
- Academic Editors
Dopaminergic neurons are constantly threatened by the thin boundaries between
functional
Parkinson’s disease (PD) is the second most common neurodegenerative disease
after Alzheimer’s disease (AD). This movement-related disorder is associated with
a progressive loss of dopaminergic neurons in the brain - particularly in the
substantia nigra pars compacta (SNpc) and in the locus coeruleus
- and with the accumulation in nerve cells of
Despite the strong effort devoted in the last decades to the investigation of PD etiology, the molecular mechanisms underlying the onset and progression of the pathology are still poorly understood. Several genes have been reported to play a role in the pathogenesis and several mutations in the AS-encoding gene (SNCA) have been linked to familial PD. However, the latter account for only 5% of the cases, while the large majority of patients develop the sporadic form. PD is, indeed, regarded as a multifactorial pathology, in which alterations in protein homeostasis, redox homeostasis, mitochondrial function, and membrane integrity are involved in a complex process leading to neuroinflammation and disease onset [3, 4, 5]. AS is known to be physiologically involved in neurotransmission and cognition, but the mechanisms of action have yet to be elucidated [6]. AS is expressed in vertebrates and particularly abundant in the brain, especially in the presynaptic terminals of dopaminergic neurons, but it can also be found in the central nervous system (CNS) and in peripheral tissues [7] or fluids, including serum [8], saliva [9, 10] and tears [11, 12]. The pathological accumulation of AS protein in specific populations of neurons and glia is the hallmark of several disorders identified as synucleinopathies. They can be classified in two major groups: Lewy body disease (characterized by LBs and LNs) and multiple system atrophy (MSA, characterized by glial cytoplasmic inclusions). Lewy body disease includes PD, PD dementia (PDD), dementia with Lewy bodies (DLB), and other neurodevelopmental and neurometabolic disorders, while MSA is subclassified into MSA with predominant cerebellar ataxia (MSA-C) and MSA with predominant parkinsonism (MSA-P) [13].
The difficulties in diagnosis and treatment of these pathologies are a mirror of the molecular complexity underlying it. The different molecular landscapes depicted in the literature suggest the involvement of distinct factors (Fig. 1) and underscore the importance of deep, individual, biochemical phenotyping for basic and translational studies [14]. Combined AS biomarkers, integrating structural, biochemical and aggregation properties, could contribute to diagnosis and typization of synucleinopathies [8]. Increasing interest is devoted to peripheral, non-invasive, AS biomarkers [8, 12, 15].
 Fig. 1.
Fig. 1.Pictorial representation of the main factors affecting
Particularly relevant to this regard is the dual-hit hypothesis, also known as the Braak’s hypothesis, stating that sporadic PD initiates in the peripheral nervous system [16]. It is hypothesized that after exposure to external insults, including viral or bacterial infections [16, 17, 18], the pathology propagates from peripheral tissues - neurons of the gut and nasal cavity - to and within the CNS [16, 18]. Dysregulation of either redox or protein homeostasis induces a toxic response in dopaminergic neurons [4, 5]. The accumulation of AS, in turn, may enhance the redox stress, thereby generating a vicious cycle that leads to cell death and neuroinflammation [19]. Thus, perturbations of redox and protein homeostasis enhance each other in degenerating neurons in PD (Fig. 1).
Proteostasis is a finely regulated process aimed at controlling the quality of protein synthesis, from transcription to folding. Alterations in this surveillance lead to the accumulation of abnormal products. Autophagy plays a key role in this context by removing dysfunctional elements and facilitating the natural turnover of cellular components. Dysfunctions in this mechanism can result in accumulation of aggregation products of misfolded AS. In turn, AS misfolding may further inhibit its degradation, generating another vicious cycle [20, 21, 22, 23, 24, 25].
Reactive oxygen, nitrogen, and sulfur species (reactive oxygen species (ROS), reactive nitrogen species (RNS) and reactive sulfur species (RSS), respectively) are produced by cell metabolism and play a fundamental role as second messengers in several metabolic processes, but also pose a threat to cellular structures and functions. Both enzymatic and non-enzymatic actors form a delicate and intricate network aimed at protecting cells from oxidative or reductive damage to DNA, lipids, and proteins. Another source of oxidative stress is dysregulation of iron metabolism, which is well-documented to be impaired in PD [26, 27]. Neurons are particularly exposed to redox stress due to dopamine (DA) metabolism. The inherent toxicity of DA and AS could be a key to understanding PD pathogenesis [6, 28].
The two crucial factors DA and AS are not only functionally but also directly linked to each other by the interactions that the protein and its aggregation products can establish with DA and its metabolites. Thus, it is of interest to elucidate the effects of such ligands on conformational and aggregation properties of AS. In this review, the recent literature on the interactions of AS with DA and its metabolites is commented, with regards to their effects on the protein conformational ensemble and aggregation process.
DA and AS are two central actors of neurotransmission by dopaminergic neurons [3]. DA transmits stimuli from SNpc to striatum to activate motor function [29], while AS is involved in storage, release and recycling of the neurotransmitter [30]. In spite of this concerted action, the two molecules have restricted interactions under physiological conditions [6, 27]. AS is a presynaptic, cytoplasmic protein. DA, too, is synthesized in the cytoplasm of dopaminergic neurons from the precursor tyrosine, through the sequential activities of tyrosine hydroxylase (TH) and aromatic amino acid decarboxylase (AADC). However, its cytoplasmic concentration is kept low by efficient loading into synaptic vesicles by the vesicular monoamine transporter 2 (VMAT2).
Cytoplasmic DA can be catabolized by potentially toxic reactions having ROS as side products (Fig. 2). One pathway involves the enzymatic oxidation of DA to 3,4-dihydroxyphenylacetaldehyde (DOPAL), with concomitant production of hydrogen peroxide, by the monoamine oxidase isoforms MAO-A and MAO-B of the outer mitochondrial membrane. The highly reactive metabolite DOPAL has long been recognized as neurotoxic. It is further converted to the non-toxic metabolite 3,4-dihydroxyphenylacetate (DOPAC) by aldehyde dehydrogenase (ALDH). The catecholaldehyde hypothesis points to DOPAL as the main trigger of neurodegeneration in PD [31, 32].
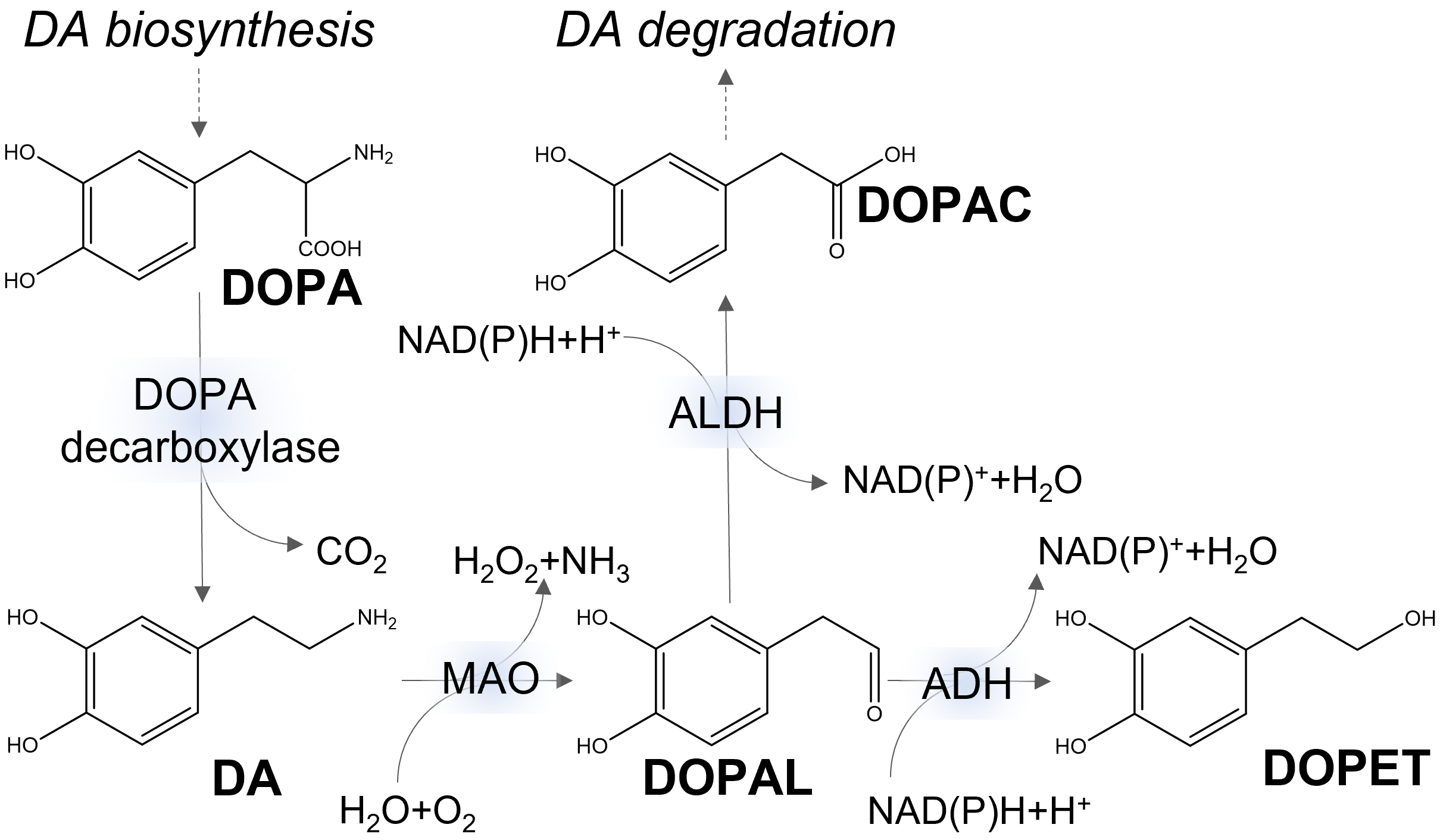 Fig. 2.
Fig. 2.Simplified metabolic pathways of dopamine (DA). DOPA, 3,4-dihydroxyphenylalanine; DOPAC, 3,4-dihydroxyphenylacetate; DOPAL, 3,4-dihydroxyphenylacetaldehyde; DOPET, 3,4-dihydroxyphenylethanol; NAD, nicotinamide adenine dinucleotide; ALDH, aldehyde dehydrogenase; MAO, monoamine oxidase; ADH, alcohol dehydrogenase.
Cytoplasmic DA is also prone to auto-oxidation, a fate that is prevented inside
synaptic vesicles by the low pH [33]. This reaction is accelerated by metal ions,
particularly Fe
A third pathway in DA metabolism is the polymerization of its auto-oxidation products and inclusion into neuromelanin (NM). NM is a pigment accumulating in dopaminergic neurons, containing DA oxidation products, iron, lipids, proteins (including AS) and different toxic compounds and metals, particularly iron, zinc, aluminum chromium, molybdenum, lead and mercury [39, 40, 41]. These components are bound to the polymer matrix and enclosed in a lipid bilayer membrane, forming intracellular NM organelles [39]. Thus, NM synthesis is thought to be protective of neurons by sequestering potentially toxic compounds from the cytosol. However, NM can also have toxic effects when released from degenerating neurons in PD, promoting microglia activation and neuroinflammation [42].
AS is an intrinsically disordered protein (IDP) composed of an N-terminal,
lipid-binding domain (residues 1–66) [43], a central, non-amyloid
 Fig. 3.
Fig. 3.Schematic representation of AS structural plasticity.
(A) Domain organization depicted on AS primary structure. (B) Main conformational
transitions of AS. NAC, non-amyloid
The soluble, disordered monomer is the most common state, in which AS is found
in the cytoplasm of neuronal cells [47]. In-vitro studies indicate that
AS forms rapidly interconverting conformers, whose nature and biased distribution
can promote specific aggregation pathways [52]. The disordered nature of
cytosolic AS has been experimentally assessed by in-cell nuclear magnetic
resonance (NMR) and electron paramagnetic resonance (EPR) spectroscopies [47].
NMR and EPR signals show that the protein remains highly flexible, transiently
interacting with various cytoplasmic components and undergoing rapid molecular
reorientation. In-vivo cross-linking (XL) coupled to mass spectrometry
(MS) is another approach that probes protein conformation inside the cell,
yielding information on intra- and inter-molecular contacts and inter-residue
distances. Such kinds of measures can feed computational modeling by experimental
constraints. This method describes AS conformational ensemble as dominated by
rather compact, globular conformations with transient secondary structure
elements. Compaction is determined by contacts between the N- and C-terminal
regions, with a transient
Multiple and transient conformations are also captured by fluorescence resonance
energy transfer (FRET), which also allows in-vivo conformational studies
on individual molecules with high spatio-temporal resolution. Time-resolved FRET
(trFRET) experiments have been performed on a set of eight AS mutants [55]. The
structural transitions simulated by molecular dynamics (MD) based on FRET
trajectories are heterogeneous and occur on the millisecond timescale.
Secondary-structure composition of AS conformers has been captured by
surface-enhanced Raman spectroscopy (SERS) coupled with in-situ optical
tweezers [56]. This experimental setting allows micro-sampling of aqueous
solutions, in which AS is present in a non-aggregated form, at concentrations
comparable to physiological concentrations in the neuronal cytosol (1 µM)
[56]. Secondary structure has been detected at quasi–single molecule level from
200 measurements performed in parallel on a same AS preparation. The majority of
molecules (
The general scheme of the protein aggregation mechanism involves misfolded conformers assembling into low-molecular weight oligomers, followed by the formation of prefibrillar aggregates and mature fibrils (Fig. 3B).
Despite the large amount of research, most of the details of these molecular events are unknown, including the structures of AS monomers and aggregation intermediates, the relationship among the different aggregation pathways reported in the literature, and the interplay between the protein structure and the cellular/extracellular contexts. Fig. 4 gives an outline of the AS aggregation pathways described in the literature, where steps modulated (triggered or inhibited) by DA and related molecules are indicated (see also the next sections).
 Fig. 4.
Fig. 4.Aggregation pathways of AS. DA* indicates events known to be modulated (triggered or inhibited) by DA and related molecules. Off-pathway intermediates and amorphous final aggregates are not shown. LLPS, liquid-liquid phase separation.
As reported in the previous section, AS monomers are highly dynamic and populate extended and partially folded conformers, whose equilibrium can be shifted by environmental factors, including ligand binding. AS is partitioned between cytosolic and membrane-bound forms. It has been shown that the presence of free monomers in solution (high AS/membrane ratio) promotes primary nucleation on the surface of vesicles already saturated with AS by up to three orders of magnitude [74].
Starting from the heterogeneous ensemble of AS conformers, two main
interconnected aggregation pathways have been described [75], both inhibited by
DA, as discussed in the next paragraph. In the classical nucleation-dependent
polymerization process [76], aggregation-prone conformers of AS [77] form
oligomeric structures, which are on- or off-pathway of fibrils formation.
Oligomers with spherical, ellipsoidal and annular morphologies have been
described. Conformational transitions from random coil to
It has been reported that the nucleation-dependent polymerization process occurs at low AS concentrations, while an increase in protein concentration above a threshold value is necessary, at least locally, for phase transition [86, 87]. PD-promoting conditions, such as AS mutations and PTMs, oxidative stress, exposure to metal ions and pesticides, have been reported to impact on AS aggregation. Under the above-mentioned conditions, to which molecular crowding is also added, a conspicuous lowering of the critical concentration for LLPS could occur [86, 87], suggesting the inclusion of PD in the list of so-called condensopathies. Pathogenic AS species deriving from these pathways spread from cell to cell, propagating amyloid aggregation similarly to prions. However, since infectivity of synucleinopathies has not been proved, AS is referred to as a “prion-like protein” or “prionoid”, rather than “prion” [88]. The AS spread is considered an important factor determining PD progression, as supported by the spatial and temporal spreading pattern of AS in the brain at different stages of the disease [89].
Intercellular AS transmission requires two fundamental steps: (1) release of seeding-competent AS species from donor cells and (2) internalization of these protein species by healthy cells where they act as aggregation nuclei for the endogenous AS monomers. Several mechanisms have been reported in the literature. Processes that have been implicated in the first step are: diffusion across intact cell membranes of AS monomers and small oligomers, diffusion through damaged cell membranes, non-classical exocytosis pathways independent of the endoplasmic reticulum (ER)-Golgi apparatus, exosomes, and tunneling nanotubes [88]. Processes that have been reported for the second step are: diffusion across intact cell membranes, diffusion through damaged cell membranes, transmembrane amyloid pore-like channels, the classic endocytosis pathway, exosome-mediated uptake, and receptor-mediated internalization. An in-depth overview of these mechanisms has been recently reported [88].
Different polymorphs and “strains” of AS could arise from these pathways with a potential role in the phenotypic heterogeneity of PD and other synucleinopathies [13, 90]. Thanks to solid-state NMR spectroscopy and cryo-electron microscopy (cryo-EM), polymorphic amyloid structures can be described at atomic resolution [91]. AS fibrils obtained in vitro using recombinant wild type (WT) and disease-associated mutants display structural differences. These observations and the implication of AS in several distinct neurological disorders support the “strain hypothesis”, where distinct pathogenic AS conformations determine the specific diseases [92]. The AS fibril structures derived from the brains of individuals affected by synucleinopathies, such as MSA, PDD and DLB, have been recently reported [1, 93]. AS inclusions from MSA brain are made of two types of filaments (each consisting of two protofilaments) that are different from those of individuals with DLB and from those obtained in vitro by recombinant AS [93]. AS filaments from the brains of PD, PDD and DLB patients, instead, are made of a single protofilament [1]. These results [1, 93] strongly support the strain hypothesis, indicating the presence of distinct AS assemblies in different synucleinopathies. Cryo-EM structures of AS in presence of lipids shows that phospholipids induce alternative arrangement of protofilaments and fill the central cavities of AS fibrils [94].
In addition to membrane destabilization by oligomers [80], several other mechanisms of AS toxicity have been reported in the literature, including synaptic dysfunction, impairment of the normal quality control systems (molecular chaperones, ubiquitin proteasome system, phagosome/lysosome system), disruption of microtubule dynamics and axonal transport, endoplasmic reticulum/Golgi dysfunction, nucleus malfunction, and microglia activation leading to neuroinflammation [95]. Among these, mitochondrial dysfunction, leading to oxidative stress, is considered one of the main mechanisms of AS-induced toxicity [95]. Indeed, AS affects mitochondrial function by several mechanisms, including impairment of mitochondrial electron transport chain complexes, dysregulation of mitochondrial calcium levels, dysfunction of the mitochondrial quality control systems, and impairment of mitochondrial protein import machinery [95].
The interest in small molecules able to bind AS is highly motivated by the goal of developing new drugs for synucleinopathies and understanding the mechanisms of disease onset and development. Such studies have been focusing on the assessment of direct or indirect interaction between AS and the ligand, identifying the structural determinants of intermolecular recognition, investigating the effects of ligand binding on protein conformation and aggregation, as well as computational modeling of supramolecular complexes and conformational ensembles.
It has long been recognized that DA and its oxidation products can inhibit and
even reverse AS fibrillation in vitro and in vivo, deviating
the aggregation pathway towards spherical, soluble oligomers [96]. This effect
does not depend on protein covalent modification and is, rather, associated to
protein conformational changes [96, 97]. DA oxidation, too, is not absolutely
required, as shown by control experiments, in which the effect is retained while
inhibiting oxidation by NaBH4. DA-induced AS oligomers slow down fibrillation in
a dose-dependent manner [98]. Circular dichroism (CD) and infrared (IR)
spectroscopy indicate that the conformational ensemble of oligomeric AS is
shifted in the presence of DA towards a predominantly disordered state with some
elements of
In-vitro characterization of serial AS deletion products and
competition experiments by designed peptide have led to first identification of
the main AS-DA interaction site within the C-terminal pentapeptide
MD simulations of AS in aqueous solutions and clustering analysis on
experimental NMR structures indicate that DA binds preferentially to the
 Fig. 5.
Fig. 5.Representative structure of the AS-DA complex from molecular dynamics (MD) simulations. Hydrogen bonds are represented by dashed lines and hydrophobic interactions by arcs with spokes (adapted with permission from [102]).
In-vivo AS conformational properties and their response to DA
concentration have been explored by the FRET-based assay for molecular proximity
called fluorescence lifetime imaging microscopy (FLIM) [104]. This technique is
based on the observation that the fluorescence lifetime of a donor fluorophore
decreases with the 6th power of the distance from a FRET acceptor. Mouse and rat
primary neuronal cultures were transfected by a doubly-tagged Myc-aSyn-V5 to
probe the intramolecular distance between N- and C-termini by FLIM, based on
immunocytochemistry with fluorescent labeled secondary antibodies. A first
interesting observation was an inhomogeneous intracellular distribution of AS
conformers, with more extended conformations in the cell body and more compact
ones in the neurites. Furthermore, DA uptake significantly reduces the average
distance, indicating that DA itself or some DA metabolite modulates AS
conformational ensemble in vivo in favor of partially structured states.
No effect was observed with DA agonists or antagonists. Very minor changes in CD
spectra have been interpreted as an increase in random-coil and loss of
DA-induced changes in AS conformational ensembles in vivo are mirrored by peculiar structural and functional features of the resulting oligomers [26]. Indeed, the oligomeric aggregates that accumulate in mice expressing the human A53T mutant, upon induction of catecholamine production, are larger (up to 122 Å) than in the control system (up to 65 Å) and do not display seeding activity on AS aggregation, as assessed by in-vitro Thioflavin T (ThT) assays and analysis of trans-synaptic spreading to motor cortex. Furthermore, AS oligomers formed in the presence of DA are known to be sodium dodecyl-sulfate (SDS)-resistant [100, 107] and this feature has been reported to be modulated by the concomitant presence of copper and DA [108]. DA-stabilized oligomers are suspected to have toxic effects by multiple possible mechanisms [109], such as inhibition of SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex [110], impairing chaperone-mediated autophagy [111], and causing mitochondrial dysfunction [112]. However, a recent article on SH-SY5Y cells and rat synaptosomes reports negligible toxicity of purified AS oligomers formed in the presence of DA [98].
In line with previous evidence, it has been shown that DA counteracts the effect
of psychosine on AS conformation and aggregation in vitro [113].
Psychosine promotes AS fibrillation in ThT assays and has a fingerprint in
Further NMR experiments have recently pointed out that
Native MS can provide important additional information in the attempt to describe disordered conformational ensembles [115, 116]. This technique captures conformational components coexisting in the original liquid sample by their different ionization propensity under mild electrospray conditions [117, 118]. The possibility to preserve non-covalent interactions, along with conformation-dependent protein ionization, makes it possible to depict conformational ensembles, discriminating conformational states by structural compactness. At the same time, non-covalent complexes are detected by their specific mass, providing a unique possibility to gather combined information on protein folding and binding [119, 120]. Native MS on AS in aqueous solutions detects four main conformational components based on charge-state distributions [121, 122]. These are referred to as compact, extended and intermediate (I and II), by comparison to the reference behavior of folded and unfolded proteins [118].
Native-MS analysis of AS-DA interactions by titration experiments at pH 7.4
[122] provides direct experimental evidence of the formation of non-covalent
complexes and shows that no chemical modification of protein or ligand takes
place in the time scale of such experiments. DA binds preferentially to a
partially structured AS conformer with stoichiometry up to 1:4 and sub-millimolar
apparent K
Another biophysical method to explore conformational ensembles is single-molecule force spectroscopy (SMFS), in which individual molecules in solution are unfolded mechanically by the cantilever of an atomic force microscopy (AFM) instrument, relating pulling force to the structural features of the molecule [125]. By randomly reiterating this procedure, it is possible to characterize the statistical distribution of distinct protein conformers. Three main conformational states of AS in buffered solutions at pH 7.4 can be detected by this method based on the strength of the intramolecular interactions [105, 126, 127]. The addition of DA shifts the conformational ensemble towards the strong interactions, with loss of the fully unstructured component [105]. These interactions must be at the level of tertiary structure, since the secondary structure content remains unaltered, as discussed above. The structural interpretation of MS and SMFS data has been corroborated by comparison to structural clusters obtained from a 73 µs-long MD simulation of the AS conformational ensemble in solution [128, 129].
AS-DA binding at near-neutral pH has been detected by other orthogonal techniques, such as isothermal titration calorimetry (ITC) and nanopore-permeability analysis. One ITC study at pH 7.8 indicates relatively weak binding, with an estimated dissociation constant of about 280 µM [106]. On the other hand, another ITC study reports too weak binding at pH 7.4 to be quantitatively evaluated [130]. Apparent dissociation constants in the sub-millimolar or millimolar range can be considered biologically relevant, since DA concentrations are of the micromolar order in the cytosol and reach the millimolar range within storage vesicles and striatal nerve terminals [99]. Nanopore-permeability assays provide additional evidence of AS-DA interaction by detecting changes in the frequency distribution of pore translocation or bumping events for the protein in solution, before and after the addition of the ligand [106]. Translocation events are only considered possible for molecules in random-coil conformation, while folded or partially folded structures, instead, bumping away on the cis side of the membrane [131, 132]. According to this interpretation, DA would stabilize a partially folded intermediate in WT AS, while stabilizing the random-coil state in the A30P mutant. Although the results are not of straightforward interpretation in terms of conformational effects, they suggest direct interaction between AS and DA [106]. Thus, the available data suggest that DA itself can bind to AS and affect its aggregation. DA oxidation would not be an absolute requirement, although, as described below, some DA metabolites, such as DA o-quinone and DOPAC, are more effective [97, 100]. Further studies are needed to understand whether apparently conflicting reports in the literature can be interpreted and reconciled by the peculiarities of the different biophysical approaches.
In order to find AS ligands that can inhibit AS fibrillogenesis and toxicity, several Authors have studied the interactions of this protein with DA-related molecules (catabolites, agonists, and analogs). DA analogues are molecules that structurally mimic DA, while DA “agonists” and “antagonists” are molecules able to activate or inactivate the biological response mediated by DA receptors.
One of the first systematic search for direct interactors has been carried out
by in-silico screening of 70 compounds and led to the identification of
5 DA analogs that, similarly to 6-aminoindole, are expected to affect the
nucleation/polymerization mechanism and possibly to bind oligomers [133] (Fig. 6). In-vitro assays by ThT and AFM has showed that 6-aminoindole and
5-hydroxyindole have the strongest inhibitory effects on AS aggregation, while
tyramine and 4-(2-aminoethyl)aniline have the poorest effects [133]. Structural
comparison between tyramine and DA suggests that the loss of just one of the -OH
groups (replaced by -H in tyramine) causes a dramatic loss in anti-fibrillogenic
activity. It has been hypothesized that a benzene ring with two -OH or quinone
groups (i.e., 1,2-benzoquinone moieties) may have great potential in AS binding.
Stacking interactions and charge interactions appear to be relevant in
determining the interaction with either soluble or aggregated AS. Below, DA
analogs showing these key structural motifs are discussed. An example of DA
analog directly interacting with soluble AS is provided by
Fasudil®, an isoquinoline substituted by a
(1,4-diazepan-1-yl)sulfonyl group at position 5 (Fig. 6). It also acts as an
inhibitor of Rho kinases and serendipitously proved to have antifibrillogenic
activity. Its use has been approved by the U.S. Food and Drug Administration
(FDA) in clinical trials of disorders such as Raynaud’s disease, atherosclerosis,
and age-related or neurodegenerative diseases [134]. Direct interaction of
Fasudil® with AS has been primarily demonstrated by NMR [135] and
analyzed by MD simulations [136]. These studies offer an atomic-level description
of a possible inhibition mechanism. Aromatic stacking and electrostatic
interactions are formed through a process termed dynamic shuttling, in
which one set of interactions breaks before another is formed nearby [136]. The
interactions preferentially involve the C-terminal region of AS, which remains
highly dynamic upon binding. Beside the tyrosine-glutamate (YE) residues
 Fig. 6.
Fig. 6.Structural formulas of the DA analogs mentioned in the text. EGCG, epigallocatechin.
The category of molecules that bind and dissolve aggregated AS includes three molecules recently isolated by in-vitro high-throughput screening based on ThT fluorescence, light-scattering and transmission electron microscopy (TEM), applied to a library of more than 14,000 compounds [137] (Fig. 6). These are named SynuClean-D [65, 138], ZPD-2 [139] and ZPDm [140], and proved effective even at sub-stoichiometric concentrations in disaggregating mature fibrils of WT AS and familial variants A30P and H50Q. All three compounds are also able to reduce the aggregation of AS in PD models of Caenorhabditis elegans but do not interact with soluble AS, as indicated by NMR studies [140, 141] (Fig. 6). Unfortunately, no structural data are available on the atomic details of the interactions between these disaggregating agents and AS fibrils. Although the three molecules have similar effects, they might have fine differences in their mechanism of action reflecting their structural differences.
For the purpose of drawing some rules about the structure and function of
disaggregating molecules, SynuClean-D has been compared with diverse and much
more complex compounds, such as EGCG [142] and the molecular tweezer CLR01 [143, 144]. The hypothesis underlying this comparison is that greater structural
complexity and larger hydrophobic contact surface (i.e., larger number of
aromatic rings) leadto stronger effects in fibril disaggregation [145]. In fact,
SynuClean-D, EGCG, and CLR01 contain, respectively, 2, 3, and 5 aromatic rings
and this ranking also reflects their disaggregation efficacy (CLR01
DA agonists include molecules that are commonly used to relieve symptoms of PD.
Fig. 7 shows the structure of some DA agonists [153, 154]. Among them, the
neuroprotectant cabergoline has been suggested to directly interact with AS by
molecular docking experiments [155]. Pergolide and bromocriptine have long been
recognized by in-vitro ThT assays to also destabilize preformed fibrils
by direct interaction [156, 157]. A small group of agonists can be considered
bifunctional, in that they also exhibit the ability to modulate AS aggregation by
either direct interaction, or by affecting its PTMs and/or turnover [158].
Carbazole-based DA agonists are a recent example of development of such kind of
multifunctional drugs [159]. Bifunctional molecules have been designed ad
hoc by introducing hydroxyl groups into the structure of known agonists, so that
their efficacy on the signaling cascade is not altered and the ability to
interact directly with AS is newly conferred. Compounds such as D-519 and D-520
(Fig. 7) prove effective in reducing symptoms in Drosophila melanogaster
and rat models of PD, having maintained their efficacy as strong agonists of
D2/D3 receptors, and showing new properties derived from their ability to
directly interact with AS [160]. In particular, D-520 can reduce the toxicity of
preformed AS aggregates against rat pheochromocytoma cell cultures and modulate
AS aggregation, as demonstrated in vitro by ThT and TEM [160].
Apomorphine (Apo) is a non-ergoline DA agonist, whose dual function is a
downside. Apo binds with high affinity to DA receptors D2, D3, and D5, and can
also directly interact with AS [160]. Apo was approved by the FDA in April 2004
for the treatment of hypomobility and dyskinesia associated with PD. Apo inhibits
fibrillation and dissolves pre-formed fibrils, as assessed by ThT assays and TEM.
Under oxidizing conditions, off-pathway oligomeric adducts, Apo-AS-O, are formed.
AS oligomerization is faster in the presence of Apo than DA. Apo-derived AS
oligomers are toxic to cultured neuronal cells, and form by recruiting innocuous
AS monomers. NMR 1H-15N-HSQC experiments show that Apo binds monomeric AS in the
 Fig. 7.
Fig. 7.Structural formulas of the DA agonists mentioned in the text.
The interaction of AS with DOPAL, the immediate metabolite of DA catabolism, has been proposed as a key factor in PD pathogenesis in the so-called catecholaldehyde hypothesis [32]. The aldehyde group generated by MAO-mediated conversion of the DA amine group makes DOPAL capable of quinonizing AS, forming covalent adducts to lysine residues or to the N-terminus by Schiff base or Michael addition, or generating a dicatechol pyrrole lysine structure [161]. ThT fluorescence experiments have shown that DOPAL destabilizes preformed AS fibrils and inhibits aggregation, generating off-pathway oligomers [162]. According to near-infrared fluorescence spectroscopy and western-blotting experiments, DOPAL induces the formation of AS oligomers more effectively than DA [163]. DOPAL-induced AS quinonization and oligomerization are enhanced in the presence of Cu(II) ions [163]. Despite some contrasting evidence [164], a consensus is emerging in the literature that many DOPAL-induced oligomers are toxic and compromise several physiological processes in neurons [165]. Blocking the DOPAL aldehyde moiety by compounds containing primary (e.g., aminoindan) or secondary (e.g., rasagiline) amines abrogates its effects on protein aggregation and cytotoxicity [165].
AS oligomers generated by DOPAL have been investigated by SEC-HPLC [166]. Detected species have been classified, according to their size, in small (dimers and trimers) and large (tens of subunits) oligomers. The relative amounts of these species are finely tuned by several factors (mutations, PTMs, etc.). Among them, DOPAL-induced oxidation of methionine residues has been shown to favor the accumulation of small oligomers, hampering the formation of larger species [166, 167]. In particular, the oxidation of the C-terminal methionine at position 127 improves the Ability of AS to scavenge ROS derived from DOPAL and prevents the assembly of large neurotoxic oligomers [167].
DOPAL-mediated oligomerization is also influenced by familial PD point mutations
of AS. In particular, the A53T, E46K and H50Q variants of AS have been shown to
stimulate formation of large (n
The DA metabolite DOPAC is the product of DOPAL detoxification by aldehyde
dehydrogenase conversion, and it is usually rapidly removed by active transport
from the cell via a sulfonylurea-sensitive transporter [169]. As its
precursor DOPAL, it can undergo spontaneous oxidation to a quinone, thus
producing H
It has been observed in vitro that DOPAC at physiologically relevant
concentrations binds non-covalently to AS and stabilizes the oligomeric state,
hindering fibrillogenesis [170]. Because of the oxidative instability of the
compound, the molecular mechanism could not be unveiled, but this effect might be
due to an alteration of the oligomer interface that plays a fundamental role in
fibril formation. The interaction has been mapped by mutagenesis to the same
N-terminal region that binds lipid vesicles [163]. At high molar excess (i.e.,
DOPAC:AS
As discussed in the previous section, DOPAL is further oxidized by mitochondrial aldehyde dehydrogenase to DOPAC, in the major oxidation pathway of DA (Fig. 2). In a minor pathway (under normal conditions), DOPAL can be, instead, reduced by alcohol dehydrogenase (ADH) to 3,4-dihydroxyphenylethanol (DOPET), also called hydroxytyrosol (Fig. 2). In addition to this endogenous source as DA metabolite, DOPET can have a dietary origin, being found in olive oil and wine. Moreover, DOPET can be produced from the hydrolysis of olive oil secoiridoids, such as oleuropein. Noteworthy, DOPET is able to cross the blood-brain barrier and exhibits antioxidant, anti-inflammatory, and anti-aggregation activities [174].
In a first in-vitro study [175], DOPET was found to inhibit AS aggregation and to destabilize preformed AS fibrils, as evaluated by ThT and TEM investigations. In particular, DOPET inhibits AS fibrillogenesis, leading to the formation of small amorphous aggregates. Moreover, DOPET was found to counteract AS-induced toxicity on PC12 cells [175].
The effects of DOPET on AS aggregation have been elucidated by employing
complementary biochemical and biophysical approaches, including ThT assay, CD
spectroscopy, TEM, native MS, limited proteolysis, intrinsic fluorescence, SEC
and electrophoretic analyses [176]. ThT assay and TEM show that a 1:1 DOPET:AS
ratio completely inhibits AS fibrillogenesis, leading to the formation of AS
oligomers, which appear structurally disordered as indicated by CD spectroscopy.
The CD analyses also suggested that DOPET does not affect the secondary
structures of monomeric AS. Chromatographic, TEM, and MS analyses of AS treated
with DOPET showed the presence of spherical oligomers and that of protein species
containing one-to-four oxidations of methionine residues, whose speed of
formation and extent of oxidation were related to DOPET concentrations. Covalent
adducts, likely involving the
The interactions of DOPET and DOPAC with WT AS and the E46K pathological mutant have been compared [177]. Both compounds partially or completely inhibit fibril formation at 1:1 or 1:5 protein:catechol ratio, respectively. In the absence of catechol, WT and E46K formed fibrils with a different morphology, as observed by TEM. DOPAC at 1:5 protein:catechol ratio induced the formation of spherical and annular oligomers of 10–20 nm and 20–40 nm for E46K and WT AS, respectively. In the presence of DOPET, mainly amorphous structures were observed under the tested experimental conditions. The formation of oligomeric species was also investigated by SEC, which showed additional peaks due to protein dimers and trimers in the presence of each compound. A higher fraction of oligomeric species was observed in the early stages of incubation for E46K, both in the absence and the presence of catechols, compared to WT AS. CD spectroscopy showed that the off-pathway species induced by DOPET and DOPAC are characterized by a predominant random-coil structure, similar to monomeric AS. In all the above investigations, DOPET appeared less active in affecting AS fibrillogenesis compared to DOPAC and the E46K mutant appeared less affected by catechols compared to the WT AS [177].
Important structural details of the catechol effects on AS fibrillogenesis were obtained by SEC, native-MS and hydrogen-deuterium isotope exchange (HDX)-MS. At the beginning of the incubation in the absence of catechols, E46K eluted at a slightly higher volume in SEC compared to WT AS suggesting increased compactness of the mutant. In the presence of DOPAC or DOPET, E46K eluted slightly earlier, suggesting a stabilization of a more extended conformer. On the contrary, catechols did not affect the retention time of the WT protein. Native-MS indicated the presence of multiple protein conformers with different compactness at the beginning of incubation, in agreement with other studies [177]. The addition of DOPAC shifted the equilibrium towards a more extended conformer, an effect that was more evident for the WT protein. Compared to DOPAC, the effect of DOPET was less evident on E46K and not significant in the case of WT AS. The most remarkable effects of both catechols were observed by native-MS at 48 hours of incubation for E46K and WT AS [177]. ThT assays and TEM indicated that DOPAC, and DOPET to a minor extent, were able to disaggregate intermediates and mature E46K aggregates obtained in the absence of the compounds. This effect was less evident in the case of mature WT AS aggregates.
On the basis of these results [176, 177], the Authors proposed the following model for the effects of the two catechols on AS aggregation. Catechol binding affects the conformational equilibrium of monomeric AS, stabilizing a conformer that is less prone to convert into amyloid fibrils. These extended monomers assemble into not-toxic, off-pathway oligomers and amorphous aggregates. AS oxidation is not required for DOPET inhibition of AS fibril formation [176, 177].
DA exerts its toxicity through multiple mechanisms related to the products of
its catabolism and to direct AS binding. DA binding to AS monomers is associated
with conformational changes in the protein that redirect the aggregation pathway
towards spherical, soluble, SDS-resistant, and toxic oligomers. DA also induces
oxidation of methionine residues, although interaction with AS and conformational
effects occur even in the absence of covalent protein modifications and DA
oxidation. At the same time, DA is also capable of disassembling amyloid
aggregates of AS, resulting in protein oligomers. Most of the DA related
molecules discussed in this review are able to affect AS fibrillogenesis by
non-covalent interactions -with the
The study of DA analogs suggests a predominant role of
The available treatments for PD are directed to symptoms without significant
effects on the molecular mechanisms of disease progression. Strategies to
directly target AS are under investigation, such as small molecules inhibiting
the accumulation of toxic AS species or immunoglobulins promoting the clearance
of AS aggregates. Relevant to this regard is the development of ligands that are
selective for AS toxic species, such as nanobodies [178] and peptides [179].
Detailed characterization of the binding properties of several antibodies
undergoing clinical trials for PD has been performed. Most of them bind to the AS
residues 102-130, which are close to or overlapping with the
In this scenario, information on the molecular mechanisms of AS aggregation and AS interaction with DA and related molecules may provide new therapeutic strategies. Indeed, it has become evident how these molecules lead to oligomers and end-point aggregates with diverse structure and toxicity. Molecules that counteract both fibrillogenesis and toxicity can be the starting point for designing new drugs. The comparison of toxic and non-toxic oligomers can provide the rationale for identifying new targets for immunotherapy.
AS existence as conformational ensembles and their variegated response to the environment represents a crucial aspect of the structure-function relationship of this protein and a major experimental challenge. Advances in experimental and computational methods are bringing us closer to the goal of modeling at atomic resolution such a difficult target in the presence or absence of ligands. Particularly relevant to this regard is the contribution of cryo-EM to the characterization of AS fibril polymorphism in the presence or absence of heparin, providing a rationale for drug design [182]. Another central challenge is to translate structural information into in-vivo protein activity. Such an in-depth understanding is required for the design of new therapeutic strategies based on small molecules or antibodies, targeting the conformers with the most pronounced fibrillogenic potential.
AN and RG designed the study. AN, SB, EP, CS, and RG contributed to writing the original draft and preparing the figures. AN, SB, EP, CS, and RG contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Given her role as Guest Editor of the journal, SB had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to Fabio Moda.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.