








 , Madeeha Shahzad Lodhi 1, Kejie Mou 3,*, Zili Liu 3, Dongqing Wei 2,4,5
, Madeeha Shahzad Lodhi 1, Kejie Mou 3,*, Zili Liu 3, Dongqing Wei 2,4,51 Institute of Molecular Biology and Biotechnology (IMBB), The University of Lahore, 58810 Lahore, Pakistan
2 Zhongjing Research and Industrialization Institute of Chinese Medicine, Zhongguancun Scientific Park, Meixi, 473006 Nanyang, Henan, China
3 Department of Neurosurgery, Bishan hospital of Chongqing Medical University, 402760 Chongqing, China
4 State Key Laboratory of Microbial Metabolism, School of Life Sciences and Biotechnology, Shanghai Jiaotong University, 200240 Shanghai, China
5 Joint Laboratory of International Cooperation in Metabolic and Developmental Sciences, Ministry of Education, Shanghai Jiaotong University, 200240 Shanghai, China
Abstract
Background: In the past 10 years, significant progress has been made in
understanding the pathogenic chain of events that causes Alzheimer’s disease
(AD). According to the most widely accepted concept, the production and
aggregation of
Keywords
- Alzheimer's disease
- mutations
- APP
- tau
- nanoparticles
In older people, Alzheimer’s disease (AD) is a progressive, slow brain disease characterized by plaque formation in the hippocampus [1]. Researchers have found that plaque formation starts about 20 years before clinical symptoms begin, so it is difficult to determine the exact trajectory of AD pathologies [2]. The incidence of AD is rising worldwide. The number of people affected by AD in 2019 exceeded 50 million; as a result, the world economy and the human workforce could be negatively affected by its increasing burden. The prevalence of AD symptoms in Americans 65 and older is estimated to reach 13.8 million by the middle of the twenty-first century. AD is the sixth leading cause of death in the United States; according to the Centers for Disease Control and Prevention, 146.2% of Americans died from AD in 2018 [1].
According to the current research on AD neuropathology, the primary
histopathologic lesions of AD are the extracellular amyloid plaques and the
intracellular tubulin associated unit (tau) neurofibrillary tangles [3]. The main
component of the amyloid or senile plaques (SPs) is a very insoluble and
proteolysis-resistant peptide fibril created by the cleavage of
Local inflammation, oxidation, excitotoxicity (excess glutamate), and tau hyperphosphorylation are some of the components of the cascade. A microtubule-associated protein called tau binds to microtubules inside cells to help the neuronal transport mechanism. Additionally, developing axons need to be stabilized by microtubules for neurons to function and development. Intraneuronic tangles are formed by abnormally hyperphosphorylated tau, generating insoluble fibrils. As a result, it separates from microtubules, prevents transport, and causes microtubule breakdown. It was initially believed that tau hyperphosphorylation happened after amyloid was deposited. Still, it’s as likely that tau and amyloid work parallel to cause AD and amplify each other’s adverse effects. The deficiency of some neurotransmitters (like acetylcholine, dopamine, and serotonin) and an imbalance among these neurotransmitters are the root causes of the cognitive impairments linked to Alzheimer’s disease (AD) [3].
AD is the most prevalent cause of memory loss in older people [6]. Cognitive
abnormalities, memory loss, and behavioral changes are all indicators of AD [7, 8, 9]. Dementia in AD is linked to neurodegeneration, which begins with synaptic
damage and progresses to neuronal death [10, 11]. Recent research has indicated
that some other components of the neurodegenerative process in AD may interfere
with the adult neurogenesis process in the hippocampus [12, 13]. Loss of synapses
in the limbic and neocortical systems is connected with cognitive impairment in
Alzheimer’s patients [14, 15]. Several lines of evidence point to AD’s
pathophysiology linked to the rising levels of A
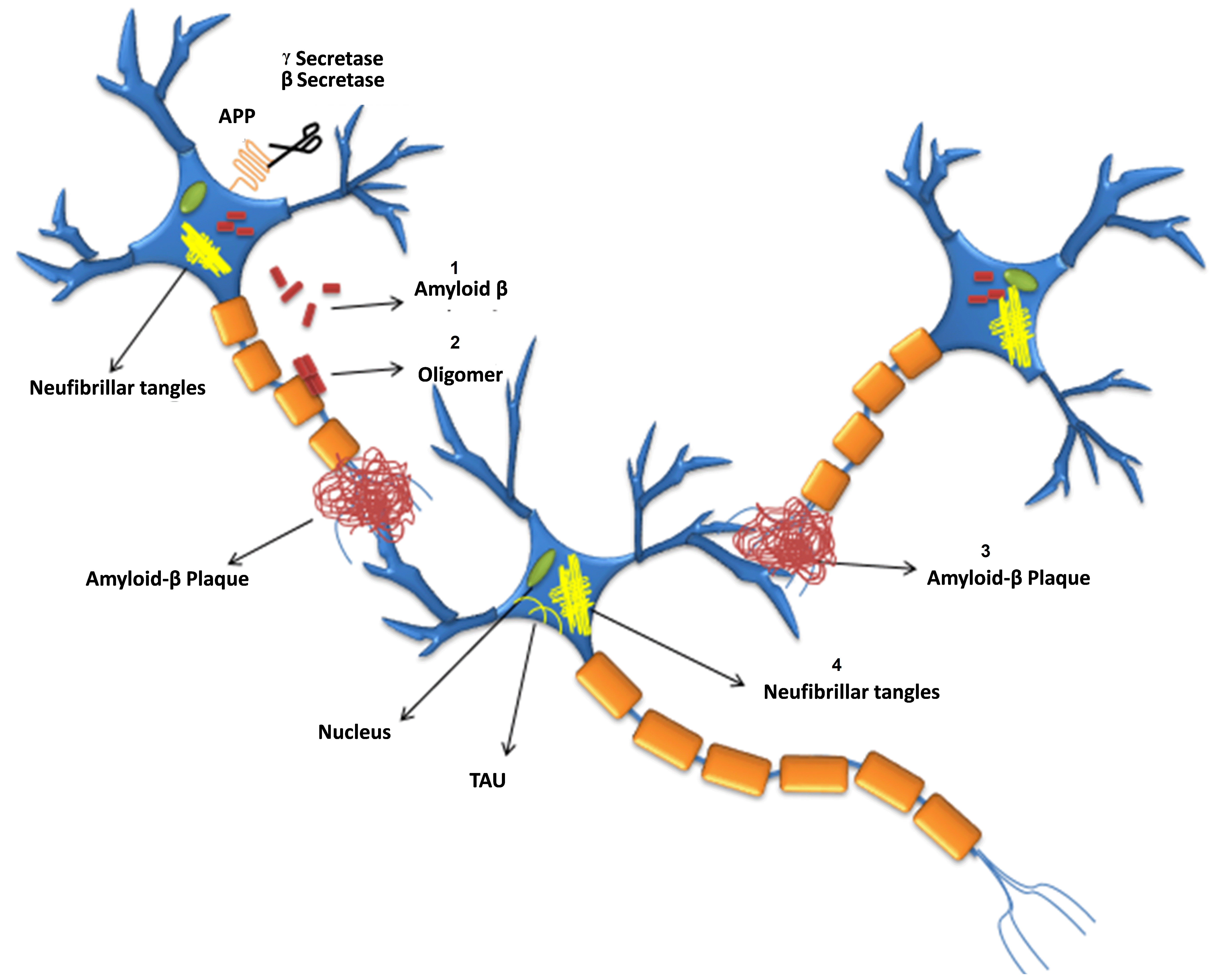 Fig. 1.
Fig. 1.Formation of
Abnormal A
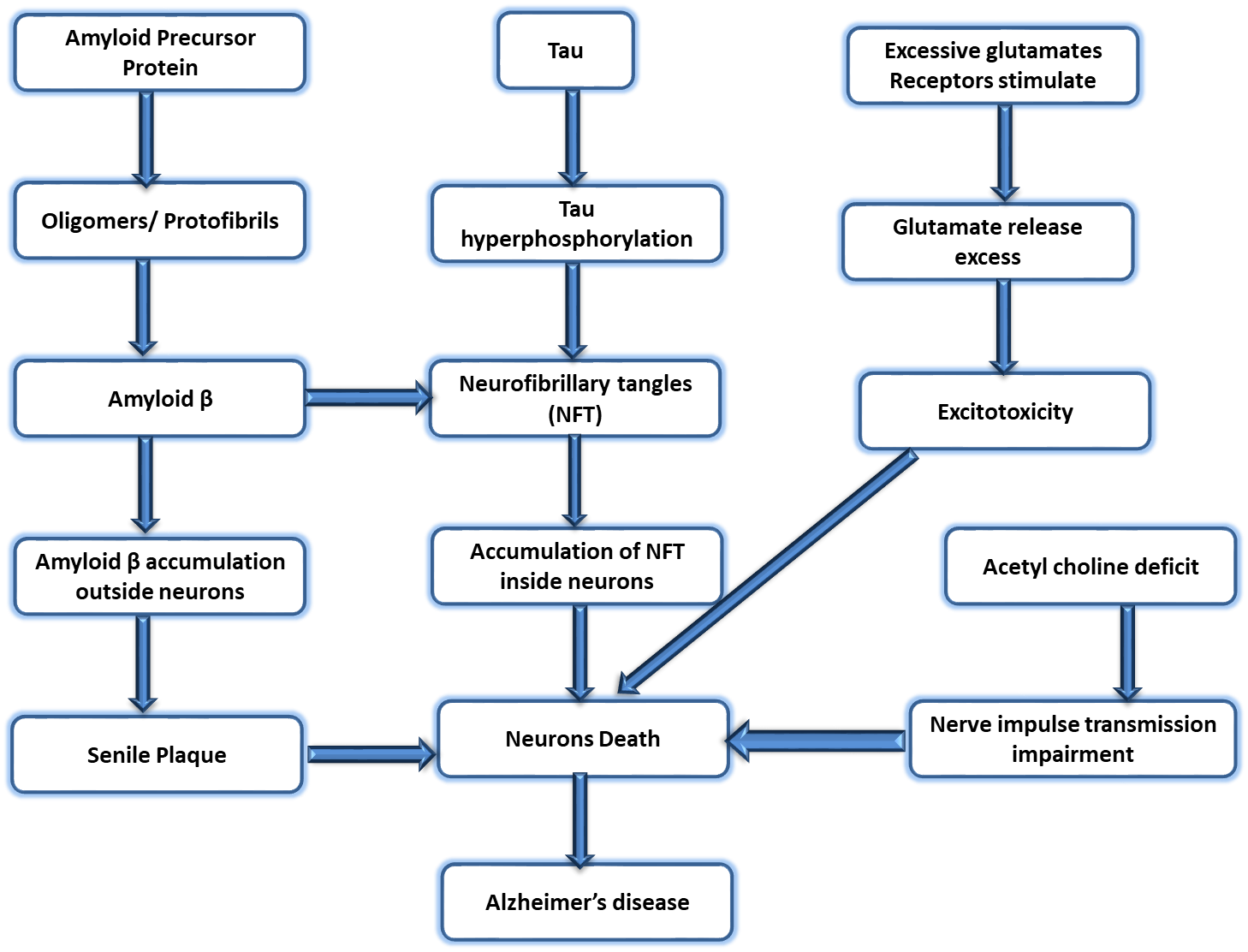 Fig. 2.
Fig. 2.Different phases of the pathophysiology of AD. AD, Alzheimer’s disease.
However, loss of synapses and axonal damage are probably crucial
neuropathological elements that cause dementia [20]. Alterations in neurogenesis
have recently been related to the neurodegenerative process in AD, along with
impairments in neuronal integrity and synaptic plasticity in mature neuronal
circuitries [21]. This shows that the double attack on the brain causes AD, the
degradation of mature neurons, and the alteration of neurogenic regions in the
adult brain [22] as the A
Patients with advanced AD experience significant memory loss, hallucinations,
confusion, and a lack of independence, finally dying from a respiratory syndrome,
infection, or starvation [27, 28]. A
Nanotechnology is a comprehensive science that involves manipulating atoms, electrons, protons, and neutrons in various ways to provide new insights into how materials might be developed to tackle numerous problems in numerous fields [31]. It has been suggested that NPs are exciting tools that may address the unmet need to improve drug transport from the blood to the brain, particularly by functionalizing their outer layer with blood-brain barrier (BBB)-targeting agents. Because they share characteristics like biocompatibility, stability, biodegradability, nontoxicity, limited antigenicity, and suitability for surface functionalization, Gold nanoparticles (GNPs), liposomes, solid lipid NPs (SLN), and polymeric NPs are the most studied NPs for brain drug delivery. They may also contain drugs that are both hydrophobic and hydrophilic, and controlled drug release is an option. Liposomes are spherical vesicles ranging from 20 nm to 500 nm. They contain an aqueous interior and a membrane bilayer typically composed of naturally occurring phospholipids and cholesterol [32]. Pharmaceutical interests were already shifting due to the potential usage of multiple biological sources for nanoparticle manufacturing and application. Silver is the most preferred metal for producing NPs because it has specific antibacterial, antifungal, anti-inflammatory, anti-angiogenic, and anti-permeability properties, among others [33].
Vegetables, nuts, whole grains, fruits, and other foods contain phytochemicals.
Combined or used alone, these phytochemicals have tremendous therapeutic
potential for treating various diseases [34]. Since ancient times, phytochemicals
have been employed to cure various human diseases. Carotenoids, catechin,
curcumin, diosgenin, quercetin, GX-50, sulforaphane, polyphenols, flavonoids, and
other chemical elements originating from different parts of plants, such as
fruits, leaves, and bark possess potential therapeutic properties. Phytochemicals
and their analogs are now widely employed as antioxidants, inhibitors,
substrates, modulators, enzymatic cofactors, and potential molecules in drug
discovery in the medical and pharmaceutical industries. Curcumin can potentially
prevent AD because it contains anti-amyloidogenic, anti-oxidative, and
anti-inflammatory effects [35]. In vitro tests have demonstrated the
anti-amyloidogenic properties of curcumin and its analog, rosmarinic acid, which
inhibit in a dose-dependent manner the elongation of neurotoxic A
A literature review was conducted utilizing various search engines, including PubMed, Science Direct, Scopus, and Google Scholar. The search was limited to papers published between 2000 and 2022, and specific search terms were employed, such as Alzheimer’s disease, neurological disorders, amyloid, nanoparticles, tau pathology, dendrimers, nanotechnology, phytochemicals, and liposomes. Only papers that appeared in the relevant search engine results were included in the review.
One to six percent of all instances of AD are early-onset (onset before the age of 60 or 65), and sixty percent of these cases are familial. Thirteen percent of these cases appear to be inherited through an autosomal-dominant mechanism [39, 40]. The distinction between early-onset family AD (EOAD), which starts before 60 or 65 years, and late-onset familial AD (LOAD), which starts after that age, is arbitrary. Early-onset cases may show up in families where the disease has a history of progressively developing [41].
AD is a chronic, irreversible brain disorder that progressively impairs memory, thought, reasoning, and other cognitive abilities. Additionally, it leads to behavioral problems that may interfere with a person’s regular activities and daily life. A patient suspected of having AD should undergo several tests, including medical and family history, neurological examination, Magnetic resonance imaging (MRI) for the neurons, and vitamin B12 lab tests [42].
For a very long time, vitamin B12 insufficiency has been linked to neurologic issues, and some studies have found that it raises the chance of AD. Elevated homocysteine levels are a specific indicator of vitamin B12 deficiency and are toxic to the brain because they increase oxidative stress, calcium influx, and apoptosis. The diagnosis of vitamin B12 deficiency may be made using measurements of serum levels of vitamin B12, complete blood counts, and serum homocysteine levels [43, 44]. The pathophysiologic process of AD starts years or even decades before clinical dementia and apparent cognitive abnormalities. As a result, the phrases “AD dementia” and “AD pathophysiologic process” are distinct.
There have been no clinical indications in preclinical AD. Measurable alterations in molecular and imaging biomarkers are being studied for pre-symptomatic evaluation.
With proper assessment, mild cognitive impairment AD can show moderate periodic changes in cognitive state and memory that do not interfere with everyday activities. It is essential to rule out any other dementia-causing factors. Genetic testing is an option in cases with early-onset familial AD to look for specific mutations.
The hallmark of Alzheimer’s dementia is a growing cognitive impairment that
makes day-to-day tasks challenging. It may be possible to confirm the diagnosis
using abnormal biomarkers such as elevated levels of tau protein in the CSF, low
levels of A
A complex neurological ailment called AD causes memory loss and progressive cognitive deterioration. For early identification, diagnosis, and the creation of efficient treatments, it is essential to comprehend the clinical stages, genetic components, and biomarkers related to AD. Here is a more thorough examination of the current state of knowledge, including any gaps and disagreements that may exist.
Diagnosing AD, tracking the course of the condition, and determining the
effectiveness of treatment depends heavily on biomarkers. Three biomarkers can be
distinguished: neurodegenerative markers, tau biomarkers, and A
The current understanding of AD has several gaps and difficulties despite significant progress. Among the crucial areas are:
3.3.1.1 Early Diagnosis
Research is still being done on developing reliable biomarkers and clinical standards for the early and precise diagnosis of AD. The timely application of disease-modifying therapies depends on early identification [47].
3.3.1.2 Biomarker Validation
To define standardized and verified biomarker cutoff values and criteria for clinical application, discussion and research are underway. This is necessary for reliable and consistent interpretation of biomarker results in various labs and clinical contexts [48].
3.3.1.3 Genetic Factors
While the APOE4 allele is a recognized genetic risk factor, other genetic variants that increase the risk of AD also need to be identified and understood. To find new risk genes, genome-wide association studies (GWAS) and other genetic research projects are ongoing [49].
3.3.1.4 Treatment Efficacy
The designing of efficient AD disease-modifying medicines is still a difficult task. There is a need for additional studies to determine the best therapy options and potential combination therapies because the results of several clinical trials targeting tau and A have been inconsistent [50].
Early-onset familial AD (EOFAD) is the term used to describe multiple AD occurrences in families. While some investigation has utilized ages 60 or 70, the average beginning age is frequently younger than 65. The average beginning age is between the 40s and early 50s, but instances of beginning in the 30s and early 60s have also been reported. Previous research shows that 41.2 out of every 100,000 at-risk individuals have EOAD in the population (between the ages of 40 and 59) [51].
Thirteen percent of patients with EOAD satisfied rigorous criteria for an autosomal-dominant inheritance, 61% had an excellent familial background EOFAD, and non-familial AD cannot be clinically distinguished, except for the age of initiation and family history. The phenotype of dementia might have a prolonged prodrome and is analogous to late-onset AD [52].
Molecular genetics: Based on the gene that causes EOFAD, at least three subtypes
(AD-1, AD-3, and AD-4) (Table 1, Ref. [53]). Provides an overview of the causal
genes and the proportional frequency of each subtype. The A
| Locus name | Percentage | Gene symbol | Chromosomal locus | Protein name | Test availability |
| AD-3 | 20–70% | PSEN1 | 14q24.3 | Presenilin 1 | clinical |
| AD-1 | 10–15% | APP | 21q21 | Amyloid |
clinical |
| AD-4 | rare | PSEN2 | 1q31- q42 | Presenilin2 | clinical |
EOFAD, Early-onset familial AD.
Finding more genes linked to the disease may be made possible by new technologies that significantly use genome-wide association studies, genetic markers, cutting-edge statistical techniques, and collaborative efforts to expand the number of study-able cases. In addition, many tools have been developed for identifying genes linked to AD.
Researchers are given access to clinical information and biological samples for their genetic analysis. The National Institute on Aging (NIA) has set up numerous systems to preserve genetic data produced by National Institute of Health (NIH) funded investigations. These data can be re-examined and used in conjunction with different datasets. Another useful tool is the AlzGene database, which has organized and examined over 900 AD association research and about 400 genes. NIA has set up numerous systems to preserve genetic data that gather current data on genetic association research carried out on AD and AD traits. In hundreds of AD-associated studies with potential AD susceptibility genes, the AlzGene database arranges and classifies these results [56].
Even though amyloid precursor protein (APP) and its derivatives have been discussed and extensively studied over the previous 20 years, it is still unclear how their normal physiological functions and metabolites work. There is proof that APP is associated with gene regulation, cell-cell, and cell-substrate interaction, brain development, and synaptogenesis. It is found in many tissues and as a membrane protein in the brain [57, 58].
APP was the first gene susceptible to AD located on chromosome 21 and encoded for a single 770 amino acid transmembrane-spanning polypeptide [59, 60].
The APP gene is duplicated due to the triplication of chromosome 21,
which may improve APP expression and A
However, 21 and 3 mutations at Exons 17 and 16 suggest that APP is a very
uncommon risk factor for AD since most pathogenic APP mutations were found close
to the regions where the
A transmembrane domain with 17 residues and a 24-amino-acid-long cytoplasmic
tail of 24 amino acids follow the mature protein’s lumenal domain, which runs
from 46 to 460. The lumenal domain of BACE1 has two active site motifs, each
containing the remarkably preserved hallmark aspartic protease sequence. These
are located at amino acids 93 to 96 and 289 to 292. BACE1 is anticipated to
function as a type I transmembrane protein, with the location of its active site
positioned on the lumenal side of the membrane. -secretase enzyme cleaves APP at
the amino terminus of A
The complex of
To cleave C89 and shortened amyloid species,
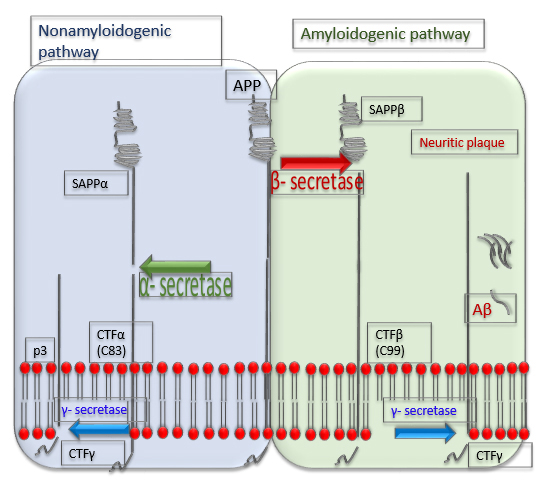 Fig. 3.
Fig. 3.Processing of a precursor protein for A
In the APP gene, there have been discovered to be about 63 coding
mutations associated with AD [69]. Most of them are risk factors for AD,
including the A673V and the Swedish mutation (KM670/671NL). They may either
encourage A
Recent research has identified an uncommon new mutation, APP S198P, found in the
ectodomain of the APP, acting as a helix-breaker rather than inside or next to
the A
What mechanisms does the APP S198P mutation use to affect APP metabolism? In the APP 695 ectodomain, involving two unique structural domains of about 160 amino acids and about 190 amino acids, referred to as the E1 and E2, respectively, serine 198 is found. This amino acid is part of a versatile and prolonged acidic-rich domain (Ala 191-Val 290) [73]. In contrast to the creation and aggregation of the wild-type serine at position 198 of full-length APPS we, from which these metabolites were generated. Researchers discovered that soluble, extracellular APPs and cellular APP C-terminal fragments (CTF) originating from full-length APP S198P precursors comprising the Swedish mutation (APPSwe-S198P) at earlier points and accumulated to elevated amounts.
Furthermore, researchers present immunoprecipitation results implying transitory
folding intermediates are generated more quickly in APPSwe-S198P-expressing cells
than in APPSwe-expressing cells. This significant discovery may prove that the
S198P variation encounters faster folding. The N-terminal E1 domain of APP has a
structural epitope that is sulfhydryl-dependent, and pulse-chase tests using the
P2-1 antibody have demonstrated that this domain does fold more quickly in cells
showing the APPSwe-S198P variation than in cells expressing APPSwe. This
discovery confirms the dependence of this structural epitope on sulfhydryl.
According to the researchers, the proline at position 198 accelerates the folding
process. It makes it easier for newly created synthetic APPSwe-S198P to migrate
from the endoplasmic reticulum to the Golgi complex late compartments, where the
enzymes BACE1 and
Compared to APPSwe, how can the folding pace of the APPSwe-S198P version be sped
up? Proline, an amino acid that either bends or breaks a helix as it lacks amide
hydrogen, is the deciding factor because of how much conformational space it
takes up due to its bulky pyrrolidine ring. Interestingly, a study of the Protein
Data Bank was used to determine the local conformations of each proline residue,
indicating that the residue before the proline is crucial for defining the
protein’s conformation. There is a high chance that the proline will adopt the
conformation when it follows an aspartate residue (
The formation of a local helix is facilitated by an Asp-Pro pair, which in turn accelerates the folding of the neighboring E1 and E2 domains and the adjacent adaptable domain. Pulse-chase experiments were carried out to support this assertion, utilizing the sulfhydryl-dependent, structural-epitope-specific antibody mAbP2-1. These investigations revealed that APPSwe-S198P binds preferentially over APPSwe [74].
The processing of APP may change as a result of mutations (Table 2, Ref. [74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123]) around the cleavage site of
| S. No | Mutation | Clinical | Pathogenicity | Biological effect | Genomic region | References |
| 1. | IVS |
AD, None | AD Benign | Unsure; deletion seems unaffected by APP splicing | Non-Cod | [75] |
| 2. | H733 |
None | AD Not Classified | Considerably increased A |
Cod Ex 17 | [76] |
| 3. | K724 |
AD | AD Not Classified | Cod Ex 17 | [77] | |
| 4. | L723 |
AD | AD Pathogenic | Rise of A |
Cod Ex 17 | [78] |
| 5. | M722 |
AD, None | AD Pathogenic | Cod Ex 17 | [79] | |
| 6. | T719 |
AD | AD Pathogenic | The T719N mutation resulted in |
Cod Ex 17 | [80] |
| 7. | T719 |
AD | AD Not Classified | Total A |
Cod Ex 17 | [81] |
| 8. | V717 |
AD | AD Pathogenic | A ratio of A |
Cod Ex 17 | [82] |
| 9. | V717 |
AD | AD Pathogenic | Increased A |
Cod Ex 17 | [83] |
| 10. | V717 |
AD | AD Pathogenic | Increased A |
Cod Ex 17 | [84] |
| 11. | V717 |
AD | AD Pathogenic | Cod Ex 17 | [85] | |
| 12. | I716 |
AD | AD Not Classified | Unidentified; in silico projected to be harmful (PHRED-scaled CADD score |
Cod | [86] |
| 13. | I716 |
AD | AD Not Classified | Unidentified, although in silico studies (PHRED-scaled CADD score |
Cod Ex 17 | [87] |
| 14. | I716 |
AD | AD Not Classified | Cod Ex 17 | [88] | |
| 15. | I716 |
AD | AD Pathogenic | Increased APP C-terminal fragments; reduced synthesis of APP intracellular domain; |
Cod Ex 17 | [76] |
| 16. | V715 |
AD | AD Pathogenic | Cod Ex 17 | [89] | |
| 17. | V715 |
AD | AD Pathogenic | Total A |
Cod Ex 17 | [90] |
| 18. | T714 |
AD | AD Pathogenic | Cod Ex 17 | [91] | |
| 19. | T714 |
AD | AD Pathogenic | Unknown, although in silico techniques suggested it would be harmful (PHRED-scaled CADD score |
Cod Ex 17 | [92] |
| 20. | A713 |
None | AD Benign | Cellular A |
Cod Ex 17 | [93] |
| 21. | A713 |
AD, None | AD Uncertain Significance | Cod Ex 17 | [94, 95] | |
| 22. | G708 |
None | AD Benign | Cod Ex 17 | [96] | |
| 23. | F690_V695 |
AD | AD Pathogenic | produces quickly aggregating A |
Cod Ex 17 | [97] |
| 24. | V695 |
AD | AD Not Classified | Unknown. Mixed findings were obtained using in silico techniques, but the integrative PHRED-scaled CADD score was more than 20, indicating a negative impact | Cod Ex 17 | [98] |
| 25. | E693 |
AD | AD Pathogenic | Unchanged A |
Cod Ex 17 | [99] |
| 26. | E693 |
AD | AD Pathogenic | Protofibrils formation by Arctic A |
Cod Ex 17 | [75, 100] |
| 27. | A692 |
AD | AD Pathogenic | Cod Ex 17 | [101] | |
| 28. | K687 |
AD | AD Not Classified | reduces cleavage of the APP by |
Cod Ex 16 | [102] |
| 29. | K687 |
AD | AD Likely Pathogenic | Unknown; however, PolyPhen-2 and SIFT suggested it would likely be harmful | Cod Ex 16 | [103, 104] |
| 30. | E682 |
AD | AD Not Classified | Cod Ex 16 | [105] | |
| 31. | D678 |
AD | AD Pathogenic | higher cytotoxicity and accelerated oligomerization kinetics compared to wild-type A |
Cod Ex 16 | [106] |
| 32. | D678 |
AD | AD Pathogenic | Cod | [107] | |
| 33. | H677 |
AD | AD Not Classified | Cod Ex 16 | [108] | |
| 34. | A673 |
AD | AD Not Classified | Incubation of mutant and wild-type A |
Cod Ex 16 | [109] |
| 35. | A673 |
None | AD Protective | reduced amyloidogenic A |
Cod Ex 16 | [110, 111] |
| 36. | KM670 |
AD | AD Pathogenic | Cod Ex 16 | [112] | |
| 37. | V669 |
AD | AD Not Classified | Unknown. Despite conflicting results, a comprehensive in silico prediction technique foresaw negative consequences (PHRED-scaled CADD score |
Cod Ex 16 | [113] |
| 38. | E665 |
None | AD Benign | A |
Cod Ex 16 | [114] |
| 39. | P620 |
AD | AD Uncertain Significance | Cod Ex 14 | [115] | |
| 40. | P620 |
AD | AD Not Classified | Cod Ex 14 | [116] | |
| 41. | S614 |
AD | AD Uncertain Significance | Cod Ex 14 | [117] | |
| 42. | V604 |
AD, | AD Uncertain Significance | Unknown. Estimated to have a negative effect in silico (PHRED-scaled CADD score |
Cod Ex 14 | [118] |
| 43. | T600 |
None | AD Benign | A |
Cod Ex 14 | [119] |
| 44. | E599 |
AD, PD | AD Benign, P.D.: Not Classified | In cells, A |
Cod Ex 14 | [120] |
| 45. | V |
None | AD Not Classified | A |
Cod Ex 13 | [120] |
| 46. | Y538 |
AD | AD Benign | Cod Ex 13 | [120] | |
| 47. | A500 |
None | AD Not Classified | In cells, A |
Cod Ex 12 | [119] |
| 48. | K496 |
AD | AD Not Classified | In cells, slight |
Cod Ex 12 | [115] |
| 49. | R486 |
AD | AD Not Classified | Unknown, however, in silico techniques estimate it will likely be harmful (PHRED-scaled CADD score |
Cod Ex 11 | [121] |
| 50. | A479 |
None | AD Benign | No discernible impact on the synthesis of A |
Cod Ex 11 | [122] |
| 51. | R468 |
None | AD Benign | Neither the synthesis of A |
Cod Ex 11 | [119] |
| 52. | E380 |
AD | AD Uncertain Significance | Unknown, but estimated by PolyPhen-2 to likely be harmful, harmful by SIFT, and neutral by PROVEAN. CADD score |
Cod Ex 9 | [123] |
| 53. | V340 |
AD | AD Uncertain Significance | Unknown, although its CADD score when scaled by PHRED was |
Cod Ex 7 | [117] |
| 54. | D332 |
AD | AD Not Classified | Unknown; however, PolyPhen-2 and SIFT suggest they will be harmful and tolerable | Cod Ex 7 | [116] |
| 55. | P299 |
AD | AD Not Classified | Unknown, however, in silico, it is considered harmful (PHRED-scaled CADD score |
Cod Ex 7 | [116] |
| 56. | T297 |
AD | AD Uncertain Significance | Unknown but expected to be unfavorable by SIFT and PolyPhen-2 (PHRED-scaled CADD score |
Cod Ex 7 | [103] |
| 57. | E296 |
AD | AD Not Classified | Unknown, although a PHRED-CADD score of |
Cod Ex 7 | [116] |
| 58. | E246 |
AD | AD Likely Benign | No considerable impact in cells on A |
Cod Ex 6 | [122] |
| 59. | D244 |
AD | AD Not Classified | Unknown, although PolyPhen-2 and SIFT suggested that it would likely be harmful. CADD score as measured by PHRED |
Cod Ex 6 | [103] |
| 60. | D243 |
AD | AD Benign | No effect on A |
Cod Ex 6 | [116] |
| 61. | A235 |
AD | AD Benign | Cod Ex 6 | [116] | |
| 62. | A201 |
None, PD | AD Benign, PDD: Not Classified | In cells, no effect on A |
Cod Ex 5 | [120] |
| 63. | S198 |
AD | AD Benign | In transgenic mice as an amyloidosis model and cultured cells, the S198P mutation boosted A |
Cod Ex 5 | [74] |
AD, Alzheimer’s Disease; P.D, Parkinson’s Disease; Cod, Coding; Ex, Exon; BACE,
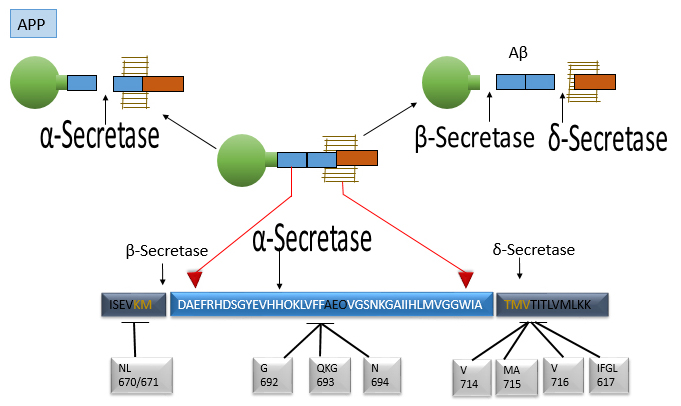 Fig. 4.
Fig. 4.
This figure displays the molecular features of the APP
gene and protein region where
Frontotemporal dementia, with or without supranuclear palsy, corticobasal syndrome, or parkinsonism, is associated with 40 mutations in the microtubule-associated tau (MAPT) gene, as reported by AlZFORUM [128]. The site of a mutation in a gene determines how it affects tau mRNA and protein as well as the disease that results. Exons 9 to 12 and exon 13 are where most coding mutations are found; however, Exon 1 mutations and intronic mutations in the intron after exon 10 that influence exon 10 splicing have also been described [129, 130].
Only two sequence changes in exon 7 of MAPT have been identified: Pro176 (silent point mutation) and Ala178Thr, although neither is harmful [131, 132]. Several types of tauopathies without mutations in the MAPT gene fall into one of several categories, such as Argyrophilic grain disease (AGD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), and neurofibrillary tangle dementia, Pick’s disease, and AD. There are also other so-called unclassifiable tauopathies. These categories are based on tau immunoreactive structures’ cellular and anatomical distribution [133, 134].
While certain MAPT gene mutations display characteristics of sporadic tauopathies, others have notable variances, such as a broader anatomical distribution and an elevated load of tau immunoreactivity. This study describes a patient with a progressive neuropsychiatric condition and a neuropathological tauopathy phenotype incompatible with sporadic disease entities. In addition, we report on the discovery of a MAPT gene variant in exon 7 [135]. Exons 2, 3, and 10 of the human genome are used for alternative splicing; this might generate up to six distinct tau isoform variations [136, 137].
The domain that binds to microtubules of tau interacts with microtubules through C terminal repeats, which explains why 4R tau isoforms are more likely than 3R tau isoforms to stimulate microtubule assembly [138]. In the mammalian brain, tau is a crucial regulator of axonal transport that is highly expressed in neurons and normally localizes largely to axons [139].
However, tau gene deletion had little effect on axonal transport, suggesting that Microtubule-associated proteins 1 (MAP1) and Microtubule-associated proteins 2 (MAP2) may make up for tau in other proteins involved in microtubule regulation or binding [140].
Although tau is involved in oligodendrocyte process outgrowth and myelination, it is still uncertain if tau regulates the physiological functions of astrocytes. Tau also plays two other physiological roles, regulating insulin signaling and iron export [141].
PSENs were discovered for the first time in 1995 while searching for
the genes causing beginning, autosomal dominant variants of familial AD (FAD)
[142, 143]. A few years after their identification, it was determined that PSENs
encode proteins that sustain the cleavage of APP by
In several tissues from mice and humans, including parts of the brain, PSEN1 and PSEN2 mRNA are conveyed equally and widely [147], with the cerebellum and hippocampus expressing at the highest levels. They can be seen in glial cells but are primarily expressed in neurons [148, 149]. PSEN-1 on chromosome-14 (14q24.3) and PSEN-2 on chromosome 1 (1q42.2). The membranes of the plasma membrane, the Golgi apparatus, the E.R, and the endosomes where PSEN-1 and PSEN-2 are 50-kDa polytopic transmembrane (PTM) proteins interacting with around 65 percent of their amino acid sequences [150].
PSENs have lately been concentrated at membranes connected to mitochondria (MAM)
[151, 152, 153]. The placenta and skeletal muscle have PSEN-2, while the brain, heart,
liver, pancreas, kidney, and placenta all include PSEN-2 isoform-2. Isoform 2
lacks amino acids 263–296 [148]. The hydrophilic, flexible N-terminus of PSENS
is organized in the cytoplasm, but the C-terminus of PSENS protrudes into the
extracellular space’s lumen. On PSENS, there are nine helical T.M. domains. PSENs
make up the catalytic core of the
Nicastrin, anterior pharynx deficient 1, P.S., and the heterotetrameric compound
with a high molecular weight (PSEN-1 or PSEN-2) make up this enzyme (PEN-2)
[155]. Voltage-dependent Na+channel subunit 2, APP, Notch, Delta1, E- and
N-cadherins, CD44, Nectina-1, and ErbB4, are also included [156]. The NTF and
CTF, formed by the endoproteolytic cleavage of the N- and C-terminal regions of
the PSEN-1 or PSEN-2 subunit, respectively, remain connected to one another in a
stable manner throughout
The immature holoprotein 7th hydrophobic domain undergoes an autocatalytic cleavage when P.S. is integrated into the enzyme complex, yielding about 20 kDa for the CTF and 30 kDa for the NTF (which is part of a long cytosolic loop). P.S. cleavage is autocatalytic; however, P.S. maturation is a saturable process and calls for additional molecular partners. Full length (F.L.), an excessive amount of which is an immature form of PSENS results in its proteasomal breakdown. Because of this deterioration, FL-PS has a much lower half-life than the mature form, 24 hours instead of 1.5 hours. FAD is caused by about 150 autosomal dominant mutations in PSEN-1 and 14 in PSEN-2 [158].
According to recent research, FAD-linked PSEN mutants are more active and
produce more A
Consequently, although this topic is still intensely debated, the initial
gain-of-function theory of loss of function has resulted from FAD-PSEN mutation.
The total amount of A
PSENs are well-known for their pleiotropic
Many families have more than one individual suffering from dementia, the majority of whom develop the disease around the age of 60 to 65. The disease lasts an average of 8–10 years; however, it can last for a long time, from 2 to 25 years. The APOE epsilon-4 (e4) allele is linked to LOFAD (Table 3, Ref. [53]). The age of onset appears to be pushed toward an earlier age by the APOE e4 allele through unknown mechanisms [162, 163].
| Locus-name | Gene-symbol | Chromosomal-locus | Protein-name | Test-availability |
| AD-2 | APOE | 19q13.2 | APOE | Clinical |
The major lipid transporter APOE, a protein with 299 amino acids, is a prominent
component of the brain. The astrocytes in the BBB are primarily responsible for
its synthesis [164]. APOE2, APOE3, and APOE4 are humans’ three main APOE
polymorphic alleles. Only two amino acids, cysteine/arginine polymorphisms at
locations 112 or 158, differentiate these proteins’ N-terminal domains from one
another. Furthermore, only 1% of instances of AD are EOFAD, the primary cause of
early-onset Alzheimer’s disease, which typically appears before the age of 65, is
an excess amount of A
The most prevalent type of AD is LOAD, which often develops in adults over 65. The primary risk factor for the pathophysiology of LOAD, which is frequently present in 15% of people, is APOE4 [167]. This study likewise found a small correlation between APOE4 and AD among Hispanics and African Americans but a larger correlation in Japanese individuals. The researchers found evidence of the APOE4 influence between the ages of 40 and 90. Still, it diminished after age 70, and the possibility of AD associated with a particular genotype varies according to gender [168]. Although APOE4 is not the primary cause of AD, only a small percentage of patients have at least one copy of the allele. In reality, only one-third of people living with AD have APOE4. It’s interesting to note that some homozygotes of APOE4 even never get AD. However, the chance of developing AD in individuals with two copies of APOE4 can be as high as 20 times. On the other hand, there is evidence that APOE2 plays a protective role in AD patients [169].
The likelihood of developing Alzheimer’s disease is greatly increased by having the APOE4 allele. However, individuals with APOE2/APOE2 and APOE2/APOE3, despite having a reduced odds ratio, are still less likely to develop AD compared to those with APOE2/APOE4, APOE3/APOE4, and APOE4/APOE4 [170]. A study, however, discovered that a variety of particular risk factors, such as high blood total cholesterol, any combination of the APOE alleles, and midlife hypertension, may collectively double the chance of developing AD in the future [171].
First, APOE is present in neuritic plaques, and AD patients with APOE4 have
higher amounts of A
Second, research has indicated that APOE3 and APOE4 attach to A
Thirdly, APP processing, A
A
Recent studies show that increasing astrocytic APOE4 expression during the
amyloid seeding stage increases amyloid deposition [179]. A
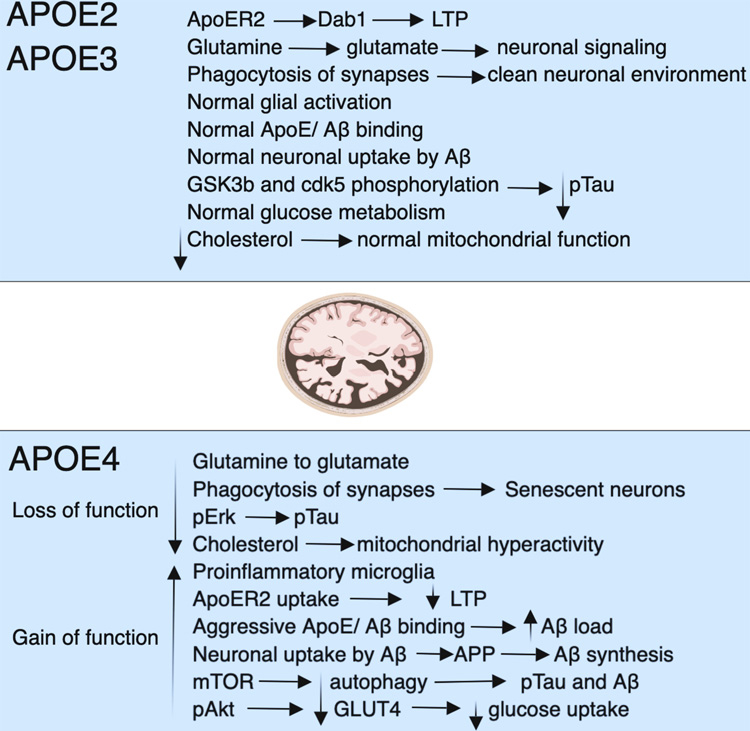 Fig. 5.
Fig. 5.A summary of how APOE4 affects the mechanistic pathways. The APOE gene has three isoforms: APOE2, 3, and 4. Neurodegeneration is more likely to occur along APOE4 routes, while APOE2 and APOE3 offer protection. Function declines and increases, especially in APOE4 carriers. Loss of phagocytosis, a decline in glucose absorption, autophagy, long-term potentiation (LTP), a rise in phosphorylated tau, proinflammatory microglia, excessive mitochondrial activity, and intense amyloid binding causes plaques are some of the symptoms. Reductions in LTP, autophagy, aggressive amyloid binding that culminates in plaques, phosphorylated tau, proinflammatory microglia, excessive mitochondrial activity, and mitochondrial respiration. The glucose transporter protein type-4 (GLUT4), mammalian target of rapamycin (mTOR), pAkt, pERK, and pTau are acronyms for specific proteins (phosphorylated tau), and all stand for “phosphorylated tau” [182].
The significance of APOE4 in A
The creation of neurofibrillary tangles (NFTs), which are a hallmark of AD and other neurodegenerative illnesses, is caused by abnormal tau protein phosphorylation. Excessive tau phosphorylation is thought to interfere with regular neuronal function and cause neuronal death [175].
According to in vitro research, different APOE isoforms may affect tau pathology differently. In the presence of sodium dodecyl-sulfate (SDS), APOE3 but not APOE4 forms a stable complex with tau. Tau’s association with APOE3 is inhibited by phosphorylation, proving that APOE3 only interacts with non-phosphorylated tau. This implies that APOE3 may inhibit aberrant tau phosphorylation and maintain the cytoskeleton of neurons [187]. Increased tau phosphorylation has been shown in studies using transgenic mice that express human APOE4 but not in astrocytes. This suggests that APOE4 has a neuron-specific impact on tau phosphorylation. Additionally, in APOE mutant mice fed a high-cholesterol diet, hyperphosphorylated tau accumulated intraneuronally. This finding suggests a connection between dietary cholesterol and APOE function in promoting tau disease [188].
The amino-terminal domain of APOE3 is assumed responsible for the protein’s interaction with tau. Furthermore, research has shown that carboxyl-terminally shortened APOE promotes tau phosphorylation and the development of intracellular NFT-like inclusions. It is still unclear how APOE gains access to the cytoskeleton of neurons. The regulation of tau kinases and phosphatases is another potential method by which APOE isoforms may contribute to tau hyperphosphorylation. To completely comprehend the molecular processes underlying the connections between APOE isoforms and tau pathology in AD, it is significant to stress that more investigation is required [189]
The main function of APOE is to redistribute lipids and keep the body’s cholesterol levels in check. APOE4 is less effective than APOE2 and APOE3 at facilitating cholesterol uptake in laboratory studies using cultured neurons. APOE4 is less effective at encouraging cholesterol clearance from astrocytes and neurons. The structural changes between various APOE isoforms may cause these functional variations [190].
There is a decline in brain cholesterol levels in AD, and mounting research indicates that cholesterol may have a role in the onset of AD. Clinical and epidemiological studies show that those with high plasma cholesterol levels are more likely to develop AD [191]. Statins, which stop the production of cholesterol, have also been demonstrated in numerous studies to slow the onset and progression of AD. Other genes related to the transport and metabolism of cholesterol have also been linked to AD risk and APOE [192].
An increased risk of developing AD has been linked to genetic variations in
cholesterol uptake receptors like the low-density lipoprotein receptor-related
protein (LRP) and very-low-density lipoprotein (VLDL) receptor as well as enzymes
that break down cholesterol like cytochrome-46 (Cyp46) [193]. Additionally, the
data point to cholesterol as a regulator of amyloid-beta (A
The location of APP and the location of the enzymes that cleave it can be
affected by the amount or distribution of cholesterol in the membrane.
A
The nervous system’s growth, regeneration, and synaptic plasticity depend on interactions between neurons and glial cells. A crucial part of these processes is the redistribution of lipids that APOE promotes. The limbic system and neocortex both experience severe neuronal loss in AD, and the synaptic disruption that results impairs the survival of neurons. It has been suggested that various APOE isoform variations contribute to synaptic plasticity and repair processes to variable degrees. Mice deficient in APOE as well as those that express APOE4 but not APOE3, have been shown to have impaired synaptic plasticity [197]
These variations in APOE isoforms have also been observed in humans, where those with the APOE4 variant demonstrate less regenerative change in the same brain regions than those without and who show inadequate compensation for the neuronal loss. These results imply that APOE4 performs less functionally in synaptic regeneration than other APOE isoforms. This deficit probably also has an impact on synaptic activity. Studies have shown that mice expressing APOE4 exhibit less long-term potentiation (LTP), a process linked to synaptic strength and plasticity, compared to animals of the wild-type and mice expressing APOE3 [198].
AD is well known for affecting cholinergic signal transduction. Comparatively to non-carriers, those with the APOE4 variation and AD show more pronounced impairments in cholinergic activity in the cortex and hippocampus [199]. Additionally, they have fewer cholinergic neurons overall, and markers of cholinergic function, such as nicotinic acetylcholine receptor binding and choline acetyltransferase activity, are reduced. However, the amounts of muscarinic receptors in AD patients with various APOE genotypes do not differ noticeably [200].
Studies conducted in vitro have shown that different APOE isoforms
affect the signaling cascade triggered by muscarinic acetylcholine receptors
differently. In response to carbachol stimulation, APOE4 affects phosphoinositide
hydrolysis, whereas APOE3 does not. Additionally, APOE3,
but not APOE4, has demonstrated the capacity to
fend off disruption brought on by the amyloid-beta protein fragment A
These findings imply that APOE4 directly impairs cholinergic signaling, which may explain why cholinergic replacement therapy is less effective in APOE4-associated AD patients. AD is well known for affecting cholinergic signal transduction. Comparatively to non-carriers, those with the APOE4 variation and AD show more pronounced impairments in cholinergic activity in the cortex and hippocampus [202]. Additionally, they have fewer cholinergic neurons overall, and markers of cholinergic function, such as nicotinic acetylcholine receptor binding and choline acetyltransferase activity, are reduced. However, the amounts of muscarinic receptors in AD patients with various APOE genotypes do not differ noticeably [203].
Studies conducted in vitro have shown that different APOE isoforms
affect the signaling cascade triggered by muscarinic acetylcholine receptors
differently. In response to carbachol stimulation, APOE4 affects phosphoinositide
hydrolysis, whereas APOE3 does not [204]. Additionally, APOE3, but not APOE4, has
demonstrated the capacity to fend off disruption brought on by the amyloid-beta
protein fragment A
The research showed synaptic and cholinergic impairments in mice carrying human APP/APOE4 before plaques developed. The synaptic and cholinergic deficits in aged mice harboring human APP/APOE4 and human APP/APOE3 were strikingly similar, despite differences in plaque load. These findings imply that APOE4 directly impairs cholinergic signaling, which may explain why cholinergic replacement therapy is less effective in APOE4-associated AD patients.
Numerous signaling pathways, some of which are pertinent to AD, have been demonstrated to be modulated by APOE. APOE has shown isoform-specific impacts on various signaling cascades in vitro studies. Different APOE isoforms activate calcium channels in different ways, which causes varying degrees of intracellular calcium rise [200]. APOE also has an isoform-specific impact on the actions of numerous proteins involved in signaling pathways, including PKC, GSK-3, pAkt, ERK, JNK, and CREB. Recent microarray studies employing hippocampus samples from AD patients have found significant variations in gene expression patterns between APOE4 carriers and non-carriers. Individuals with the APOE4 gene tend to express tumor suppressors and negative regulators of cell development more frequently, which could promote APOptosis. On the other hand, they have decreased expression of genes linked to mitochondrial energy metabolism, neuronal outgrowth, synaptic plasticity, and neurotransmitter receptors [206].
A person with APOE4 may develop resistance to several pharmacological treatments
because of changes in neurotransmitter receptors and downstream signaling
pathways. Additionally, it appears that APOE4 carriers experience both a loss of
function in cell defense mechanisms and a gain in function in pathways leading to
the formation of A
By toxically impacting neurons, APOE may also directly contribute to neurodegenerative processes. Vitro studies have demonstrated that APOEderived fragments and lipid-free APOE, notably APOE4, are hazardous to neurons [208]. However, this toxicity is prevented when APOEcontaining lipoproteins, which are more physiologically relevant, are utilized instead of delipidated APOE. APOEcontaining lipoproteins have shown that they can, in an isoform-specific manner, enhance cell survival and neurite outgrowth (APOE3 being more advantageous than APOE4) [209].
APOpE3/E3 mice are more protected against age-induced neurodegeneration than APOE4/E4 animals, according to in vivo studies employing transgenic mouse models expressing human APOE3, APOE4, or both. This shows that APOE4 acts as a major negative component, interfering with APOE3’s positive function and offering less neuroprotection than APOE3 [210].
The neurotoxic effects of APOE have been attributed to its numerous proteolytic fragments, with studies demonstrating that both the N- and C-terminal fragments cause neurodegeneration and impair neuronal function. Importantly, APOE C-terminal fragment levels were greater in AD patients’ brains than controls, especially in conjunction with neurofibrillary tangles. The neurotoxicity and aggregation of APOE may be influenced by the susceptibility to cleavage and the existence of lipid-bound or lipid-free APOE [188].
According to recent studies, APOE aggregates into soluble fibrils in vitro. The aggregation rate varies amongst isoforms, with APOE4 having the highest propensity. It has been discovered that these APOE fibrils are more hazardous to cultured neuronal cells than APOE tetramers. Nevertheless, further research is still needed to determine whether APOE fibrils and isoform-specific differential fibrillation exist in the AD brain. Gaining knowledge of the role of APOE in neurodegenerative processes requires understanding both the elements that lead to APOE neurotoxicity (such as lipid association, proteolytic cleavage, and fibrillation) as well as its neuroprotective properties [204].
It can activate several routes for downstream kinase signaling, resulting in dephosphorylation in Thr231, Ser235, and Ser396. Although the link between APOE and phosphorylation has been proven, more research into the isoform-specific effects of APOE on protein is still needed. Because APOE can influence regulation and modulate AD pathophysiology positively and negatively, Future studies should concentrate on how each APOE isoform regulates activity, causing microtubular and cytoskeletal instability that aids in disease development [211].
Nanotechnology studies and manipulates matter with dimensions ranging from one to one hundred nanometers. Materials create unique phenomena at these dimensions, allowing for novel uses that have only recently been investigated in research and medicine [212]. Nanotechnology is not new; this discovery was the impetus for developing nanomedicine. It is a relatively new field focusing on all human biological systems and their detection, control, creation, repair, defense, and improvements [212]. Nanotechnology is not new; evidence reveals that Egyptians, Greeks, and Romans used nanocrystal-contain hair colors [213].
Nanotechnology is a comprehensive science that involves manipulating atoms, electrons, protons, and neutrons in various ways to provide new insights into how materials might be developed to tackle numerous problems in numerous fields [31]. Nanotechnology has emerged as one of the most promising science-related technologies. Nanotechnology involves studying materials at the nanoscale and developing system architectures, devices, and materials with distinct properties and functions that arise from their nanoscale dimensions and characteristics [212]. Nanotechnology-produced metal nanoparticles (NPs) have gotten much attention because of their wide range of uses in the medicinal and agricultural industries [214]. The application of microorganisms and plants in synthesizing metal nanoparticles has gained substantial attention as a sustainable and efficient method of utilizing these organisms as practical nano producers [215].
Nanotechnology focuses on producing nanomaterials with a range of forms (from 1 to 100 nm) and their applications in numerous industries [214]. A DNA double helix has a diameter of roughly 2 nm, and the length of a C-C bond is approximately 0.12 nm [214]. Pharmaceutical interests were already shifting due to the potential usage of multiple biological sources for nanoparticle manufacturing and application. Silver is the most preferred metal for producing NPs because it has specific antibacterial, antifungal, anti-inflammatory, anti-angiogenic, and anti-permeability properties, among others [33].
Nano-medicine [216] applies nanotechnology in medicine, focusing on nano systems for treatments, diagnostics, and imaging [212]. Synthesizing nanoscale particles to sizes compatible with biological molecules such as proteins and nucleic acids can be utilized as potential probes, delivery platforms, carriers, and devices. This presents new opportunities for disease detection, therapy, and prevention [217]. Moreover, NPs can potentially improve the pharmacokinetic and pharmacodynamics profiles of well-studied treatments, making them appealing carriers for classical anti-cancer drugs [218]. Nano medicine has recently led to the creation of nanoparticle carriers for drug/gene delivery [219], imaging [220], and theranostics (diagnostics and therapies) [217].
Liposomes, specifically their phospholipid bilayer, are a potential solution for transporting medication across the BBB, but the BBB is not easily penetrable. Therefore, numerous surface modifications have been made to enhance the transport of liposomal carriers across the BBB [221]. The BBB surface contains proteins, peptides, antibodies, and other ligand receptors, which can aid in transcytosis. Ligands present in these compounds facilitate the movement of cationic liposomes through the BBB. To enhance their movement throughout the body, glucose and other nutrients are incorporated into the liposomes [222]. The passive diffusion mechanism is started by the brain’s passive efflux when the liposomes reach the brain [222]. The substance releases at the same rate as before. New tactics have been developed that adapt to changes in the patient’s physiological parameters and control the drug’s release. pH changes, changes in enzyme activity, or changes in glutathione levels can all cause liposome drug release [223]. Acetylcholine (ACh) is bound by amyloid peptides, causing the dissolution of amyloid plaque and reduction of inflammation in the brain. This supports the preservation of healthy neurons and serves as a treatment option for AD once it has developed. To facilitate active internalization, liposomes can be modified with ligands due to the negative charge of the BBB and the occurrence of electrostatic forces [224].
To improve the pharmacokinetic profile of liposomes, PEG11 or polysaccharides can be added to the particles’ surface, extending their circulation time and improving their distribution into the brain by inhibiting rapid clearance via the reticuloendothelial system (RES). However, there is no guarantee that liposomes will cross the BBB, even with “stealth” liposomes that can significantly reduce circulation time. Several emerging strategies to enhance the stealth liposomes include incorporating peptides, antibodies, or small molecules that specifically bind to receptors or transporters overexpressed on brain endothelial cells. Among the various methods of delivering liposomes to the brain, receptor-mediated transcytosis is the most efficient and commonly used approach due to the precise interactions between receptors and ligands [225].
The transmembrane glycoprotein TfR13 is a frequently targeted receptor due to its high concentration in brain endothelial cells. However, the binding of endogenous Tf inhibits the binding of TfR-targeting ligands to the receptor. To overcome this, experiments involving Tf14-functionalized liposomes have utilized antibodies that bind to unique sites on the TfR receptors to reduce ligand antagonism and enhance the effectiveness of the liposomes in crossing the BBB [226, 227]. A different approach, mammalian iron-binding glycoprotein lactoferrin, has also functionalized liposomes. Lactoferrin interacts with Lf15 receptors and is widely dispersed throughout the BBB. Using receptor-mediated transcytosis, lactoferrin-modified liposomes were developed as modified nanocarriers for BBB crossing [228].
To determine whether combining the two BBB-piercing peptides may have a synergistic effect and whether curcumin interfered with the activity of the BBB-targeting ligands, the previous research was coupled with curcumin and two BBB-invading peptides [229].
The surface of multifunctional liposomes is modified by introducing two
BBB-binding ligands and a curcumin derivative, which targets transferrin and LDL
receptors. Researchers studied the inhibitory effects of these surface-modified
liposomes on A
Gold nanoparticles (GNPs) have several advantages over other metal NPs, including chemical inertness, biocompatibility, and lack of cytotoxicity. Humans have been using gold internally for 50 years [232]. The nano size, surface area, and photothermic nature directly impact the antibacterial activity of GNPs [233]. Another accepted theory is that GNPs connect intracellularly to the Sulphur base found in cells as the thiol group in enzymes, causing an abrupt disruption of the respiratory chain by forming many free radicals in death. GNPs, on the other hand, lower Adenosine tri-phosphate (ATP) activities by reducing t-RNA and ribosome interaction [234]. GNPs also decrease bacterial growth by blocking transmembrane hydrogen efflux; yet, at lower concentrations, GNPs can reduce bacterial growth by 250-fold. Because GNPs are smaller than bacterial cells, they attach to pathogen cell walls and cause cell death by delaying cell processes [234].
GNPs apply to several industries, including electronics, energy, optical, and health care. GNPs are studied for multiple purposes, like ultra-sensitive biomarkers [235], targeted heat therapy, and drug delivery vectors [236]. Gold has a higher radio density compared to contrast agents already in use. GNPs have exhibited more potent contrast qualities than those now used as a contrast agent for CT scanning. GNPs can also be used as a drug delivery vector for various reasons. For example, the same GNPs can be linked to several functional groups, enabling customized drug delivery [234]. Nanoscale particles are particularly well suited to this purpose due to their high surface area to volume ratio; additionally, light absorption by the plasmon of GNPs can be exploited to induce the heat-sensitive release of medicinal chemicals. The plasmonics of GNPs also enables localized heat therapy through the absorption of specific wavelengths of light [237]. The absorbed energy is then diffused onto the surrounding region. Combining this method of cancer tumor attack with the production of heat-sensitive anti-carcinogens is wildly successful [238].
GNPs have been investigated to boost the efficiency of solar panels in energy technology, and their utility is also derived from their plasmonic characteristics. Specific wavelengths of light are required for solar panels to function. Nevertheless, as the plasmon of NPs can be altered through regulated synthesis, Developing NPs that can both absorb light at frequencies that the solar panel cannot use and release light at those frequencies is possible [238]. Other metallic NPs have been studied using similar methods. Another notable application of GNPs is wiring in nano-circuitry [238]. Since the formulation of Moore’s law in 1965, there has been a lot of interest in making circuits smaller and smaller, eventually leading to courses on the nano-scale [239].
The administration of immunotherapy doses to target amyloid plaques for
Alzheimer’s disease treatment results in significant adverse outcomes, including
meningoencephalitis [240]. Nanoparticles coated with antibodies that target
specific proteins can detect and dissolve protein clumps in brain cells,
potentially reducing the harmful side effects of immunotherapy for Alzheimer’s
disease. One technique to accomplish this is using metal oxide nanoparticles
coated with antibodies that bind to AD-associated proteins, which can be detected
using secondary ion mass spectrometry [241]. Polyethylene glycol (PEG)
nanoparticles coated with specific antibodies can degrade A
Iron oxide nanoparticles (NPs) are commonly used in biomedical research, and
there is ongoing research to explore the possibility of creating new theranostic
drugs for Alzheimer’s disease using ultra-small superparamagnetic iron oxide
nanoparticles and a phenothiazine-based near-infrared (NIR) fluorescent dye.
These particles have the potential to offer NIR fluorescence and magnetic
resonance imaging of A
A
| S. No | Nanoparticles | Combinations | Effects | References |
| 1. | Liposomes | Curcumin + liposomes (Liposomes inner |
curcumin-loaded liposomes |
[221, 229] |
| 2. | GNPs | A |
[248] | |
| 3. | Antibody Decorated NPs | W20/XD4-SPIONs are composed of a superparamagnetic iron oxide nanoparticle, a class A |
W20/XD4-SPIONs preserved the anti-A |
[249] |
| 4. | Iron Oxide Nanoparticles (IONPs) | Anti- A |
used for imaging-based detection of amyloidogenic plaque or fibril depositions on the surface of cells | [250] |
| 5. | Quantum Dots | Tramiprosate (aminosulfonate compound) linked covalently with GQDs | shown synergistic inhibition of A |
[251, 252] |
*NPs, Nanoparticles; GNPs, gold nanoparticles; GQDs, graphene quantum dots.
Tramiprosate covalently coupled with graphene quantum dots (QDs) decreased
A
The distinct characteristics of QDs make them an attractive alternative to
traditional imaging techniques and dyes. QDs can detect A
In gene therapy, which was initially proposed in the 1960s, the barrier of gene delivery is overcome due to NPs. These challenges include cell-specific targeting, protecting genetic cargo from deterioration, preventing reticuloendothelial system (RES) clearance, and lysosomal and endosomal escape [256].
Inorganic NPs can be engineered to enhance gene delivery effectiveness. An ideal gene delivery method should possess the following characteristics: the capability to enter the cell’s plasma membrane, disrupt the endosomal membrane, bind and condense genetic material, safeguard the nucleic acid cargo, ensure specific delivery to the intended target, remain stable in the bloodstream and evade immune system detection [256, 257]
Extensive research into the underlying disease mechanisms of neurodegenerative disorders has identified specific genetic abnormalities that play a crucial role in disease onset. Gene therapy offers a means to deliver various types of genetic material, including messenger RNA (mRNA), small interfering RNA (siRNA), guide RNA (gRNA), and microRNA (miRNA). Scientific investigations have shown that RNA interference (RNAi), a technique that reduces the production of specific mRNA molecules using siRNA and miRNA, is highly effective for silencing genes involved in disease progression [258].
Synthetic double-stranded siRNAs, approximately 21–25 nucleotides long, are highly precise for targeting specific mRNA sequences and silencing genes within human cells [259]. The advent of RNAi has revolutionized the field of therapeutics, providing a new pathway for treating a wide range of diseases, including cancer and neurological disorders [260]. However, to practically implement siRNA-mediated gene silencing in medicine, it is crucial to have an efficient and safe delivery method, preferably a nanocarrier, to transport the siRNA to the desired site effectively. Recent advancements in gene therapy, such as genome editing, hold significant promise in specifically targeting abnormal genetic modifications in diseased areas [261].
The accumulation of misfolded proteins, such as amyloid-beta oligomers and alpha-synuclein, is a key factor in various diseases. These proteins lead to endoplasmic reticulum (ER) stress and trigger ER-associated degradation, making them potential targets for gene therapy [262]. When these proteins aggregate within the ER lumen, they disrupt ER calcium homeostasis and disturb the unfolded protein response (UPR) signaling. As a result, neurons undergo apoptotic cell death through pro-apoptotic reactions [263, 264]. In the context of diseases like Parkinson’s disease (PD), it has been possible to improve protein folding and preserve dopaminergic neurons’ survival and motor function through gene therapy. Specifically, gene therapy has been employed to suppress the overexpression of the BiP gene, also known as glucose-regulated protein 78, which is associated with a reduction in the unfolded protein response. In such cases, gene silencing techniques can effectively address the pathology [265].
In AD, PD, and Huntington’s disease (HD), aberrant signaling of the rapamycin (mTOR) pathway has been noted in addition to the deregulation of epigenetics, autophagy, and dysfunctional microglia and astrocytes. As the situation worsens, specific types of dysfunction are displayed by each disease mechanism. Therefore, to provide the best care, it is essential to pinpoint the precise mechanism underlying how a patient present with these disorders. Gene therapy is a viable strategy for treating neurodegenerative diseases because it has effectively treated various problems. This is especially noteworthy given the findings on genetic anomalies in PD and AD patients [266].
Between 2017 and 2020, numerous nanoparticles were used in central nervous system (CNS) gene therapy, as shown in Table 5 (Ref. [267, 268, 269, 270, 271]). Our knowledge of targeting particular genes that are defective or aggregated proteins linked to neurodegenerative illnesses has greatly benefited from these investigations. Approaches for safe and efficient delivery using nanoparticle-based gene therapy are particularly promising in overcoming biological barriers like the BBB. The natural synthesis of NPs offers several advantages besides gene therapy, mainly when using certain extracts that can work together with therapeutic genes [267].
| NPs | Disease | Therapeutic | Therapeutic effect | Cell entry | References |
| Gold | PD | Nerve growth factor (NGF) and exogenous interfering RNA (RNAi) were integrated into the pDNA | substantia nigra striatum dopaminergic neuronal death and PC12 cell inhibition | Endocytosis of nerve growth factor (NGF) | [268] |
| Gold | AD | L- and D-glutathione at 3.3 nm | preventing A |
Crossing of the BBB via chiral nanoparticle endocytosis | [267] |
| Silver | PD | Citrate cap | Increased production of H |
Silver’s organic characteristics enter the brain | [269] |
| Silver | AD | Plant extract- from Lampranthus coccineus - and Malephora lutea - F. Aizoaceae | As a plant-based anti-Alzheimer medication, anticholinesterase and antioxidant action | Silver’s organic characteristics enter the brain | [270] |
| Selenium | AD | Curcumin–loaded nanospheres | Amyloid- plaques in AD lesions are less prevalent | Curcumin forms intermolecular hydrogen bonds with A |
[271] |
pRNA, packaging RNA; BBB, blood-brain barrier.
Applications for diagnosing and treating AD using nanotechnology are promising. Targeting particular AD-related biomarkers with functionalized nanoparticles enables non-invasive imaging approaches for early detection and diagnosis [272]. By encapsulating therapeutic chemicals and boosting their stability, bioavailability, and targeted distribution to the brain, nanoparticles can help improve drug delivery and targeted therapy [273, 274]. Additionally, functionalized nanoparticles can support neuroprotection and regeneration by providing neurotrophic factors and reducing neuroinflammation and oxidative stress [275].
However, there are challenges to overcome in translating nanotechnology into AD.
Biocompatibility and safety: To assure the long-term safety of nanomaterials, thorough studies are needed to evaluate their possible toxicity and clearance pathways [276].
Blood Brain Barrier: Effective distribution of nanoparticles across the BBB is still difficult. To increase BBB penetration, strategies like surface alterations, functionalization with targeted ligands, and focused ultrasonic techniques are being investigated [277].
Scalability and manufacture: For clinical translation, scalable and repeatable synthesis and manufacturing processes for nanoparticles are required [278].
Various foods, such as fruits, whole grains, nuts, and vegetables, contain phytochemicals. These phytochemicals offer tremendous therapeutic promise for treating a variety of ailments, either individually or in combination [34]. Phytochemicals with nutraceutical properties contained in food are crucial due to their favorable effects on human health. Cancers, coronary heart disease, diabetes, high blood pressure, inflammation, microbial, viral, and parasitic infections, depression, anxiety, erratic circumstances, ulcers, osteoporosis, related disorders, and other diseases are all protected by them [279].
Curcumin, found in the rhizome of turmeric (Curcuma longa), is used in Indian
cuisine to add flavor and preserve food [280] (Fig. 6). An intriguing observation
is that the incidence of AD among individuals aged 70–79 in India is four times
lower than that in the United States, implying that a diet high in curcumin among
older Indians could be accountable for the reduced risk of AD [281]. The
anti-amyloidogenic, anti-inflammatory, and anti-oxidative properties of curcumin
have been backed by robust in vitro and in vivo studies, which
suggest that it can potentially prevent Alzheimer’s disease [35]. Curcumin and
its derivative rosmarinic acid exhibit anti-amyloidogenic properties in
vitro, inhibiting the formation of neurotoxic A
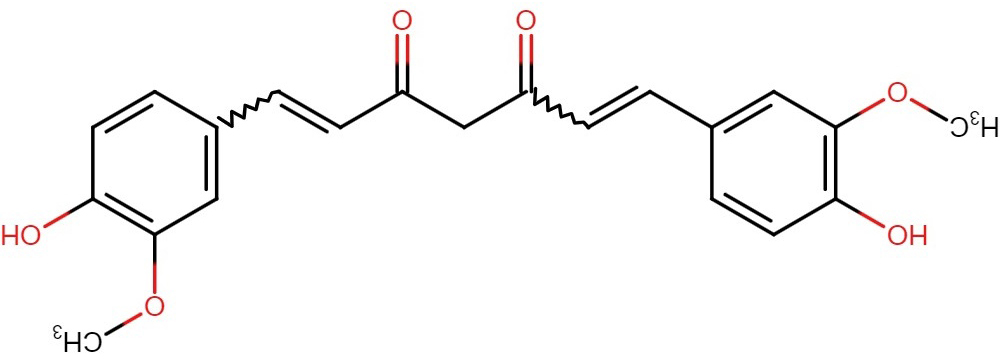 Fig. 6.
Fig. 6.Structure of curcumin.
Curcumin, a substance that can target amyloid pathology, has drawn much
attention for its potential as a treatment for AD. Its use in diagnostics using
imaging techniques is one area of focus. When human neuroblastoma SK-N-SH cells
were subjected to H
Athymic mice fed with the NanoCurcumin formulation showed decreased levels of
H
In male Lacca mice, an oral curcumin lipid nanoformulation has demonstrated
encouraging results in correcting the adverse effects of aluminum (AlCl
The compound
N-[2-(3,4-methoxyphenyl)ethyl]-N-[2-(3,4-methoxyphenyl)ethyl]-3-phenylacrylamide,
also known as GX-50, has been found in Sichuan pepper (Zanthoxylum bungeanum) and
has been recognized as a potential therapeutic agent for AD [289]. Furthermore,
in vitro studies showed that GX-50 can disintegrate A
The possibility of treatment approaches that target this receptor for treating
the condition is highlighted by the function of TLR4 in the onset of AD. Numerous
studies have shown that blocking TLR4 can halt the course of AD. The clearance of
A
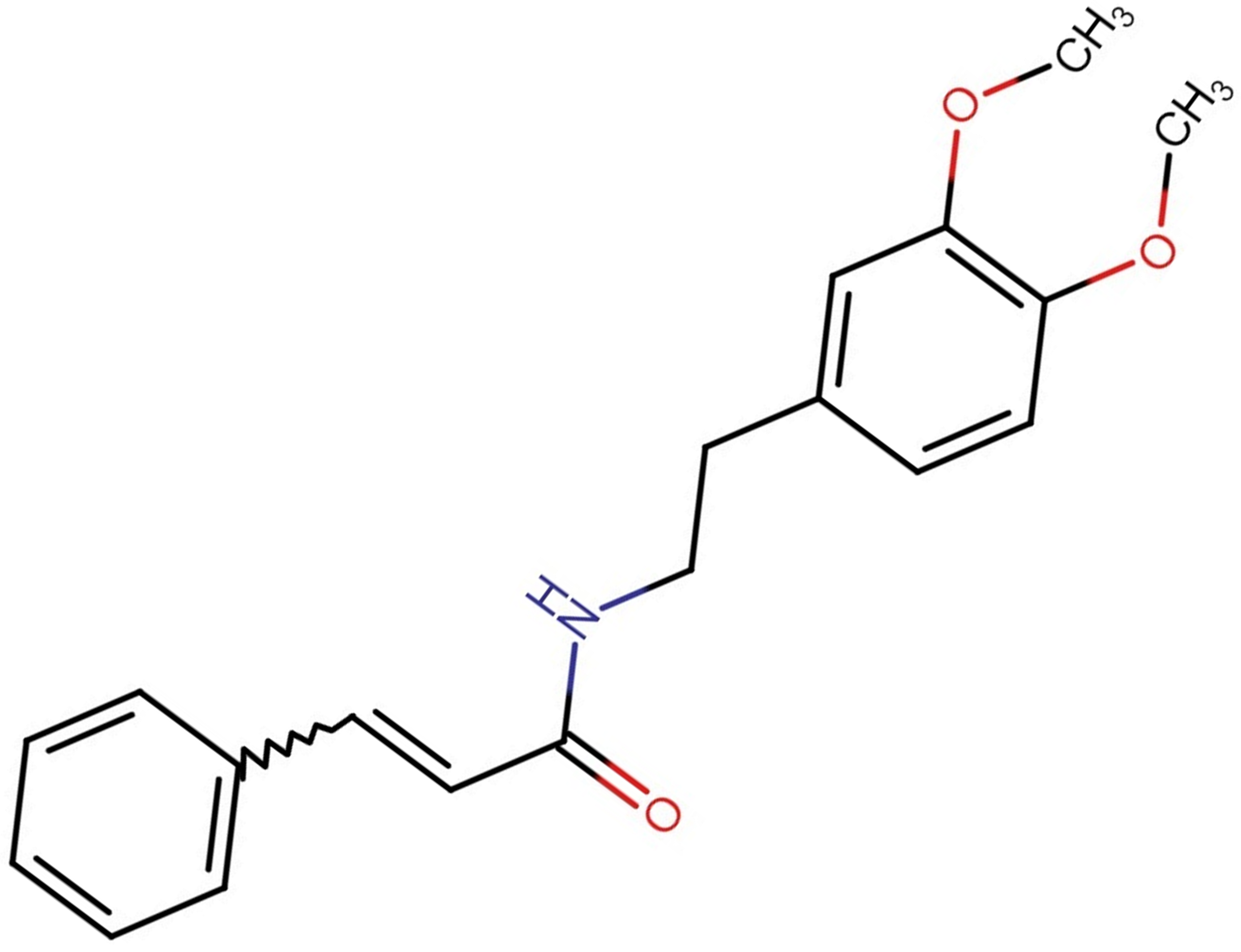 Fig. 7.
Fig. 7.Structure of GX-50 molecule.
The possibility of GX-50 inhibiting A
Resveratrol, a polyphenolic phytoalexin found in berries, grapes, and red wine,
has been associated with several biological and pharmacological effects [280].
Numerous epidemiological studies have reported an inverse association between
wine consumption and the development of AD, indicating that resveratrol may
contribute to wine’s beneficial effects in treating Alzheimer’s patients.
Resveratrol as shown in (Fig. 8), like curcumin, can readily penetrate the intact
BBB and enter the brain tissue [300]. Recent research has demonstrated that
resveratrol possesses considerable neuroprotective properties in vivo
and in vitro experiments. It has been found to hinder neuronal cell
death and reduce brain damage caused by ischemia/hypoxia, trauma, and
excitotoxicity [301, 302]. In various cell lines expressing the Swedish mutant
APP695, resveratrol has been demonstrated to have anti-amyloidogenic properties
by decreasing the secretion or intracellular accumulation of A
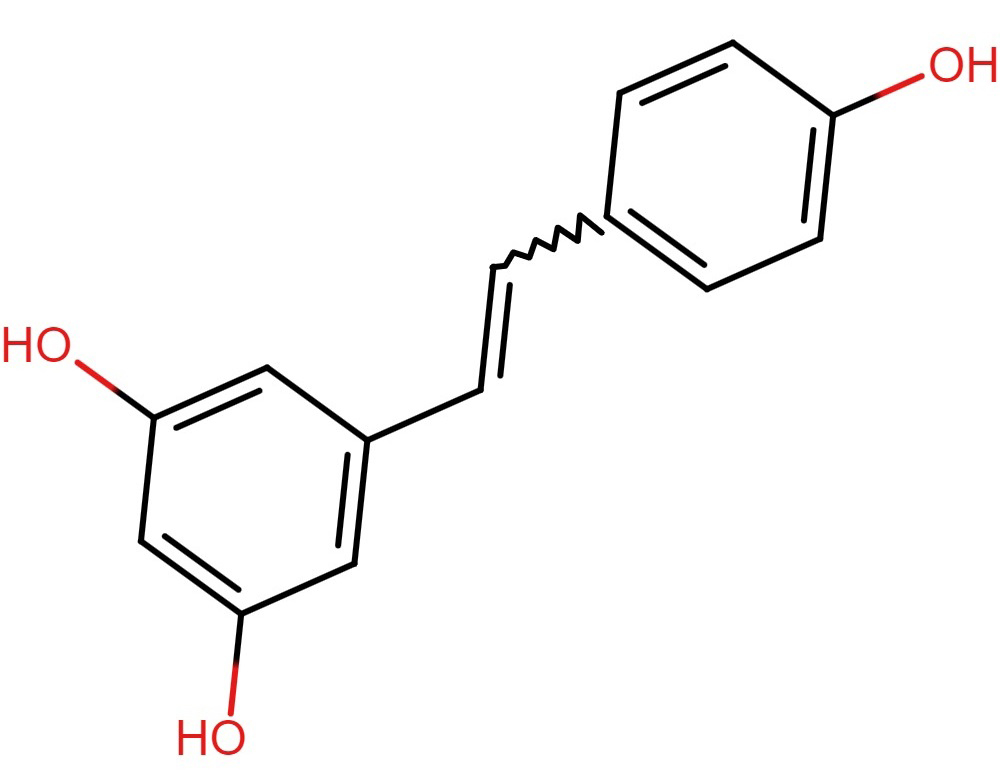 Fig. 8.
Fig. 8.Structure of resveratrol.
Resveratrol-Selenium Nanoparticles (RSV-SeNPs) demonstrated a potent anti-inflammatory and antioxidant impact against neurotoxicity. Additionally, because RSV-SeNPs have an anti-amyloidogenic capability, they improve the clearance of misfolded proteins. RSV-SeNPs affect several signaling pathways implicated in AD development, alleviate cholinergic deficiencies, and enhance neuropathology and neurocognitive functions. RSV-SeNPs may be utilized to treat AD in general. However, more research is needed to pinpoint the precise underlying pathways [304].
Acetylcholine (ACh) is well known for playing a critical part in memory and
learning processes. A negative feedback loop that controls A
Based on this knowledge, cholinesterase inhibitors are efficient AD treatments, supporting Davies and Maloney’s (1976) original concept on the role of cholinergic deficiencies in AD pathology. Tacrine, Donepezil, Rivastigmine, Galantamine, Xanthostigmine, Para-aminobenzoic acid, Coumarin, Flavonoid, and Pyrroloisoxazole analogs are only a few of the drugs that have been created and explored for the treatment of AD [307, 308]. Drugs like Rivastigmine, Donepezil, and Galantamine that have received Food and Drug Administration (FDA) approval improve cholinergic function by preventing the activity of acetylcholinesterase, which breaks down acetylcholine. The brain’s ACh levels rise as a result of these drugs. Acetylcholinesterase inhibitors are often well tolerated except for tacrine; side effects are typically dose-dependent. A monoamine oxidase A and B inhibitor called Ladostigil (TV3326), currently in phase II clinical studies, also exhibit antidepressant benefits [309, 310].
It is generally known that glutamate-induced excitotoxicity causes cells to become overloaded with calcium, experience a mitochondrial malfunction, produce more nitric oxide, and make a lot of oxidants, all of which contribute to neuronal apoptosis. However, NMDA receptor antagonists like memantine can lessen the harmful effects of glutamate. Memantine, which the FDA licensed in 2003 for use in patients with moderate-to-severe AD, has demonstrated some modest cognitive advantages in mild-to-moderate AD [311, 312].
Memantine protects neurons by inhibiting the activity of glycogen synthase kinase-3 (GSK-3), which lowers tau phosphorylation. Memantine is an acetylcholinesterase inhibitor that functions as a noncompetitive antagonist of the glutamatergic NMDA receptor. It can be used alone or in conjunction with other medications. It’s important to remember, though, that as compared to monotherapy, combination therapy may not result in as many positive changes [307, 313].
The APP cleavage by
The
Inhibitors have been developed to target the transmembrane protease BACE1, although early attempts based on molecular docking techniques to target the enzyme’s inaccessible catalytic core failed. Compared to other ADAM proteases, BACE1 inhibition has fewer adverse consequences [317, 318].
A multisubunit protease complex called
To stop the production of A
The effects of
Significant research efforts have been made in recent years to create cures that
aim to stop the development and aggregation of the A
Another strategy uses artificial peptides called “sheet breakers” generated from
the iA5p sequence. Zetidine-2-carboxylic acid, 3-phenyl azetidine-2-carboxylic
acid, proline, and sulfonyl proline are a few peptides that have demonstrated
promise in reducing the cellular harm brought on by A
Additionally, stemazole has demonstrated defense mechanisms against
A
Neurofibrillary tangles, which are collections of hyperphosphorylated tau protein in the form of paired, helical-twisted filaments, are one of the therapeutic targets in treating them. Axon’s Alzheimer’s Disease vaccine (AADvac1) was the first vaccination evaluated in clinical immunotherapy trials; currently, trials for ACI-35, another vaccine based on liposomes, are being conducted [308].
It has been investigated to prevent tau proteins from becoming phosphorylated, which is a factor in the aberrant aggregation of these proteins. However, testing of tideglusib, an irreversible inhibitor of the protein kinase GSK-3 (which is implicated in tau phosphorylation), failed to produce any statistically significant advantages. Cyclin-dependent kinase 5 (CDK5), which is similarly involved in tau hyperphosphorylation, is a potential therapeutic target [309].
Several compounds are being tested in clinical studies for their potential to prevent tau aggregation. One such example is the ability of methylene blue (MB) and its metabolites azure A and azure B to enhance protein degradation and inhibit the activities of caspase-1 and caspase-3 [325]. Additionally, in mouse models genetically modified to express tau mutations associated with AD, leucomethylthioninium with a suitable counterion (LMTX, in phase III clinical trials) and methylthioninium chloride, or MTC (in phase II clinical trials) have demonstrated the capacity to decrease tau aggregation and reverse behavioral deficits [326]. Additionally, they can reduce the course of the disease in AD patients. LMTX and MTC have been shown to have neuroprotective effects in vivo, although the precise mechanisms by which they do so are still not well known. N-phenylamines, anthraquinones, phenyl thiazolyl-hydrazides, rhodanines, benzothiazoles, and phenothiazines are further interesting tau aggregation inhibitors [327, 328].
Delivery of therapeutic medicines into the brain is a significant barrier to treating neurodegenerative disorders because of the BBB obstructive nature [329, 330]. Different methods have been formulated to release medication into the brain effectively. Chemical drug and prodrug modification, temporary disruption of BBB, local distribution into the brain by nanoparticle-mediated transport. However, osmotic pressure causes the BBB to open when tight junctions are temporarily disrupted. In general, using NPs for drug delivery has numerous benefits, including being non-invasive, inexpensive, having strong biodegradability and long-term stability, being simple to make, having high targeting effectiveness, and having high controllability to load and release conjugated pharmaceuticals across the BBB. Conjugating these phytochemicals may have promising therapeutic effects against diseases such as AD. Evidence suggests nanoparticle assisted medication transport across the BBB is a relatively effective technique [331].
Numerous in vivo studies have investigated how nanoparticle size affects the biodistribution of those particles in mice. Because differing GNP sizes impact how well they are cleared from the blood, insulin-targeted gold nanoparticles (INS-GNPs), which can target BBB insulin receptors, are a successful in vivo technique for imaging and therapeutic purposes. Eliminating nanoparticles by renal excretion is preferred since it involves less catabolism, largely excretes the particles in their original state, and minimizes the risk of adverse effects.
AD is one of the leading causes of dementia in many individuals worldwide. The
pathophysiology of AD is explained by many ideas. Although precise causes of the
disease have not yet been discovered. Patients with AD’s most severe form won’t
be able to carry out even the most basic physical duties, and they’ll be forced
to rely on others for practically all of their daily activities. They could even
struggle with simple tasks like swallowing when the illness is severe. As a
result, caring for an AD sufferer is quite expensive. Usually, 20 years after the
A plaques started to aggregate, patients with AD started to show apparent
cognitive problems. As a result, most people suspected of having AD have suffered
significant neuronal damage. This makes a mechanism for AD early detection
extremely important. The detection techniques should also be capable of picking
up on changes in the course of the disease. Thorough testing while the medicine
is administered would allow researchers to expedite the discovery of drugs for
AD. There is currently no treatment for AD. Even though there is a wealth of
knowledge about this complex condition, there are few alternatives for managing
it. Unfortunately, the therapy options currently available (AChEIs and memantine)
only address the symptoms of the disease rather than its root cause. So, there is
now more optimism for cutting-edge treatments that tackle the underlying causes
of the disease and have the potential to halt the steady buildup of A
We suggested GX-50 and curcumin conjugated with GNPs would be a potential
candidate medication for AD treatment. It investigated the impact of GX-50 on
A
The data is available inside the manuscript. There is no other data. generated nor used.
KM, MTK, MSL, DW Conceived and designed the study. MM, DW collected data. DW, KM, ZL, MM completed the data analysis and interpretation. MM, MTK drafted the article. KM, DW, MSL modified content. DW, ZL completed editing and data validation. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.