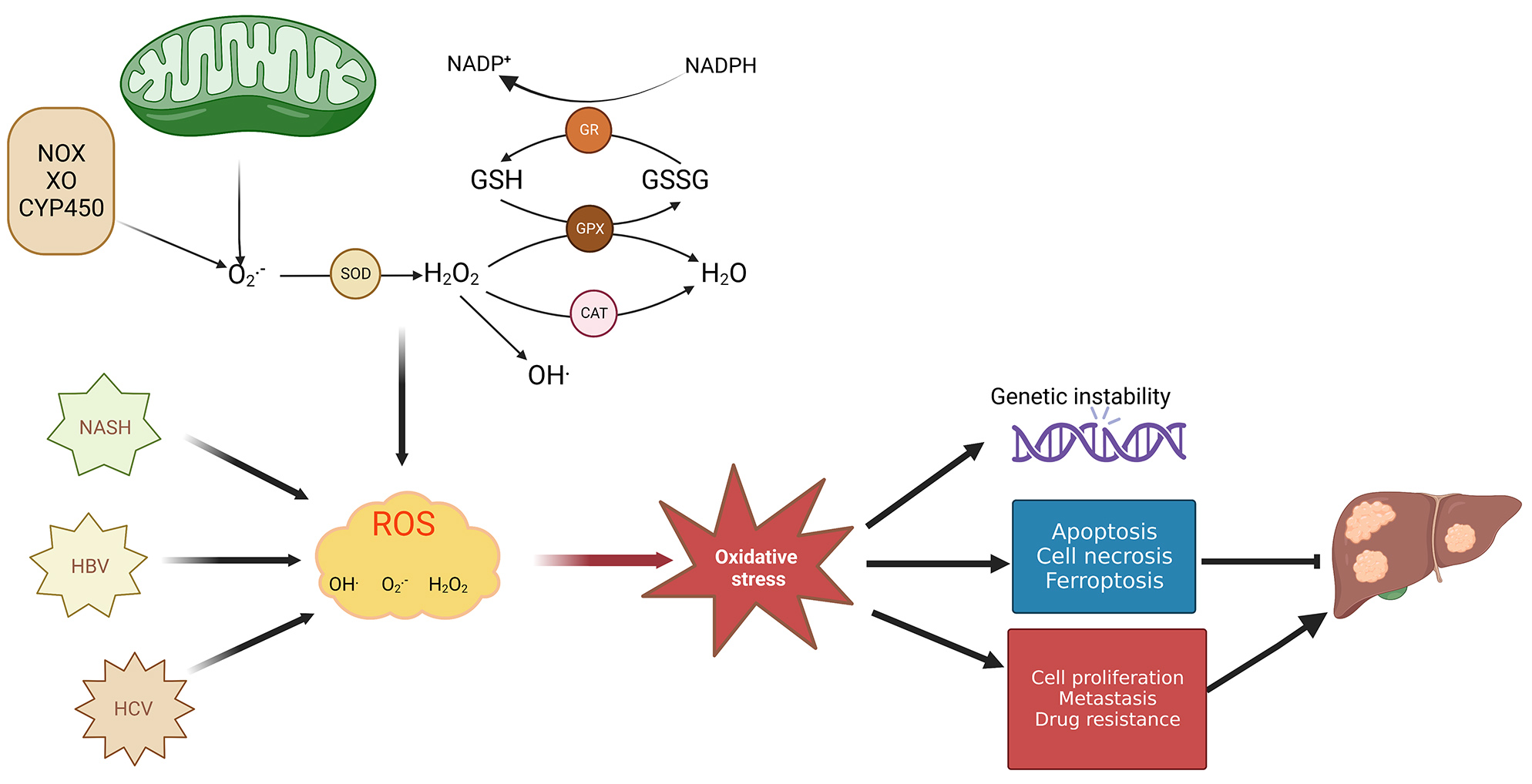
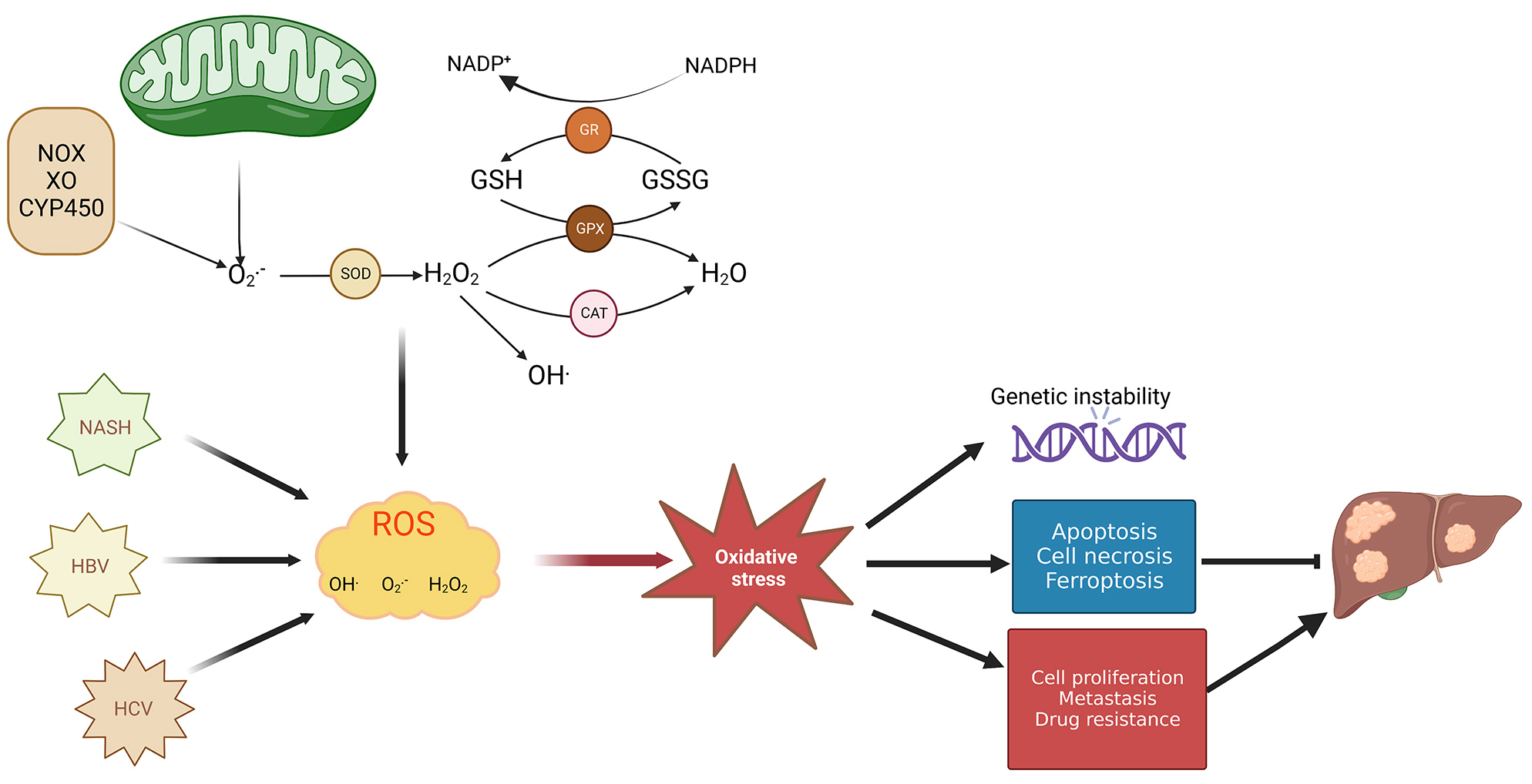 Fig. 1.
Fig. 1.Mechanism of redox balance–related HCC development. Both
mitochondrial electron leakage and oxidase (NOX, XO, and CYP450) enzyme activity
can produce superoxide anions (O
In different stages of cancer formation such as during tumor development, metastasis, and apoptosis, ROS activates various functions depending on cellular location, concentration, and duration within particular subcellular structures [21]. ROS’s dual roles in cancer progression encompass ROS-driven malignant transformation and cell death induced by OS. ROS are capable of promoting alterations in genetic materials which help drive cancer occurrence, growth, and progression of both tumors and drug resistance. However, long-term overproduction of ROS is toxic to cells, and increased ROS may trigger apoptotic signals and consequently lead to cell death [22]. Particularly, ROS production at high levels is accompanied by cellular hyperproliferation within the tumor; however, tumor cells are capable of thriving under circumstances where the oxidative burden pushes redox balance away from a reduced state. This adaptability is achieved by tumor cells due to an enhanced antioxidant status, which optimizes ROS-fueled growth while avoiding ROS levels that would induce senescence, apoptosis, or ferroptosis [23] (Fig. 1). For example, oxidative stress may enhance glutaminolysis to promote synthesis of GSH and reduce oxidative stress [24]. However, studies have shown that excessive GSH promoted HCC tumor formation and growth [25]. Recent research demonstrates that tumor cells are capable of both glycolytic and OXPHOS metabolism, which renders them resistant to oxidative stress through enhanced antioxidant response and enhanced detoxification capacity [26]. In addition, changes in mitochondrial metabolism are closely related to the progression and metastasis of HCC [15]. In this review, we will highlight the effect of OS exerts on gene expression, signaling pathways, transcription factors, and tumor microenvironment (TME) in HCC development. Additionally, we will explore how OS functions in therapeutic treatment of liver cancer.
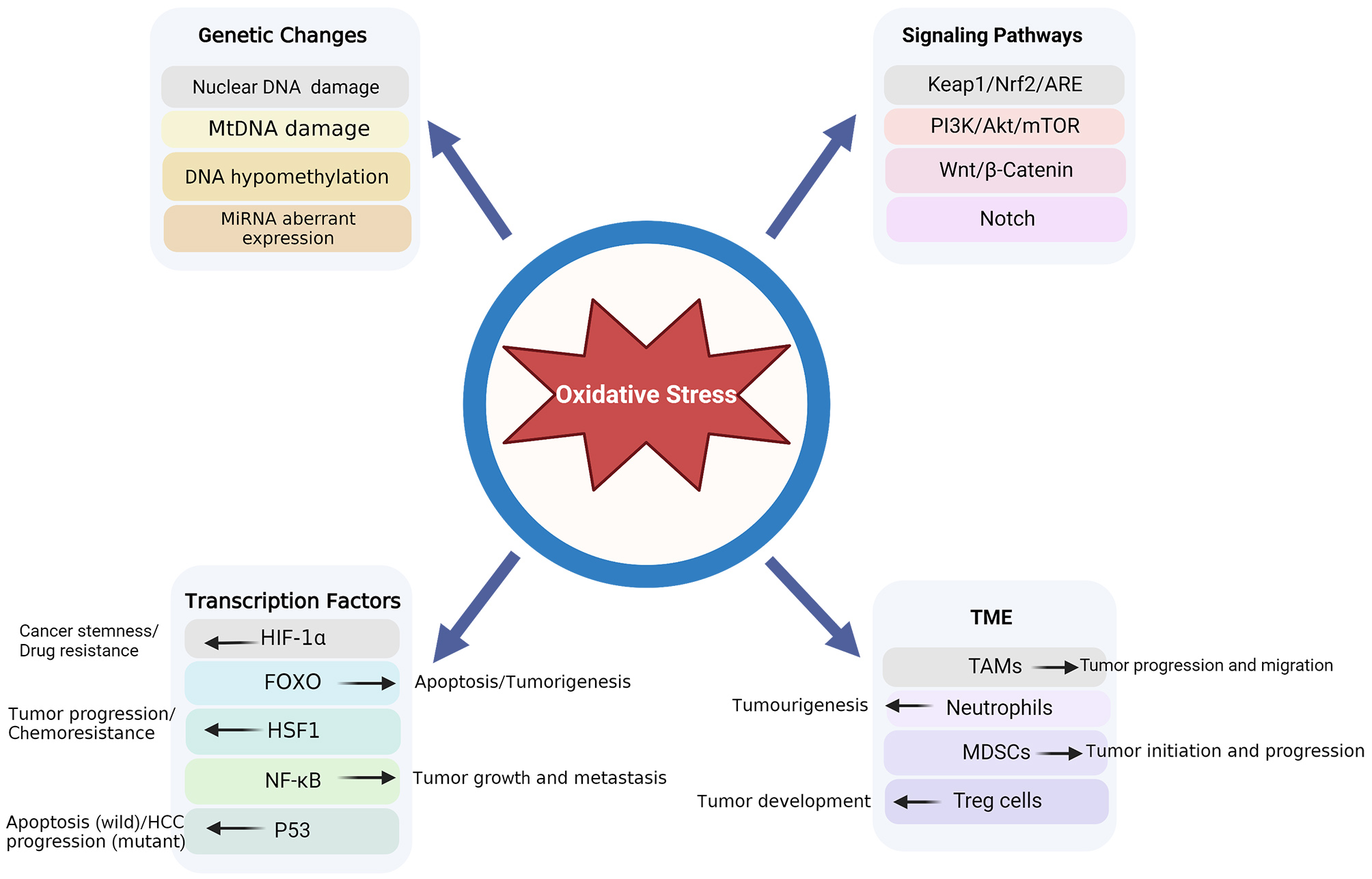
Four aspects are involved in the process via which OS results in HCC development: genetic alterations, signaling pathways, transcription factors, and the TME (Fig. 2).
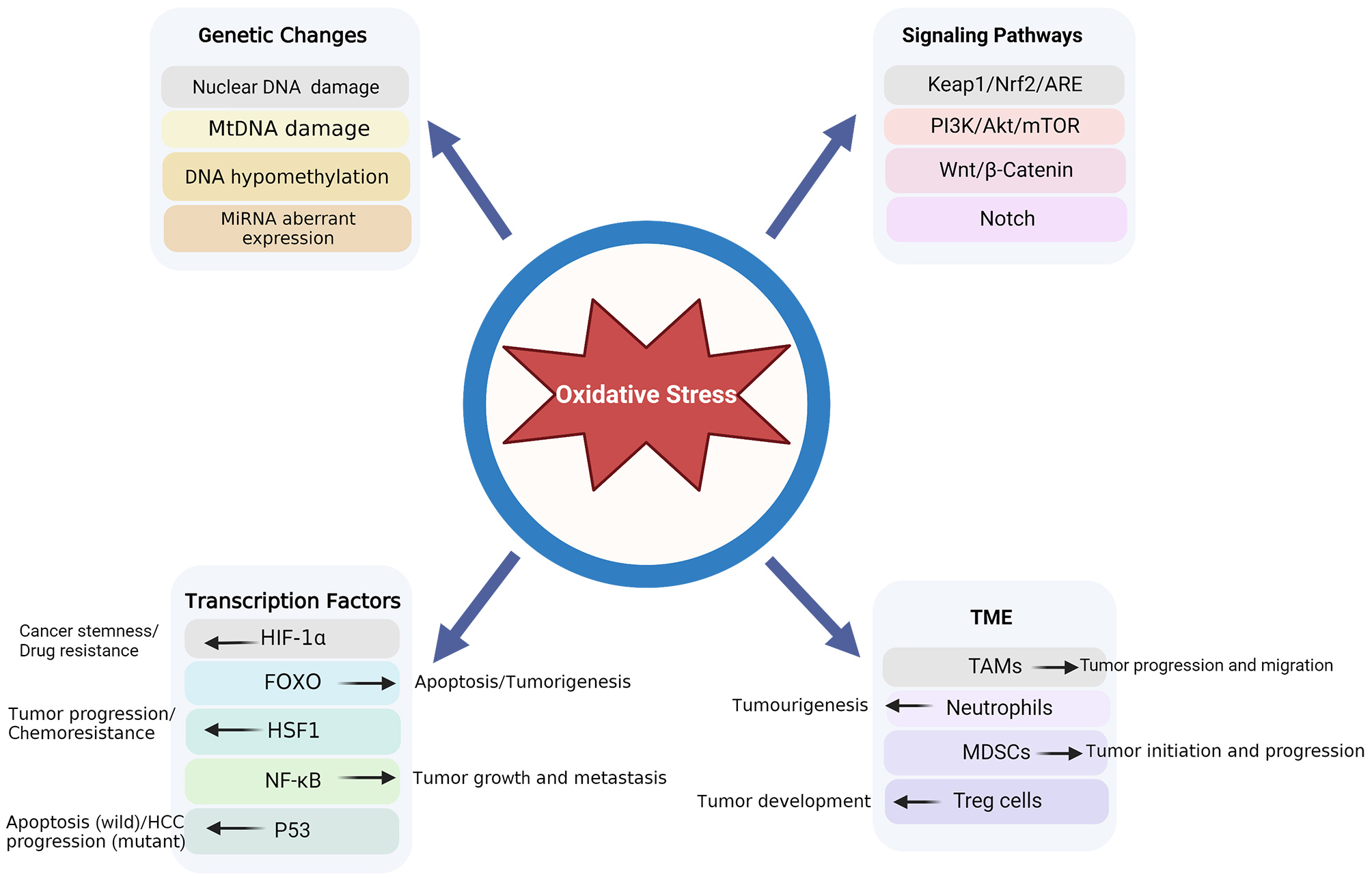 Fig. 2.
Fig. 2.OS-associated pathogenesis in HCC. HCC, hepatocellular carcinoma; OS, oxidative stress.
HCC can be induced by OS-associated genetic changes, including both oxidative nuclear and mitochondrial DNA damage, DNA hypomethylation, and aberrant microRNA expression. ROS-induced DNA damage and genetic instability are crucial developments in HCC onset and advancement [27, 28]. 8-Hydroxydeoxyguanosine (8-OHdG), an oxidative DNA adduct, is a widely accepted marker for OS [29]. Ma-On et al. [30] studied the cellular response to OS and the level of DNA damage in human HCC tissues attributable to oxidation. They found that the 8-OHdG expression level was elevated in HCC tissues, particularly in HCC cell nuclei, and was linked to poor patient survival. In vitro experiments demonstrated that oxidative stress was induced by ROS, which substantially increased 8-OHdG expression levels in HCC cells. These outcomes indicate that ROS fosters an oxidative microenvironment in HCC cells, crucially promoting tumor progression.
Mitochondria can be impacted by various damaging factors, such as drugs, viruses, fat accumulation, and carcinogens [31]. When these factors exceed the mitochondria’s coping ability, abnormalities in mitochondrial structure and function occur, for example, ROS accumulation following a disruption in the ROS scavenging system [18, 32]. MtDNA, which lacks histone protection, is vulnerable to damage caused by ROS [33, 34]. Mitochondrial dysfunction contributes to ROS accumulation and mtDNA damage, potentially leading to HCC advancement. Shetty et al. [35, 36] reported that mitochondrial ROS levels doubled at the time of the disease progression of nitrosodiethylamine-induced HCC in mice, resulting in DNA damage and proto-oncogene activation, and ultimately promoting tumorigenesis.
A high-calorie diet can cause fat accumulation, leading to metabolic stress and
abnormalities in liver mitochondria [16]. When the condition progresses to NASH,
the adaptability and flexibility of the mitochondria are reduced. As previously
described, OS, mitochondrial malfunction, and oxidative mtDNA may facilitate the
progression of NASH to HCC. Xie et al. [37] introduced the hepatitis B
virus x (HBx) gene into the human liver cell line HL7702 and discovered
that, in the presence of OS conditions, HBx triggered the inflammasome NLRP3 in
HL7702 cells, as well as promoted pyroptosis via the mtROS pathway under OS
conditions. The binding of the HBV protein HBx to the mitochondrial outer
membrane enhances its permeability, resulting in the breakdown of mitochondrial
membrane potential and increased ROS generation [38]. In addition, HBx directly
binds to cytochrome c oxidase, thereby disrupting the mitochondrial respiratory
chain and inducing increased ROS production [39]. HCV induces OS in liver cells,
resulting in increased levels of ROS, including O
DNA methylation represents a significant epigenetic process
that modulates gene expression, and is linked to the onset and advancement of
differing cancer types [42]. This mechanism involves the covalent bonding of a
methyl group, catalyzed by DNA methyltransferases (DNMTs), to the carbon at the
5th position of cytosine residues within CpG dinucleotides [42]. ROS can hinder
this mechanism resulting in global DNA hypomethylation [43, 44, 45]. A link between
chromosomal region abnormalities and DNA hypomethylation at repetitive DNA
sequences has been observed, suggesting that global DNA hypomethylation could
trigger chromosomal instability, ultimately contributing to hepatocarcinogenesis
[46]. OS can change DNA methylation status by influencing the function of other
enzymes that maintain epigenetic status including histone methylases, and histone
deacetylases (HDACs). Inducing OS in the HCC cells using hydrogen peroxide
(H
Micro RNAs (miRNAs) are responsible for regulating oncogenes and tumor suppressor genes in HCC, establishing a mechanistic link between epigenetics, inflammation, viral infection, and OS [49]. Both miRNAs and OS participate in the process of development of acute and chronic liver diseases, including viral hepatitis and oncogenesis, by affecting various signaling and metabolic pathways [50]. For example, miR483-3p may promote HCC metastasis via OS stimulation, uncovering a newly-discovered function of epigallocatechin-3-gallate for protection from miR483-3p–regulated HCC metastasis through the epigenetic modulation of miR483-3p [51]. Additionally, miR-33 deficiency improves mitochondrial function and reduces OS, and may represent an effective therapeutic approach for disease progression at different stages of HCC [52]. Overexpression of miR-92 and activation of telomerase activity are linked to the accumulation of ROS-mediated oxidative DNA damage during the development of chronic liver injury in HCC [53].
Numerous studies, including those on the Keap1/Nrf2/ARE, PI3K/AKT/mTOR,
Wnt/
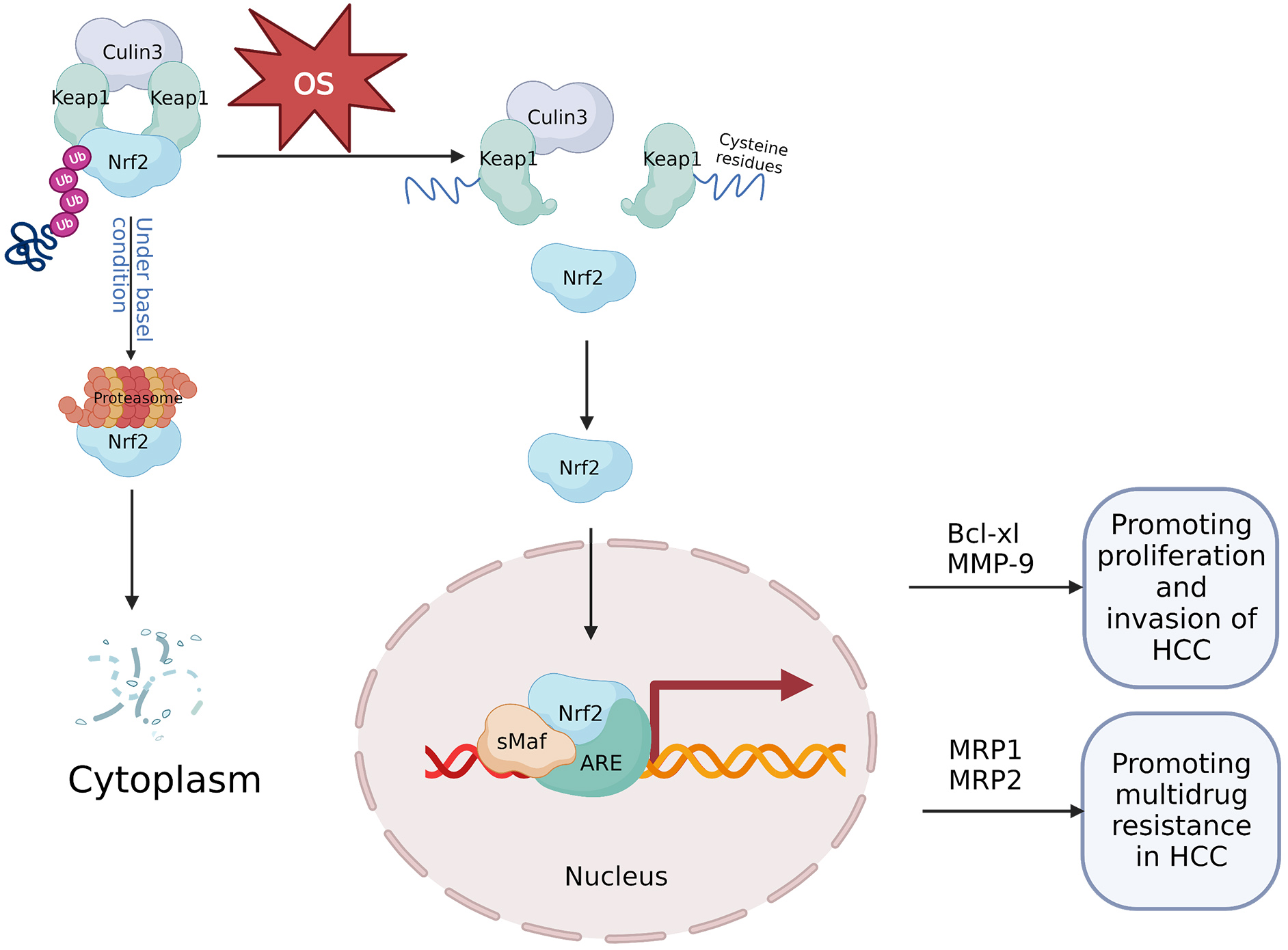
The Nrf2 protein is a critical regulator of antioxidant response [54]. Under normal physiological circumstances, Keap1 functions as a Nrf2 inhibitor, maintaining basal cytoplasmic Nrf2 levels through ubiquitin-proteasomal system degradation of Nrf2 [55]. Conversely, during stress conditions, and most notably OS, Nrf2 expression increases. Cysteine residues within the Keap1 intervening region domain serve as redox sensors, and upon their oxidation, Nrf2 is liberated from its regulatory complex [56]. Further, under OS, the Nrf2 protein stabilizes and initiates a multistep activation process, including nuclear translocation and heterodimerization with its partner, small v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog (sMaf) proteins including MafG. Subsequently the complex recruits transcriptional co-activators and later attachment to target gene antioxidant response elements (AREs) [57]. The Nrf2-sMaf heterodimers occupy specific ARE sites within the genome, recruiting other transcriptional activators and inducing further Nrf2 transcription [58]. As Nrf2 shields cells from stress damage, it is formally classified as a tumor suppressor [59]; however, persistent oxidative stress during cancer development can result in Nrf2 hyperactivation, thus enhancing cancer cell survival [60] (Fig. 3).
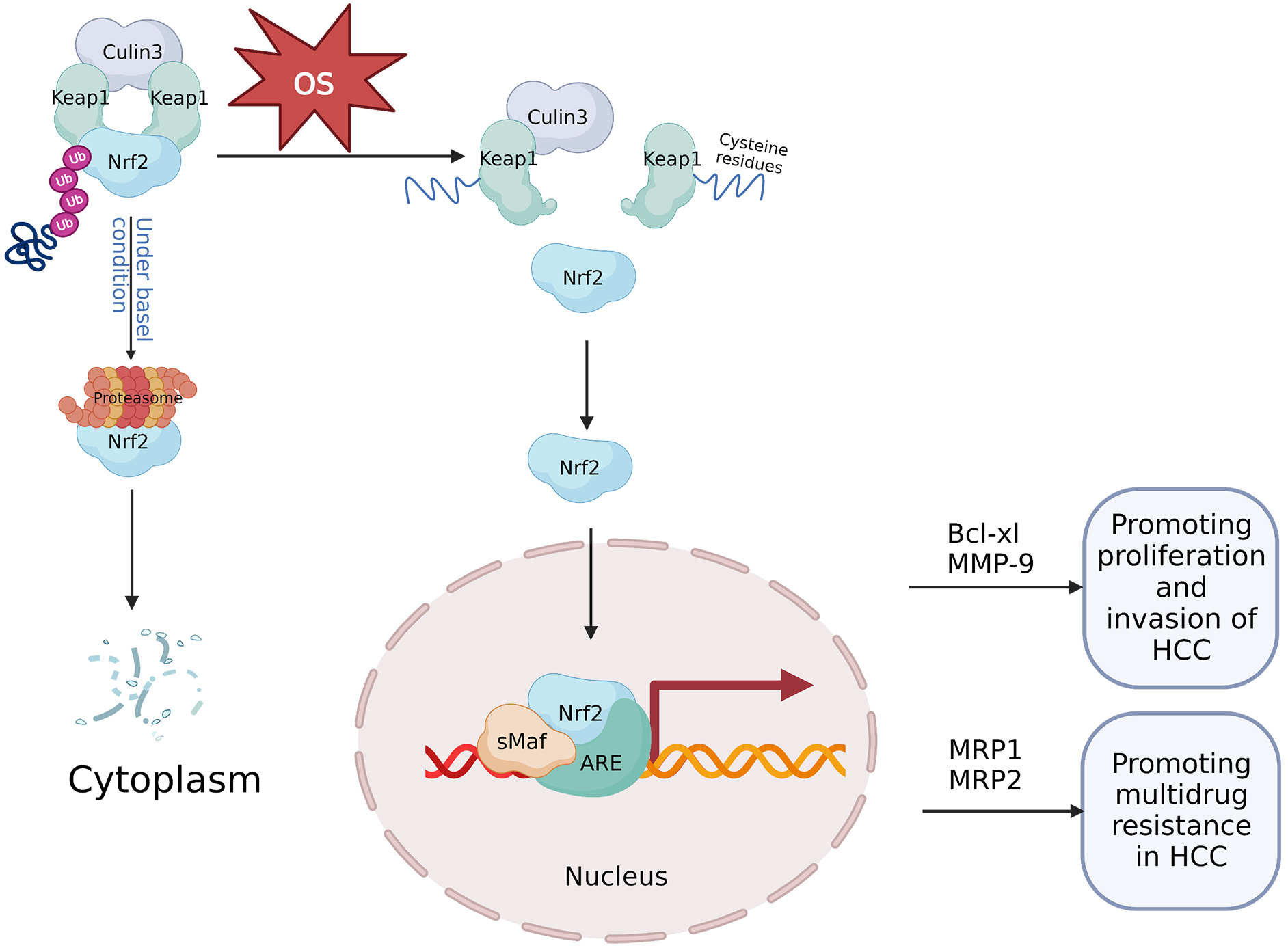 Fig. 3.
Fig. 3.Mechanism of the OS-mediated Keap1/Nrf2/ARE signaling axis in HCC. Under normal physiological circumstances, Keap1 binds to Nrf2 and the ubiquitin ligase culin3, and forms a cytoplasmic complex. Keap1 reduces the stability of the Nrf2 protein through the ubiquitin-proteasome degradation, thereby negatively regulating Nrf2 and maintaining low cellular levels of Nrf2. However, during OS, cysteine residues within Keap1 are oxidized, Nrf2 is freed from its regulatory complex, resulting in stabilization of the Nrf2 protein and translocation to the nucleus where it forms a heterodimer with sMaf. Subsequently, Nrf2-sMaf heterodimers occupy ARE in the genome and activate target genes which contribute to cancer development. For example, Nrf2-dependent activation of Bcl-xl and MMP-9 can enhance growth and invasiveness of cancerous hepatic cells. Further, increased expression of MRP1 and MRP2 mediated by Nrf2 can promote multidrug resistance in hepatocellular carcinoma. ARE, antioxidant response element; HCC, hepatocellular carcinoma; MMP, matrix metalloprotein; MRP, multidrug-associated protein; OS, oxidative stress.
In HCC, Nrf2 expression is associated with cell survival and various clinicopathological factors [61]. Nrf2 is abundantly expressed in cancerous cells and functions in tumor cell division and invasion. ROS significantly increases Nrf2 expression in liver cancer cells and creates an oxidative microenvironment in HCC, thus promoting further tumor development. The advancement of HCC due to ROS may be partially mediated through the activation of Nrf2 [62]. This suggests that stimulation of the Nrf2/Keap1/ARE signaling axis improves response to OS, which is advantageous for tumor cells in promoting liver cancer progression [30]. Thus, Nrf2 could be a potential prognostic indicator, and contribute to human HCC tumor cell proliferation and invasion partially by modulating Bcl-xL and matrix metalloproteinase-9 (MMP-9) expression [62] (Fig. 3).
The Keap1/Nrf2/ARE signaling axis is associated with chemotherapy resistance [62]. ROS-induced activation of Nrf2 controls sorafenib resistance in liver cancer cells [63]. Recent findings indicate that Nrf2 is overexpressed in chemoresistant HCC cells, thus contributing to the development of chemoresistance [64, 65, 66]. Ma et al. [67] demonstrated that Nrf2 expression was observed in three HCC cell lines, including HepG2, HepG3B, and SMMC-7721, and was associated with cisplatin (DDP) chemoresistance. Inhibiting dysregulated Nrf2 activation, particularly when combined with anticancer chemotherapeutics, is thought to be an effective strategy for overcoming multidrug resistance (MDR) and inhibiting tumor progression [68]. Nrf2 involvement in the MDR of HCC cells includes anti-apoptotic mechanisms and overexpression of Nrf2-regulated efflux transporters such as MDR-regulated proteins (MRPs), which enhance chemotherapy drug efflux and strengthen ROS defense systems. Niture et al. [69]reported that Nrf2 regulates Bcl-xl (an anti-apoptotic gene) expression by occupying a promoter in close proximity to Bcl-xl. This suggests that Nrf2 might contribute to anti-apoptotic protein expression and, thus, promote chemoresistance in HCC cells [69]. The expression of Nrf2-regulated MRPs, including MRP1 and MRP2, also increases, further contributing to chemoresistance in HCC cells [70] (Fig. 3). Gonzalez-Sanchez et al. [70] determined that Nrf2 activation reduced ROS levels stimulated by adriamycin, cisplatin, sorafenib, and SN-38 (an irinotecan metabolite with antineoplastic activity), leading to anti-apoptosis and reducing the effectiveness of chemotherapy against HCC. Nrf2 is upregulated in response to low doses of ROS, and Nrf2 has been shown to increase telomerase activity, decrease oxidative stress, and promote the survival of liver cancer cells through regulating human telomerase reverse transcriptase (hTERT) gene expression [71]. Mitochondrial hTERT promotes drug resistance of tumor cells by reducing ROS production and Mitochondrial DNA damage, and playing a protective role on mitochondrial respiratory chain [72]. Recent clinical immunohistochemistry analysis of hTERT expression in 135 liver cancer tissues found that hTERT is mostly expressed in the cytoplasm of these tissues and is directly connected to oxidative stress [73].
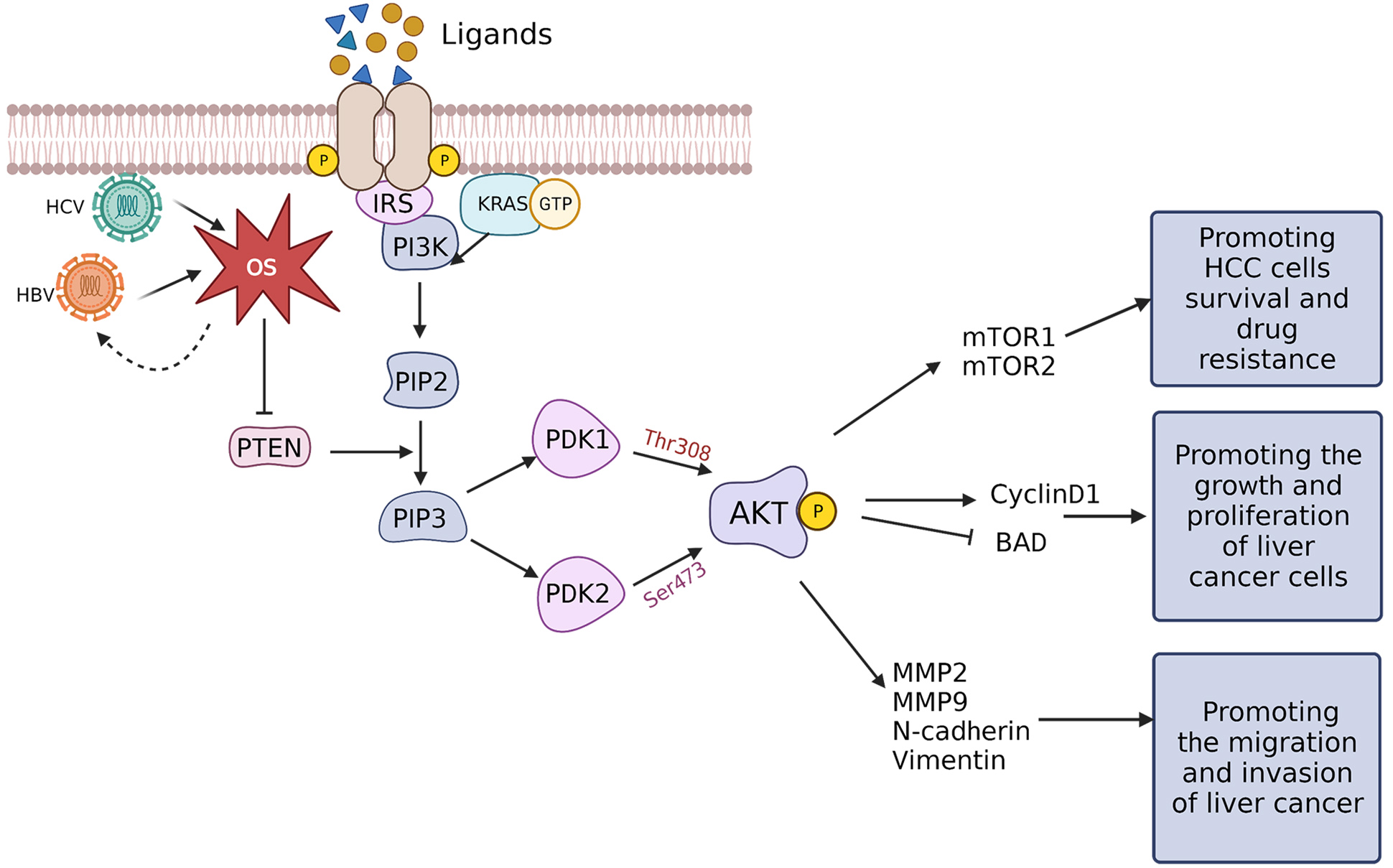
The PI3K/AKT/mTOR pathway is a key intracellular signaling pathway that participates in regulating various cellular mechanisms, including cell cycle progression, cell proliferation, death, metabolism, and angiogenesis. It is stimulated in different cancer types due to dysregulation of receptor tyrosine kinases (RTKs) [74]. RTK monomers serve as high-affinity cell surface receptors for growth factors, cytokines, and hormones. Upon ligand binding, RTK monomers activate and dimerize, triggering autophosphorylation and subsequent activation of the PI3K/AKT/mTOR signaling axis [75, 76]. Activated RTKs activate the lipid kinase PI3K, which catalyzes phosphorylation of phosphatidylinositol present within the internal leaflet of the plasma membrane. Direct activation of PI3K is achieved through binding to the regulatory region of RTKs or indirectly through adaptor molecules including insulin receptor substrates. Activated PI3K phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) to form phosphatidylinositol-3,4,5-triphosphate (PIP3) [77, 78]. Subsequently, PIP3 interacts with phosphoinositide-dependent protein kinase 1 (PDK1) and AKT and recruits them to the plasma membrane. For AKT activation to be complete, Thr308 and Ser473 residues within the AKT regulatory domain must be phosphorylated by PDK1 and PDK2, respectively [79]. The tumor suppressor phosphate and tensin homolog (PTEN), is a phosphatase that catalyzes dephosphorylation of PIP3 to generate PIP2, which also regulates AKT. Two complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), are formed when several proteins bind to mTOR and activate it [80] (Fig. 4).
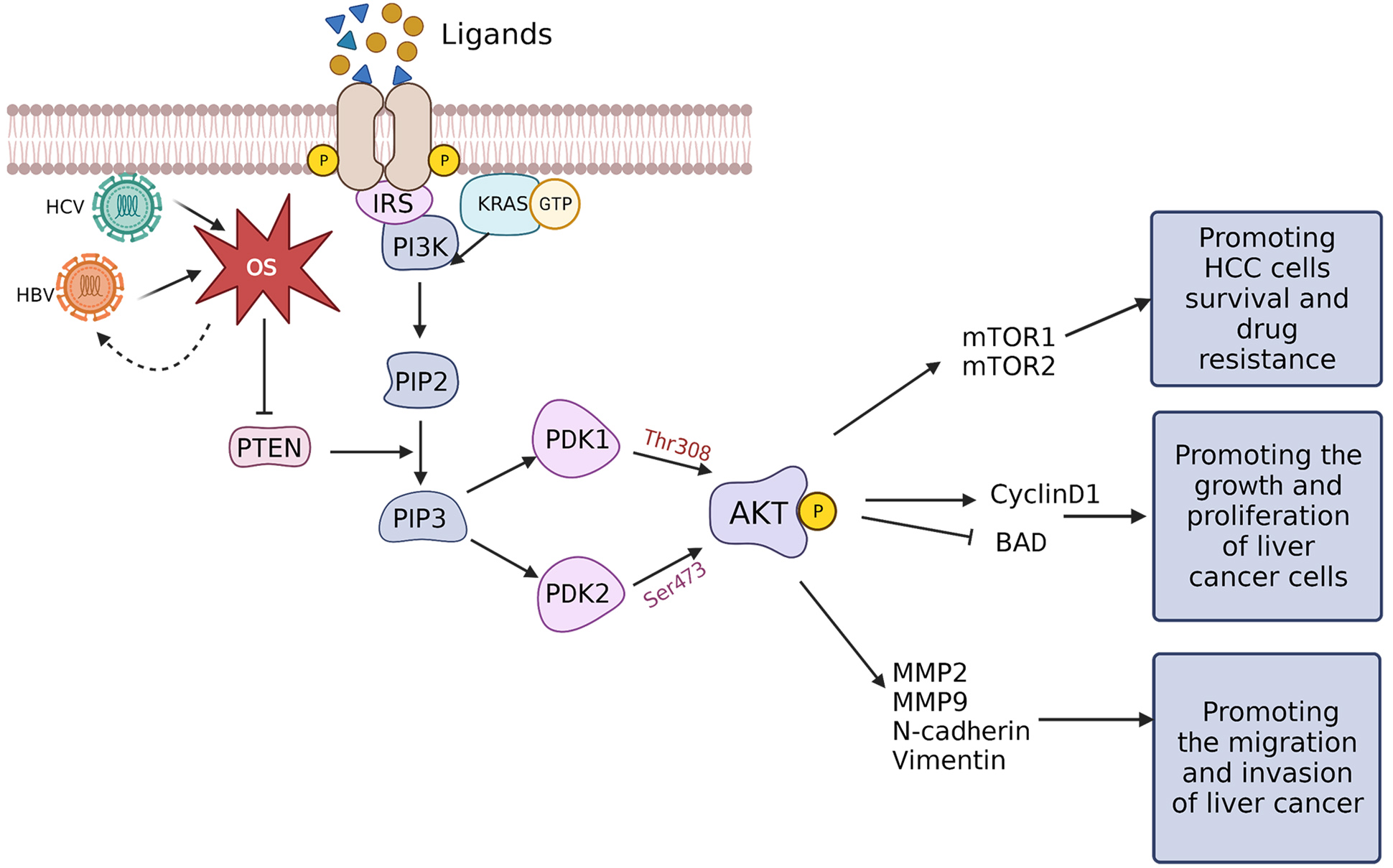 Fig. 4.
Fig. 4.Transduction mechanism of OS-controlled PI3K/AKT/mTOR signaling in HCC. When ligands, including growth factors, cytokines, and hormones, bind RTKs the RTKs undergo autophosphorylation and form homodimers. The activated RTKs can subsequently recruit PI3K, undergo direct activation via the regulatory subunits that bind to the G-protein KRAS, or are indirectly activated by IRS. Activated PI3K can phosphorylate PIP2 to form PIP3 which can, in turn, recruit PDK1 and PDK2. PDK1 and PDK2 phosphorylate Thr308 and Ser473 of the AKT regulatory region, respectively, thereby phosphorylating AKT. PTEN can dephosphorylate PIP3 to block AKT activation, while HBx and HCV core proteins can downregulate PTEN through ROS-mediated pathways to activate the PI3K/AKT/mTOR signaling. AKT can promote the survival and drug resistance of liver cancer cells by activating mTOR1/2 and can enhance the growth and proliferation of cancerous hepatic cells by upregulating cyclin D1 and blocking BAD. Furthermore, AKT can also upregulate EMT-related proteins such as MMP-2 and 9, N-cadherin, and vimentin, promoting liver cancer cells migration and invasion. BAD, Bcl-2-associated death; EMT, epithelial-mesenchymal transition; HBx, hepatitis B virus x; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; MMP, matrix metalloproteins; mTOR, mammalian target of rapamycin; OS, oxidative stress; PTEN, phosphate and tensin homolog; RTK, receptor tyrosine kinase.
Stimulation of the PI3K/AKT/mTOR signaling pathway is linked proliferation, migration, invasion, and drug resistance of HCC cells [81, 82, 83]. The mTOR pathway is the major tumor-initiating pathway in HCC, and mTORC1 is upregulated in liver cancer, which is a possible pharmacological target for mitigating drug resistance in liver cancer [83]. Among the major factors for the abnormal stimulation of the AKT/mTOR signaling pathway is PTEN expression. Strong evidence for the crucial function of mTORC2 during liver tumor formation comes from how PTEN loss combines with c-Met to increase the development of HCC via the mTORC2 pathway [84].
The primary regulatory protein in HCC advancement is HBx, which is encoded by
the HBV genome [85]. The oxidative inactivation of PTEN by HBx-stimulated ROS
production activates the AKT pathway. Moreover, the positive regulatory loop that
HBx and ROS maintain, which, through cyclin D1, contributes to hepatocellular
carcinogenesis [86]. Hep3B cells and 293T cells overexpressing HBx stimulate AKT
to phosphorylate I
HCC cell invasiveness induced by ROS is also believed to be mediated through the epithelial-mesenchymal transition (EMT), and is considered among the key regulatory factors for activating EMT [95]. Furthermore, EMT plays a significant part in HCC progression [96]. Stimulation of the PI3K/AKT/mTOR signaling pathway can induce EMT to promote HCC progression through MMP2, MMP9, N-cadherin, and vimentin [97] (Fig. 4).
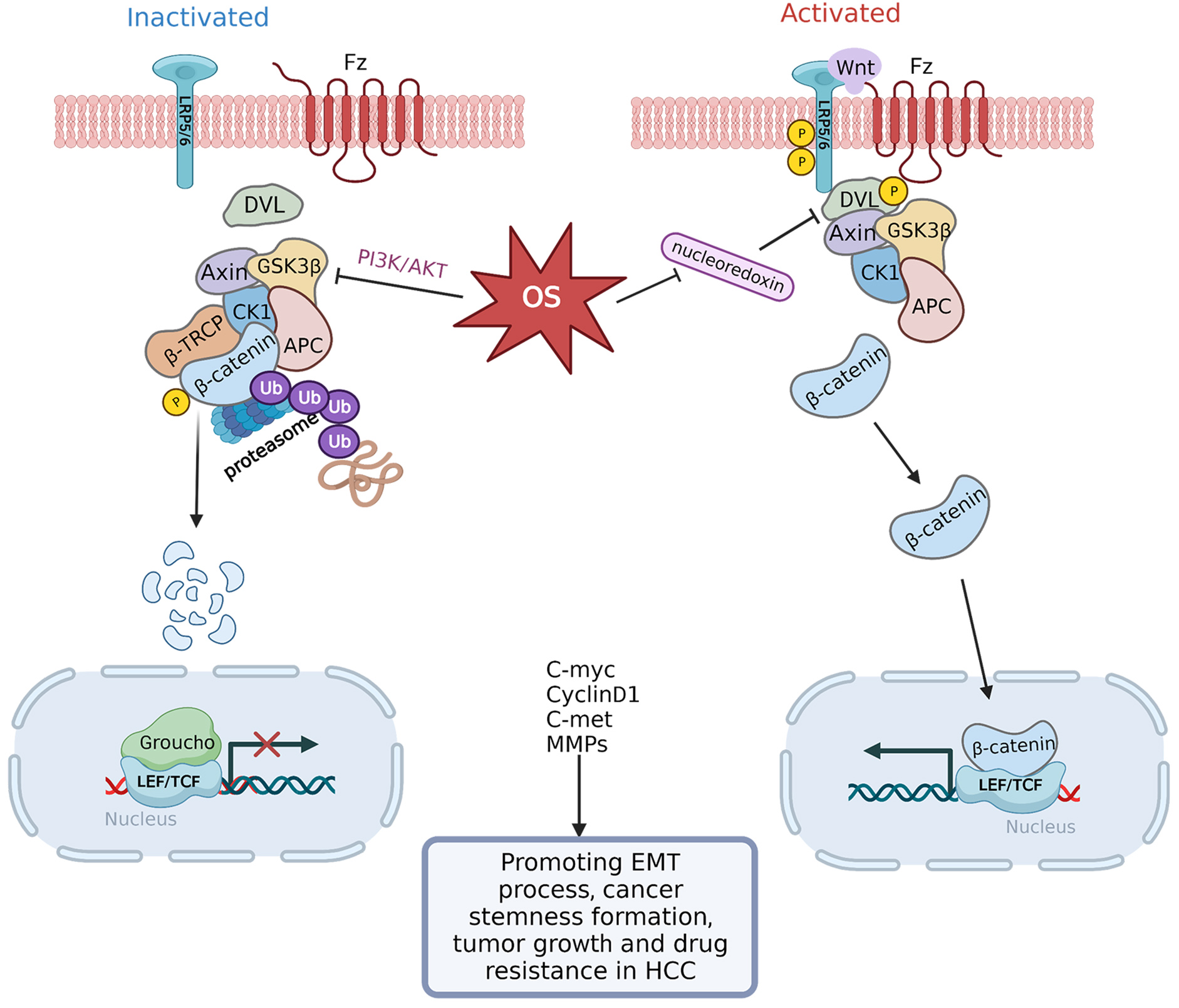
The Wnt/
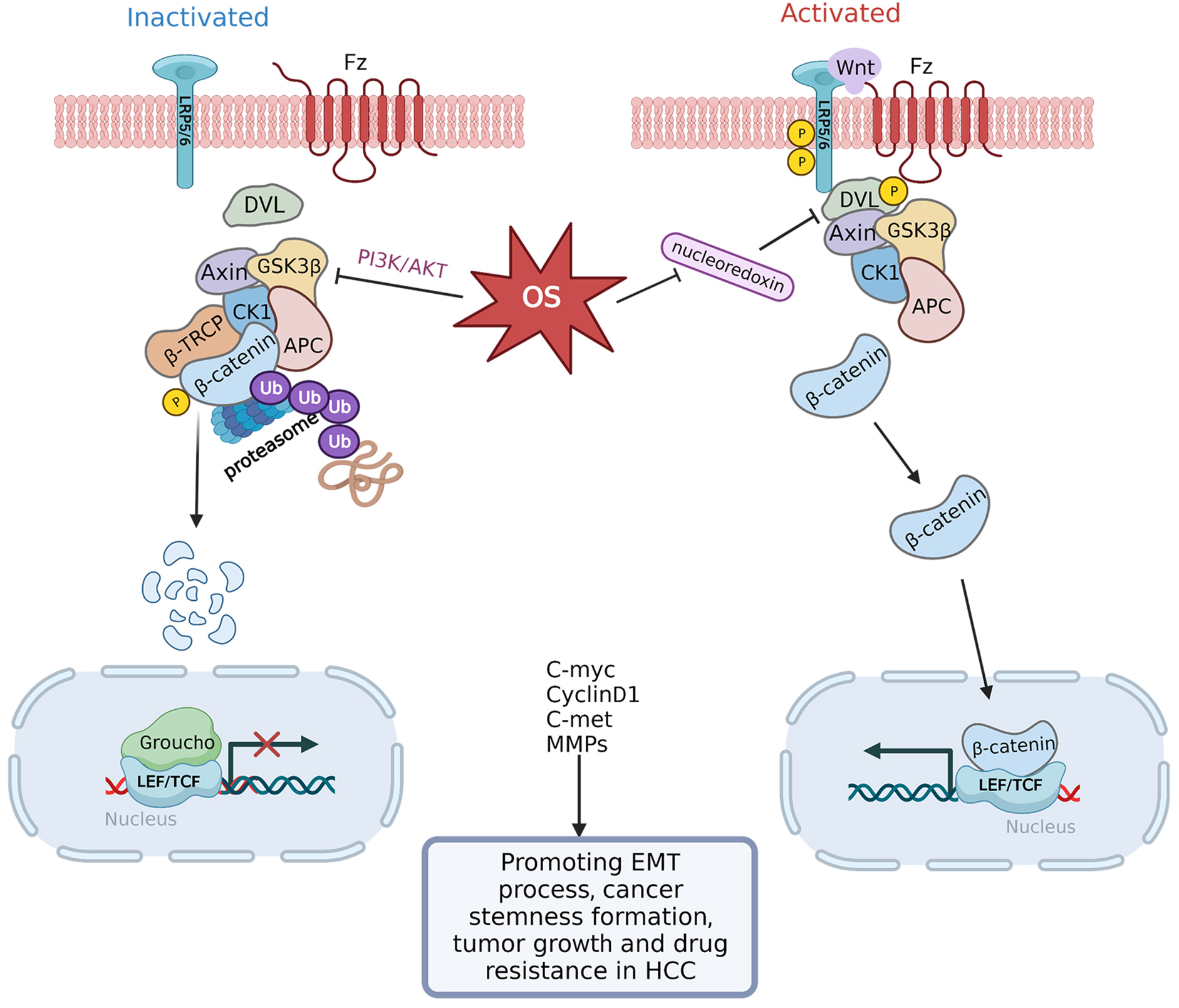 Fig. 5.
Fig. 5.Transduction mechanism controlling OS-regulated
Wnt/
The activation of EMT is a crucial step for HCC cells to acquire a malignant
phenotype [108]. Among the primary signaling pathways that control the onset of
EMT is the Wnt/
Notch signaling has a link to steatosis and OS, specifically, H
ROS stimulates several transcription factors such as hypoxia-inducible factor-1
alpha (HIF-1
FOXO family members including FOXO1, FOXO3, FOXO4, and FOXO6 are involved in supporting cellular homeostasis through various mechanisms [120]. These molecules enhance antioxidant status by inducing genes that eliminate ROS, improve mitochondrial redox, and inhibit levels of free transition metal ions by increasing metallothionein and ceruloplasmin abundance [121]. FOXOs respond to ROS, nutritional status, as well as genes implicated in apoptosis and cell cycle arrest like GADD45 [120]. ROS can activate AKT to directly phosphorylate FOXOs, which leads to the expulsion of FOXOs from the nucleus and suppression of their transcriptional activity. Inhibition of FOXO transcriptional activity could trigger apoptosis in HCC cells [122]. Since FOXOs cause cell cycle arrest and apoptosis, they were once characterized as tumor suppressors; however, mounting evidence indicates that FOXOs, particularly FOXO3, actually promote carcinogenesis. According to Lu et al. [123], FOXO3 expression accelerated carcinogenesis caused by hepatotoxin and the positive feedback loop between AKT and mTORC2 activation was activated when FOXO3 induced OS and DNA damage. It bears consideration that FOXO3, triggered by both ROS-promoting and ROS-eliminating systems, is linked to the activation of the pentose phosphate pathway (PPP).
Hypoxic response is largely regulated by hypoxia inducible factor-1 alpha
(HIF-1
Heat Shock Transcription Factor 1 (HSF1) responds to protein misfolding–induced
stressors through induction of genes encoding heat shock protein chaperones. HSF1
specifically responds to ROS via oxidation of internal Cys-35 and Cys-105
residues, and subsequent stimulation of antioxidant gene expression [132, 133].
Upregulated HSF1 expression commonly occurs to protect tumor cells against
diverse types of stresses [134] and increased expression of HSF1 activates a
metabolic phenotype switch and chemoresistance in cancer cells. For example,
HSF1/AMP-activated protein kinase
By triggering cytokines, chemokines, and receptors that promote tissue healing,
NF-
P53 increases the antioxidant status of cells via transactivation of genes which
encode proteins that scavenge ROS, support GSH production, enhance NADPH
synthesis, detoxify xenobiotics, and promote pro-oxidant enzymes such as NO
synthase 2 (NOS2) and cyclooxygenase 2 (COX2) [144, 145]. Improvement in NADPH
synthesis triggered by p53 occurs as a result of upregulation of the p53-induced
glycolysis and apoptosis regulator TIGAR, which promotes DNA repair and reduces
ROS to maintain redox balance [146]. Conversely, p53 also exhibits pro-oxidant
activities through upregulation of p53-inducible genes (PIGs), such as PIG3 a
quinone oxidoreductase/
P53 promotes antioxidant genes under moderate OS conditions to aid in adaptability; however, under harsher circumstances, p53 modulates cancer cell death by promoting the generation of ROS. PIG3 and PIG6 are two p53 target genes that promote apoptosis, and mitochondrial proline dehydrogenases generate ROS via a mechanism that supplies carbon to various other mitochondrial dehydrogenases, which in turn produce ROS [148, 150]. Genes encoding BAX, PUMA, and p66Shc are induced when p53 is activated by high levels of ROS. These genes disrupt the functioning of mitochondria, release cytochrome c, and enhance the generation of ROS [151]. Moreover, SOD2 and numerous Nrf2 target genes are repressed when p53 is active in proapoptotic scenarios [152]. OS induces changes in p53 activity and can upregulate p53 in HCC cells thereby inducing apoptosis of cancerous hepatic cells and inhibiting tumor proliferation and growth [153]. Furthermore, chronic infection with HBV and HCV contributes to ROS and RNS production, which can cause mutations in the p53 gene and subsequent promotion of liver cancer [154].
HCC is a form of cancer associated with inflammation, and most cases of HCC occur due to liver damage and chronic inflammation [155]. Prolonged and unresolved inflammation leads to the infiltration of immune cells into the liver which facilitates tissue remodeling [156]. The infiltration of immune cells results in a disturbance in the generation of chemokines and cytokines and a rise in the synthesis of ROS and RNS, which promote the development of fibrosis, cirrhosis, and, ultimately, the malignant transformation of liver cells [157]. The genesis, development, and therapeutic response in HCC tumors are all influenced by TME remodeling. By influencing various factors such as tumor-associated macrophages (TAMs), neutrophils, myeloid-derived suppressor cells (MDSCs), and Treg cells within the TME, OS plays a prominent role in modulating HCC development and progression.
TAMs within the TME mainly exert their function through secreted factors. Such
factors include epidermal growth factor (EGF), cytokines (IL-6 and
TNF-
Neutrophils are also becoming recognized as a significant player in the pathophysiology of HCC. Current evidence indicates that neutrophils are key mediators of the immunosuppressive environment as well as being drivers of tumor progression [164]. Growing evidence indicates that neutrophils function during the initial phases of carcinogenesis because they directly interact with HCC cells, mostly by producing ROS which leads to genomic instability [165]. S100A9, which belongs to a class of proteins termed damage-associated molecular patterns (DAMPs), displays upregulated expression when induced via HBV and has a direct impact on HCC cells by facilitating HCC progression and metastasis [166]. S100A9 affects neutrophil recruitment in acute and chronic hepatic damage in a mouse model, as well as promoting neutrophil stimulation and degranulation [166]. S100A9, which is also regulated by HBV, has an indirect impact on HCC malignancy by promoting the proliferation of neutrophil extracellular traps and activating neutrophils.
MDSCs induce the differentiation and expansion of Tregs during tumorigenesis, as
well as producing OS [167]. The expansion of MDSCs can suppress immune function,
as demonstrated by increased levels of arginase, NO, and ROS [168]. MDSCs also
exhibit consistent alteration following a rise in the synthesis of ROS in
response to therapy with Pam3CSK4 and Ro5-3335 [169]. Maintaining the
immunological TME is crucial for MDSCs and promoting their polarization will
enhance cancer immunotherapy. The immunosuppressive TME that enhances tumor onset
and advancement can be alleviated by preventing the formation and recruitment of
MDSCs within the liver. For example, long-term metformin therapy can prevent the
accumulation of MDSCs in Ncoa5
Factors involved in chronic liver disease influence Treg cells. Alcohol,
high-fat diet, gut microbiota, and metabolites are some examples of environmental
stressors that can promote hepatic damage [18]. OS considerably contributes to
the downregulation of Treg stimulation and their population, resulting in
steatosis and fibrosis [171]. The progression of NASH toward tumor development
increases the population of Tregs to establish a pro-tumorigenic TME. The
development of NAFLD-linked HCC is aided by Tregs and neutrophils facilitating
communication between the innate and adaptive immune systems. In work conducted
by Hang et al. [172], isoallo-LCA increases FOXP3 expression levels
which in turn triggers mitochondrial ROS generation and encourages Treg
development. Fe-MnO
These results suggest that a rise in ROS triggers alterations in the TME that promote carcinogenesis by changing the roles of MDSCs and TAMs and, coordinately, generating alterations in Tregs that may inhibit immunological reactions against tumor cells.
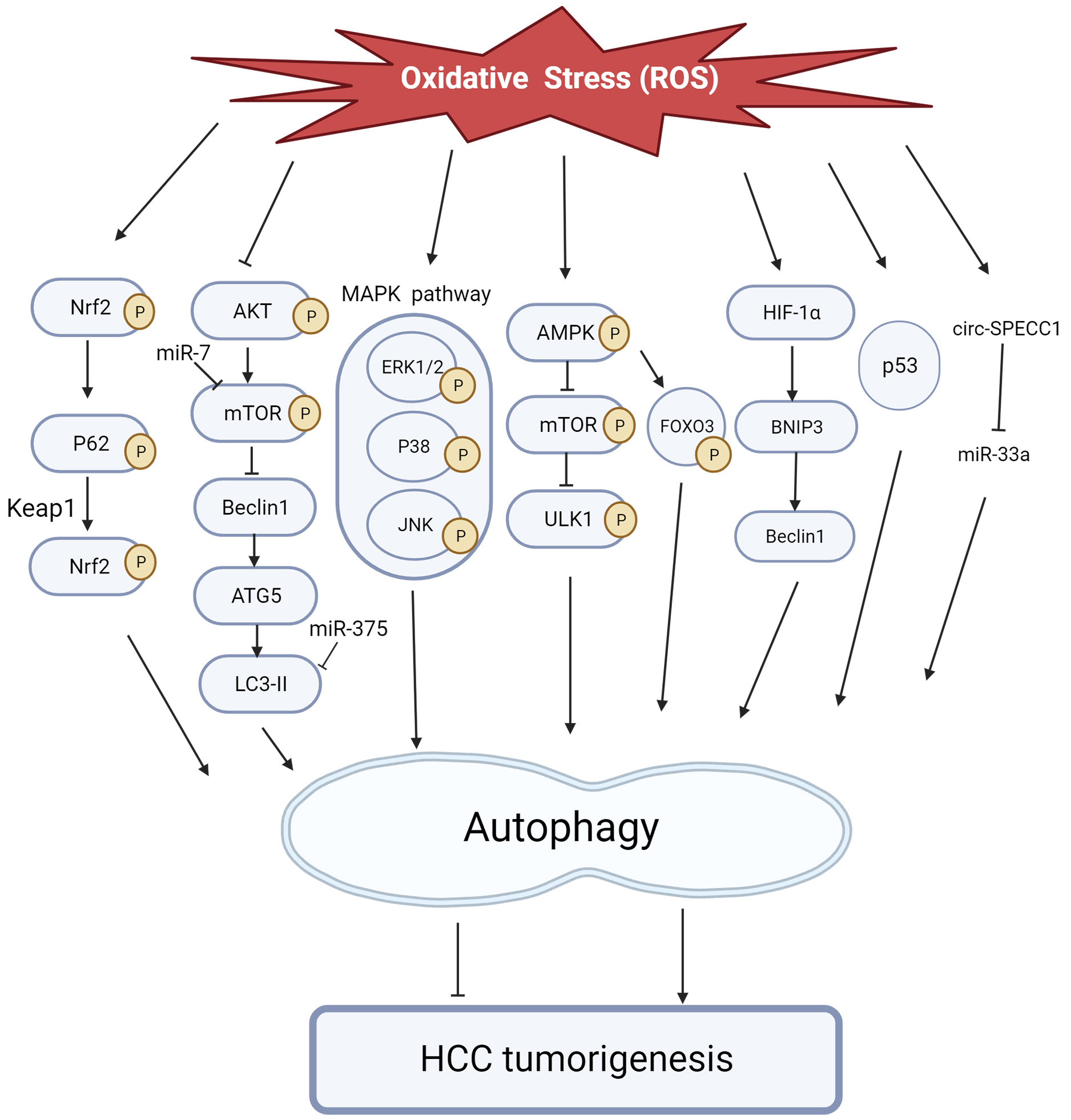
Autophagy, a catabolic process, eliminates defective and damaged cellular material via lysosomes. It is activated in response to a variety of stresses, including nutrient deprivation, hypoxia, and oxidative stress [174]. ROS can initiate the formation of Autophagosome and act as a cell Signaling molecule for autophagic degradation. Conversely, autophagy reduces oxidative damage and ROS levels by removing protein aggregates and damaged organelle such as mitochondria [175]. Additionally, autophagy has a dual influence, initially inhibiting the formation of tumours. Mitochondrial autophagy eliminates damaged mitochondria, preventing the accumulation of ROS and thereby reducing the effect of ROS on tumour formation [174]. But promoting cancer cell proliferation in already formed tumors, especially after chemotherapy with drugs, enhances autophagy, which may lead to tumor resistance and disease recurrence [176].
Under oxidative stress, Nrf2 activates and induces the autophagy pathway in
cells, ultimately leading to the proliferation, migration, and invasion of HCC
[177]. It is known that autophagy related protein p62 plays a key role in the
modulation of Nrf2-Keap1 pathway [177, 178]. In addition to Nrf2 activation, the
carcinogenic signal transduction of p62 protein may affect other and multiple
pathways, such as NF-
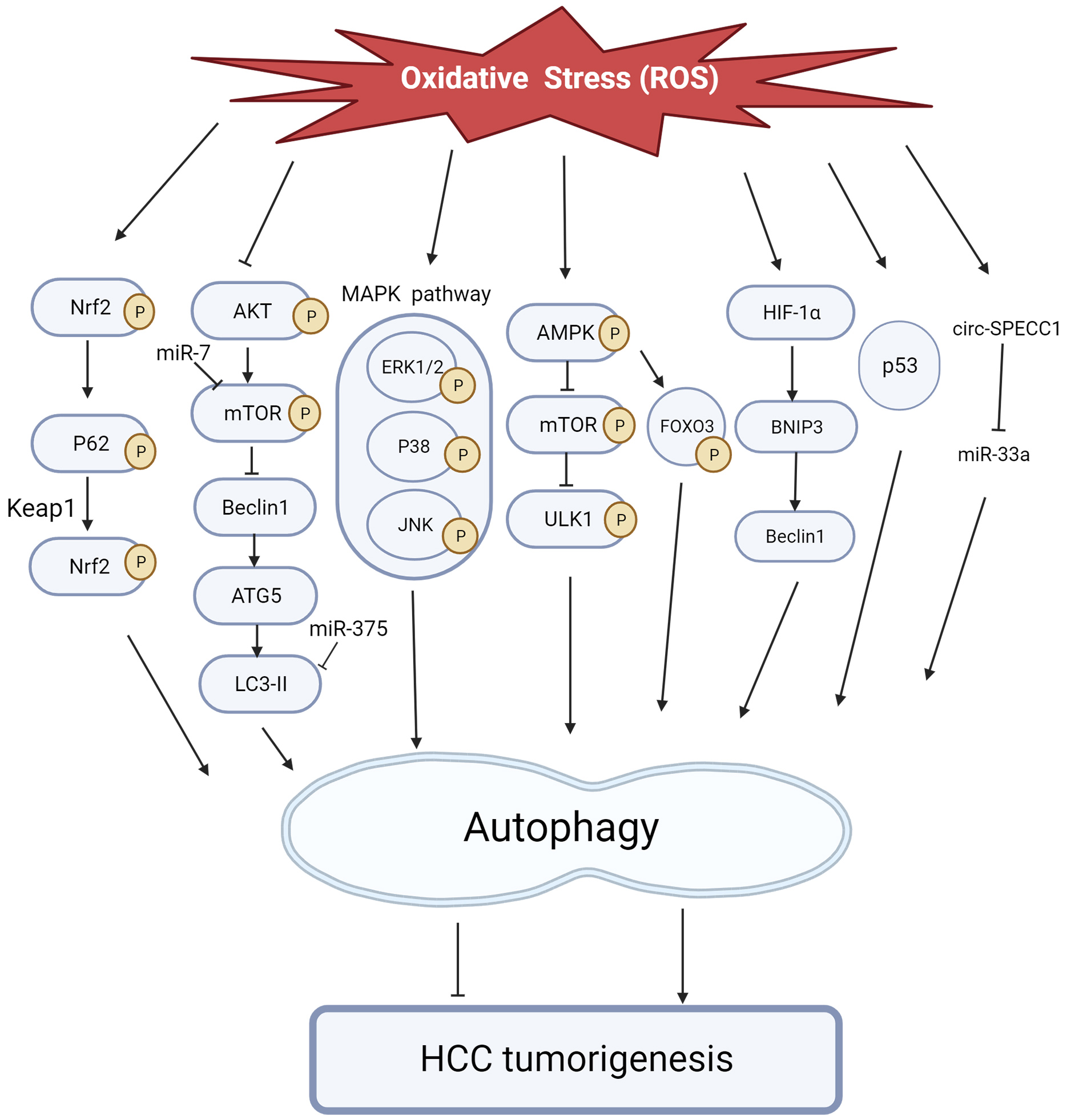 Fig. 6.
Fig. 6.The role of ROS-induced autophagy in HCC tumorigenesis. ROS regulates autophagy via the pathways, transcription factors, and non-coding RNAs depicted in the figure above, initiating or inhibiting liver cancer development.
Considering the importance of OS in promoting HCC, restoring the balance between
oxidants and antioxidants is of significant therapeutic significance in HCC, and
drugs that control OS are a key option for effective HCC therapy.
Chemotherapeutic medications, organic compounds, traditional Chinese medicine,
and nanoparticles have all recently drawn interest as these drugs can tackle
abnormal OS in HCC in various ways. The majority of these substances, including
vitamin C and troxerutin, enhance OS resulting in oxidative harm to DNA and a
triggering of apoptosis in HCC cells. A few of these drugs, including
19-
| Category | Materials | Cell lines/Animals | Trial | Targets | Mechanism | Effects | Refs. |
| PX-12 | HepG2 and SMMC-7721 cells | Cleaved PARP |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting proliferation | [192] | ||
| Chemotherapeutic Drugs | APN inhibitor 4cc | PLC/PRF/5, HepG2, and H7402 cells | Bax |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting tumor growth | [193] | |
| Itraconazole | HepG2 and Bel-7405 | Hedgehog pathway |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting proliferation | [194] | ||
| Raddeanin A | QGY -7703 cells | P53 |
ROS-dependent apoptosis | Inducing apoptosis | [195] | ||
| Caffeine | SMMC-7721 and Hep3B cells | Cleaved PARP |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting proliferation | [196] | ||
| Allicin | SK-Hep-1 and BEL-7402 cells | Cleaved Caspase-3 |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting proliferation | [197] | ||
| Galangin | HepG2 and Hep3B | Bax |
ROS-dependent apoptosis | Inducing apoptosis | [198] | ||
| Kaempferol | HepG2 | Bax/Bcl-2 |
ROS apoptosis | Increasing apoptosis | [199] | ||
| Vitamin C | HCC-LM3 and Huh7 | Cleaved-Caspase-3 |
ROS-induced apoptosis and DNA damage | Inducing apoptosis, inhibiting proliferation, eradicating liver CSCs | [200] | ||
| Naringenin | HepG2 | P53 |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting proliferation | [201] | ||
| Troxerutin | Huh7 | cleaved Caspase-3 |
ROS-induced DNA damage | Inducing apoptosis, inhibiting proliferation | [202] | ||
| Siegesbeckia Orientalis | Hepa1-6 and HepG2 | IL-6 |
ROS-induced apoptosis | Promoting inflammatory response, inducing apoptosis and inhibiting migration | [203] | ||
| Natural compounds and Chinese medicines | Giganteaside D | HepG2 and Bel-7402 | Cleaved Caspase-3, -9 |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting proliferation | [204] | |
| Oleanolic acid | Huh7 and HepG2 cells | Cleaved PARP |
ROS-induced cell death | Inducing apoptosis | [205] | ||
| Linalool | HepG2 | MAPK pathway |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting proliferation | [206] | ||
| Apigenin | SK-Hep-1 and BEL-7402 cells | Cleaved Caspase-3 |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting tumor growth | [207] | ||
| Alnustone | BEL-7402 and HepG2 | PI3K/AKT/mTOR pathway |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting tumor growth | [92] | ||
| Coptisine | Hep3B cells | PI3K/AKT/mTOR pathway |
ROS-mediated mitochondrial dysfunction | Inducing autophagic cell death | [181] | ||
| C0818 | HepG2 and SK-Hep-1 | P21 |
ROS-dependent cytotoxicity | Inducing apoptosis, inhibiting proliferation | [208] | ||
| Koumine | Huh-7 and SNU-449 | NF-κB pathway |
ROS-dependent apoptosis | Inducing apoptosis, inhibiting proliferation | [209] | ||
| Sanguinarine | Bel-7402 and Bel-7404 cells | APAF-1 |
ROS‑dependent mitophagy and apoptosis | Inducing apoptosis, inhibiting proliferation | [210] | ||
| Artesunate | PLC/PRF/5, HuH7, HepG2, Hep3B and HCCLM3 cells | P-ERK |
ROS apoptosis | Inducing apoptosis, inhibiting proliferation | [211] | ||
| Butyrate | Huh7 cells | AKT/mTOR pathway |
ROS -induced autophagy | Inhibiting cell proliferation | [180] | ||
| Notopterol | HepJ5 and Mahlavu cells,Female BALB/c nude mice | JAK2 |
OS-associated cell death | Attenuating HCC cell migration and invasive capacity, inhibiting HCC tumor development | [212] | ||
| Chamaejasmenin E | Hep3B and SMMC-7721 cells, Male BALB/c nude mice | C-Met/Ras/Raf/MEK/ERK pathway |
OS-regulated cell death | Inhibiting the proliferation of HCC cells, reducing HCC cells migration, triggering apoptosis and inhibiting tumor growth | [213] | ||
| HEG | Huh7 and HepG2 cells | SOD |
OS |
Inhibiting tumor growth | [214] | ||
| Polydatin | SMMC-7721 and HepG2 cells | SOD |
OS |
Inducing apoptosis, inhibiting proliferation, invasion and migration | [215] | ||
| Coenzyme Q10 | NCT01964001 | SOD |
OS |
Protecting against the development of HCC | [216] | ||
| Vitamin B-6 | NCT01964001 | Plasma homocysteine |
OS |
Protecting against the progression of HCC | [217] | ||
| BCAA | Korenaga M et al. (2015) | BAP/dROM |
OS |
Inhibiting HCC development in patients with cirrhosis | [218] | ||
| MCTs | Mice | Lipid peroxidation |
OS |
Inhibition of oncogenesis and tumor growth | [219] | ||
| CLT | Mice | AST |
OS |
Inhibiting HCC growth | [220] | ||
| Decalactone | Swiss Wistar rats | CAT, GSH, GPX and SOD |
OS |
Suppressing the progression of HCC | [221] | ||
| Daphnetin | Swiss albino Wistar rats | AFP, AST, ALP and ALT |
OS |
Suppressing the development of hepatic cancer | [222] | ||
| Genistein | Rats | Versican/PDGF/PKC signaling pathway |
OS |
Suppressing HCC development | [223] | ||
| Bevacizumab and CCR2 Inhibitor Nanoparticles | HepG2 and Huh7 cells | CCR2 |
ROS-mediated apoptosis | Inducing apoptosis | [224] | ||
| Cisplatin and Curcumin Co-loaded Nano-liposomes | HepG2 cells | P-ERK1/2 |
ROS-dependent apoptosis | Inducing apoptosis | [225] | ||
| Nanoparticles | Cisplatin-oleanolic acid co-loaded calcium carbonate nanoparticles | HepG2 cells | P53 pathway |
ROS-mediated apoptosis | Inducing apoptosis, inhibiting proliferation | [226] | |
| Silver Nanoparticles | HepG2 cells | AKT, MAPK and p53 signaling pathways |
ROS-induced apoptosis and DNA damge | Inducing apoptosis, inhibiting proliferation | [227] | ||
| ZnO nanoparticle-ferulic acid conjugate | Huh-7 and HepG2 | ROS-induced apoptosis and DNA damage | Inducing apoptosis, inhibiting proliferation | [228] |
PX-12, 1-methylpropyl 2-imidazolyl disulfide; PARP, poly ADP-ribosepolymerase;
APN, aminopeptidase N; 8-hydroxydeoxyguanosine, 8-OHdG; TNF-
Chemotherapeutics cause OS, inflammation, apoptosis, and aberrations in neurotransmitter metabolism, all of which contribute to their toxicity. Oxazoline anthracyclines have drawn interest as potential signaling mediators of proliferation, differentiation, and death of cancer cells because they produce ROS and RNS [229]. High amounts of ROS are produced by platinum-containing complexes, alkylating agents, camptothecins, and arsenic agents. In contrast, low quantities of ROS are produced during response to taxanes, vinca alkaloids, nucleotide analogs, and antimetabolites such as antifolates and nucleosides [230]. 1-Methylpropyl 2-imidazolyl disulfide (PX-12), a thioredoxin 1 (Trx1) inhibitor, has been studied in various cancer types [231, 232, 233]. PX-12 therapy upregulates Bax expression and downregulates Bcl-2 expression, indicating that PX-12–mediated apoptosis occurs in a mitochondrion-dependent manner. PX-12 also synergizes with 5-fluorouracil to significantly inhibit tumorigenicity both in vitro and in vivo [192]. The aminopeptidase N inhibitor 4cc synergistically acts with 5-fluorouracil to exert antitumor effects on cancerous human hepatocytes though ROS-regulated drug resistance, inhibition, as well as simultaneous triggering of the apoptotic mitochondrial pathways [193]. In addition, itraconazole exposure activates apoptosis, blocked cell cycle advance, and downregulated MMP in HepG2 cells [194]. Additionally, a link between intracellular ROS production and accelerated senescence triggered by cisplatin could be utilized as a prospective HCC target [234].
HCC has been treated with various natural chemical extracts and Chinese herbal
medications. For example, alnustone increased the synthesis of ROS in BEL-7402
and HepG2 cells, and downregulated proteins linked to apoptosis and
PI3K/AKT/mTOR/p70S6K signaling [92]. Coptisine promotes autophagic cell death via
the downregulation of proteins linked to PI3K/AKT/mTOR signaling and upregulation
of ROS-regulated mitochondrial dysfunction in Hep3B cells [181]. Koumine
suppresses HCC proliferation and enhances apoptosis. NF-
Due to their distinctive characteristics, including its significant drug-loading capacity, inherent anticancer actions, integrated diagnostic and therapeutic functions, and ease of surface engineering with targeting ligands, nanomedicines have received significant attention when attempting to discover and develop effective therapies for HCC [235]. Bevacizumab and CCR2 inhibitor-containing nanoparticles sensitize doxycycline-treated Huh-7 cells by activating ROS-stimulated apoptosis [224]. Lipid-coated cisplatin/oleanolic acid calcium carbonate nanoparticles (CDDP/OA-LCC NPs) were reported to induce tumor cell apoptosis via upregulation of proapoptotic proteins, downregulation of proteins associated with the P13K/AKT/mTOR pathway, and upregulation of proteins linked to p53 proapoptotic functions [226]. Cisplatin and curcumin-loaded nano-liposomes (CDDP/CUR-Lip) act by elevating ROS levels in HCC cells [225] and can be used to co-deliver CDDP and CUR in vitro and in vivo to improve HCC treatment through synergistic effects with minimal toxicity.
Silver nanoparticles (AgNPs) have recently come to light as a cutting-edge method of treating tumors, particularly hepatocarcinoma. AgNP-induced apoptosis is dependent on ROS overproduction as well as the effects of MAPKs, AKT signaling, and DNA damage-mediated p53 phosphorylation to trigger HepG2 cell apoptosis [227]. ZnO nanoparticle–ferulic acid conjugate (ZnONPs-FAC) triggers cells to undergo apoptosis and results in the suppression of diethylnitrosamine (DEN)-induced HCC. ZnONPs-FAC can cause significant mitochondrial damage and generate ROS production, and it can also induce oxidative DNA damage in HCC cells [228]. There is little doubt that nanomaterials will play a significant role in treating HCC when moving forward, particularly when paired with therapeutic drugs.
A growing body of research highlights the vital function of OS in the onset and
advancement of HCC. This occurs through genetic modifications, changes in
signaling pathways, transcription factor alterations, and effects on the TME.
Genetic changes induced by OS, such as oxidative damage, damage to nuclear and
mitochondrial DNA, DNA hypomethylation, and irregular miRNA expression all can
induce HCC progression. OS-mediated signaling pathways, including
Keap1/Nrf2/ARE, PI3K/AKT/mTOR, Wnt/
This review also focuses on four therapeutic approaches targeting abnormal OS in
HCC: chemotherapeutic agents, natural compound extracts, Chinese herbal
medicines, and nanoparticles. Their mechanisms of action may involve influencing
oxidative DNA damage, modulating signaling pathways such as PI3K/AKT/mTOR and
Wnt/
YL and YY designed, wrote and revised the manuscript. YL prepared figures of the manuscript. YY contributed to making table of the manuscript. YL and YY participated in collecting data of the manuscript. LY and RW made significant revisions and proofread the manuscript. LY and RW also provided support for the publication of the manuscript. All authors contributed to the article and approved the final version.
Not applicable.
Figures created with BioRender.com (https://www.biorender.com).
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.