


 , Xiaochun Peng 2,4,*
, Xiaochun Peng 2,4,*1 PET Center of Nuclear Medicine, Department of the First Affiliated Hospital of Yangtze University, and School of Medicine of Yangtze University, 434000 Jingzhou, Hubei, China
2 Department of Pathophysiology, School of Basic Medicine, Health Science Center, Yangtze University, 434023 Jingzhou, Hubei, China
3 Department of Oncology, Jingzhou Hospital Affifiliated to Yangtze University, 434023 Jingzhou, Hubei, China
4 Laboratory of Oncology, Center for Molecular Medicine, School of Basic Medicine, Health Science Center, Yangtze University, 434023 Jingzhou, Hubei, China
5 Department of Rehabilitation Radiology, Beijing Rehabilitation Hospital, Capital Medical University, 100144 Beijing, China
†These authors contributed equally.
Abstract
Aging and related diseases significantly affect the health and happiness index around the world. Cellular senescence is the basis of physiological aging and is closely related to various senile diseases. AMP-activated protein kinase (AMPK) is associated with both the regulation of cellular energy metabolism and the regulation of cellular senescence. Another set of proteins, sirtuins, has also been demonstrated to play an important role in cell senescence. However, it is not clear how AMPK and sirtuins coordinate to regulate cellular senescence. Herein, we summarized the role of AMPK and sirtuins in regulating metabolism, repairing DNA damage, and even prolonging human life. We have provided a detailed explanation of the clinical trials relating to the AMPK and sirtuins involved in aging. Systematically analyzing individual senescence genes and developing functional reference notes will aid in understanding the potential mechanisms underlying aging and identify therapeutic targets for both anti-aging interventions and age-related illnesses.
Keywords
- AMPK
- sirtuins
- aging
- mTOR
- senescence
Globally, there has been a consistent increase in the aging population, as depicted in Fig. 1. In China, the population aged 60 years or older comprises around 264 million people, accounting for 18.7% of the total population, according to the 7th national census data. China has the largest elderly population in the world. Aging diseases also place a considerable economic burden on China [1]. Age-related diseases, such as cardiovascular diseases, coronary heart disease, cancer, diabetes, and arthritis, significantly impact the population’s health [2]. Furthermore, aging exacerbates the severity of various diseases [3]. Clinical research is increasingly focused on understanding how to counteract cellular senescence to enhance the quality of life.
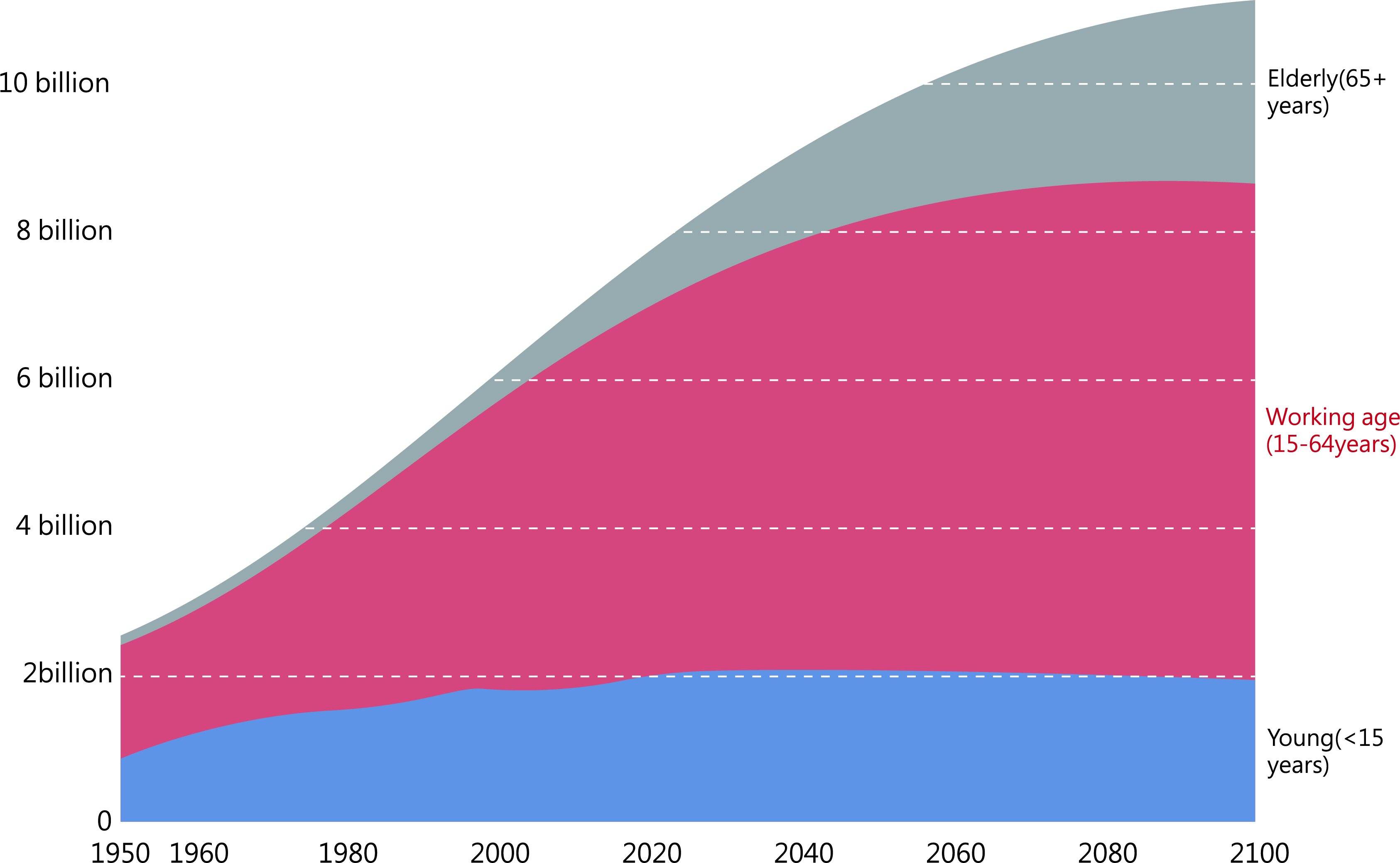 Fig. 1.
Fig. 1.Population size of young, middle-aged, and older people, 1950–2100. Historical estimates for 1950–2015 and estimates based on United Nations (UN) medium-term scenario projections to 2100. Source: United Nations Century Population Prospects (2017).
Cellular senescence is defined as a stress response in which cultured cells irreversibly lose their ability to proliferate, leading to cell cycle arrest, also known as the onset of senescence [4]. While aging and cellular senescence are distinct processes, they are intricately interconnected [5]. As an organism undergoes senescence, its overall functionality diminishes, consequently heightening the risk of mortality [5]. Previous research has indicated that there are numerous mechanisms of cellular senescence, including autophagy of cells, DNA damage, abnormalities in telomerase function, and genetic abnormalities related to carcinogenesis and oncogenesis [6].
Telomere shortening is a predominant factor in cell cycle progression. Cellular
autophagy is an important process in eukaryotes that regulates
homeostasis in vivo. Autophagy is associated with various causes,
including inflammatory and immune mechanisms, oxidative stress, caloric
restriction (CR), mitochondrial function or structure disruption, and cellular
hormonal disorders. These factors can influence autophagy and, consequently,
cellular senescence [7]. In the context of anti-aging research, cellular
senescence can be regulated indirectly or directly by various substances, such as
the insulin/insulin-like growth factor signaling pathway (IIS), The Mammalian Target of Rapamycin (mTOR), nicotinamide adenine dinucleotide
(NAD
AMPK regulates energy metabolism throughout the body and plays a role in the cellular senescence process by maintaining intracellular energy balance and vital activities [9]. The sirtuin family is involved in cellular biological activities, such as metabolism, inflammatory responses, DNA repair, and more. Furthermore, the sirtuin family is also involved in the cell cycle and apoptosis, which significantly affects cellular senescence [10]. In recent years, clinical research has demonstrated that the AMPK and the sirtuin family are highly influential in the progression of senescence [11]. This review focuses on their impact on cellular senescence progression as well as their interactions and regulation to enhance cell resistance to senescence.
AMP-activated protein kinase (AMPK) is an ATP-dependent protein kinase closely
related to biological energy metabolism. AMPK is a heterotrimeric complex
composed of three subunits:
AMPK is essential for providing energy for cellular activities as it is
sensitive to changes in AMP/ATP and ADP/ATP ratios [13]. AMPK is widely
distributed in all eukaryotes and can be activated in response to hypoxia,
ischemia, or nutritional deficiency. This leads to a decrease in ATP
concentration and a rapid increase in 5′-AMP levels. The interaction of
5′-AMP with the
AMPK has been shown to increase metabolism [15], promote cellular autophagy, reduce oxidative stress, and inhibit inflammation and endoplasmic reticulum stress [16], all of which can have a significant impact on the progression of cellular senescence through various pathways (Table 1, Ref. [16, 17, 18, 19, 20, 21, 22]).
| AMPK-regulated substances | Role | AMPK on its direction of action | Experimental subjects | References |
| CRTC-1/CREB signaling | AMPK can inhibit the CRTC-1/CREB pathway to achieve life-extending effects | Inhibition | Caenorhabditis elegans | [16, 17] |
| mTOR | Suppression of autophagy | Inhibition | Mice | [18] |
| FOXO axis | Increased antioxidant effect | Promotion | Caenorhabditis elegans | [16] |
| p53 | Enhancing self-efficacy | Promotion | Mice | [16] |
| NF-κB | Associated with immune response, inflammatory response, etc. | Inhibition | Mice | [16, 19] |
| Nrf2/SKN-1 | Improving cellular defense function under oxidative stress | Promotion | Caenorhabditis elegans | [16, 20] |
| ULK1 | Increase in self-longing | Promotion | Mice | [21, 22] |
AMPK, AMP-activated protein kinase; CRTC-1, CREB-regulated
transcriptional coactivator-1; CREB, cAMP-response element binding protein; mTOR,
The Mammalian Target of Rapamycin; FOXO, Forkhead box class O; NF-
cAMP-response element binding protein (CREB) is a well-studied regulatory protein that plays a crucial role in gene transcription and has been associated with memory. Low CREB activity can lead to long-term memory impairment, whereas high activity may improve age-related deficits in neuronal excitability and cognitive performance [23].
Studies have demonstrated that the cyclic adenylate response element binding protein (CREB)-regulated transcriptional coactivator-1 (CRTC-1) is the primary long-lived target of AMPK and calcineurin in Caenorhabditis elegans. The phosphorylation status of CRTC-1 is regulated in an antagonistic manner by both AMPK and calcineurin, which modulate their activity and exert effects on cellular senescence. AMPK and calcineurin share a common signaling pathway that interacts with CRTC-1 and is involved in mediating longevity. Calcineurin is a protein phosphatase that activates CRTC-1 by dephosphorylating it, thereby directly antagonizing AMPK. Conversely, CRTC-1 can be directly phosphorylated by AMPK. Our research shows that CRTC-1 is a direct target of AMPK and associates with the CREB homolog-1 (CRH-1) transcription factor in vivo. Activation of AMPK or inactivation of calcineurin reduces the activity of CRTC-1 and CRH-1. Several studies have shown that activation of AMPK or inactivation of calcineurin slows down senescence in C. elegans and that both regulate cellular senescence through post-translational modifications of CRTC-1 [17].
Studies have demonstrated that AMPK activation promotes the transcriptional reprogramming of metabolic genes through the CRTC-1/CREB pathway, which is associated with cellular senescence. Interestingly, the AMPK/AAK-2 signaling pathway is also closely linked to cellular senescence. CRTC-1 is a direct phosphorylation target of AAK-2/AMPK in C. elegans, and studies have shown that suppressing the CRTC-CREB pathway through AAK-2/AMPK signaling prolongs the lifespan of C. elegans. These findings suggest that factors inhibiting CRTC-induced CREB activation are critical in regulating the senescence progression and lifespan [16].
The Mammalian Target of Rapamycin (mTOR) is a serine-threonine kinase that exists as the catalytic subunit of at least two protein complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). When activated, mTORC1 regulates metabolism and cellular physiological processes by promoting protein translation and cell growth. mTORC1 also regulates autophagy, the ability to degrade cells and maintain cellular homeostasis when energy levels are low. mTORC2 is involved in cytoskeletal reorganization and cell survival [24].
AMPK can directly inhibit mTORC1 through phosphorylation of Raptor, and by suppressing the inhibitory effect of mTORC1 on cellular autophagy, a lifespan extension effect can be achieved [18]. AMPK can also indirectly inhibit mTORC2 by activating tuberous sclerosis 2 (TSC2) [25].
In addition, mTOR inhibition reduces mRNA translations, protein toxicity, and oxidative stress, thereby delaying senescence. Activating cellular autophagy can lead to the removal of damaged proteins, thereby extending lifespan [26]. Moreover, inhibition of mTOR activity facilitates the self-renewal of mouse hematopoietic stem cells. It has also been reported that activation of the mTOR pathway could inhibit sirtuins family (SIRT1), leading to impairment of cellular autophagy and counteracting effects on resistance to cellular senescence. Studies have shown that inhibition of mTOR activity enhanced SIRT2 activity, thereby extending the lifespan of yeast. Resveratrol also suppresses mTOR activation and improves cell aging [27].
Forkhead box class O (FOXO) is an insulin-sensitive protein that plays
a critical role in cellular physiological processes such as cell proliferation,
apoptosis, reactive oxygen species (ROS) response, cellular senescence, cancer,
and regulation of the cell cycle and metabolism [28]. Studies have shown that
under dietary restriction conditions, AMPK/aak-2 and FOXO/daf-16 can
increase the lifespan of worms [29]. AMPK
SIRT1 can increase resistance to oxidative stress in mammalian cells by regulating FOXOs. SIRT2 enhances FOXO3 by deacetylating FOXO3, leading to cell cycle arrest and resistance to oxidative stress [27]. FOXOs can inhibit cellular triglyceride accumulation via the sirtuin pathway [30].
p53 is a transcription factor that regulates the transcriptional
process of target genes [24]. As a well-known anti-oncogene, its main functions
are to prevent rapid cell proliferation and induce apoptosis [31]. p53
is a mediator of aging mainly through regulating cellular autophagy [6].
Activated AMPK can increase p53 expression and induce autophagy by
acting in conjunction with mTOR [31]. Moreover, AMPK promotes
p53 activity through phosphorylation at Ser-15 [32]. Forced
expression of AMP
During CR, AMPK activation leads to the activation of p53, causing cell cycle arrest and promoting cell survival. Continuous activation may lead to irreversible cell cycle arrest and promote the occurrence of senescence [33]. In addition, when p53 is activated, it inhibits mTOR and therefore suppresses senescence [34].
SIRT1 deacetylates p53, reducing DNA damage and stress-mediated cellular senescence. The stimulation of p21 by p53 inhibits the activity of cyclin-dependent kinases (CDKs) and regulates cell cycle arrest during cellular senescence. Studies have shown that SIRT1 can inhibit cellular senescence by decreasing the expression of p53 and p21, thereby promoting the differentiation of adipose cells [27]. The p53 gene can also prevent DNA damage, promote DNA repair, and prevent mutation.
The nuclear factor kappa-light-chain-enhancer of activated B cells
(NF-
Nuclear factor-erythroid 2-related factor-2 (Nrf2) and C. elegans skinhead family member 1 (SKN-1) play a crucial role in the oxidative stress response of cells [16]. Nrf2 is a transcription factor that protects against oxidative damage and is involved in the prevention of age-related diseases [20]. It is also associated with the nervous system and has been shown to enhance neuronal transcription and alter neurodegenerative lesions [36]. Additionally, Nrf2 may enhance cardiac function stability in the context of abnormal hemodynamics [37] and can protect against inflammation-related diseases [38].
Activation of the Nrf2/SKN-1 signaling pathways has been shown to prolong lifespan in various organisms, and AMPK-mediated activation of the pathways promotes cellular antioxidant and anti-inflammatory responses, potentially contributing to lifespan extension [16]. However, elucidating the exact mechanism requires further research.
ULK1 is a serine/threonine protein kinase that is a direct mammalian homolog of yeast Atg1. ULK1 has five homologs: ULK1, ULK2, ULK3, ULK4, and serine/threonine kinase 36 (STK36), with ULK1 and ULK2 thought to be involved in signaling associated with autophagy. Cellular autophagy can be severely affected when ULK1 is absent, indicating the important role of ULK1 in autophagy [21]. Phosphorylation of ULK1 by AMPK can increase autophagy and prolong the lifespan [22]. Furthermore, the AMPK/ULK1 pathway may play a role in tumor suppression and regulation of cellular metabolism in physiological processes. It has also been demonstrated that AMPK can indirectly inhibit ULK1 by suppressing mTOR, which inhibits autophagy and leads to anti-aging effects [39].
Nicotinamide adenine dinucleotide (NAD
In cell biology, NAD
Oxidative stress can result in an accumulation of ROS, which can cause damage to
cellular proteins, lipids, and DNA, ultimately leading to cellular damage.
Cellular damage can contribute to the development of various pathologies such as
aging, cancer, neurodegenerative diseases, cardiovascular diseases, and diabetes.
The levels of NAD
In mammalian SIRT1–7, each member has a catalytic domain that
binds NAD
SIRT1 is a well-known long-lived protein and plays a key role in mediating cell death, viability, and cardiovascular disease. SIRT2 is essential for the regulation of the cell division cycle. Improved expression of SIRT3 has been shown to be associated with human lifespan and also protects cardiomyocytes from senescence and oxidative stress while inhibiting myocardial hypertrophy. Recently, SIRT6 has also been shown to reduce myocardial hypertrophy, while SIRT7 is thought to regulate apoptosis and stress responses in the heart [44].
Thus, the sirtuin family is not only linked to antioxidant and oxidative stress responses but also to longevity, mitochondrial function, DNA damage repair, and metabolism. In the following sections, we explore the function of SIRT1–7 in cells and the relationship between the sirtuin family and aging in more detail.
Sirtuins are often referred to as longevity proteins, and their activity is
dependent on NAD
| The sirtuins | Positioning | Role | Experimental subjects | References |
| SIRT1 | Nucleus | Anti-aging effects on smooth muscle and blood vessels, slowing down brain aging and participating in physiological activities such as cell survival and apoptosis | Mice | [27, 46] |
| SIRT2 | Nucleus, cytoplasm | Anti-vascular senescence, regulates the cell cycle and improves autophagy | Mice | [47, 48] |
| SIRT3 | Mitochondria | Antioxidant effect, metabolic control | Mice | [50] |
| SIRT4 | Mitochondria | Regulation of metabolism | Mice | [49] |
| SIRT5 | Mitochondria | Regulating metabolism and enhancing autophagy | Mice | [49, 51] |
| SIRT6 | Nucleus | Genome and telomere stabilization, DNA repair, anti-aging effects on smooth muscle cells, endothelial cells | Mice | [45, 52] |
| SIRT7 | Nucleus | Anti-aging, DNA damage repair through enhanced stress resistance | Mice | [49] |
SIRT1–7, Members of the sirtuin family.
SIRT1, the most extensively studied of the sirtuin family, is located
on chromosome 10q21.3 and consists of eight introns and eleven
exons. The protein structure of SIRT1 contains 747 amino acid residues
and a catalytic core region surrounded by variable NH
SIRT1 is involved in regulating several proteins, including
FOXO, p53, nuclear factor-
SIRT1 also inhibits NF-
Mitochondrial cell function is frequently disrupted with age, which promotes the
production of ROS and increases oxidative stress, which severely affects cellular
senescence. One of the major transcriptional coactivators regulating
mitochondrial function is peroxisome proliferator-activated receptor
coactivator1
Overall, SIRT1 is involved in a range of pathophysiological processes such as cell differentiation, proliferation and metabolism, DNA repair, regulation of inflammatory responses, oxidative stress, and immune responses [46].
The SIRT2 gene is on chromosome 19q13. 2 and comprises 389 amino acids, is 21 kb long, contains 17 exons, and has a catalytic center of 323 amino acids. SIRT2 was identified in a yeast model and is present in the nucleus [47]. It has been shown to regulate the cell cycle and a range of biological processes, including telomere and ribosomal DNA (rDNA) silencing, information transduction of various substances, and metabolism through the deacetylation of histones and various transcription factors and cofactors [27].
A previous study identified an association between SIRT2 and cellular
senescence, whereby inhibition of SIRT2 leads to an increased cellular
oxidative stress response. This augmentation further promotes the development of
cellular senescence [48]. Additionally, SIRT2 deficiency has been shown
to lead to myocardial hypertrophy in mice [55], while it can control macrophage
inflammatory responses by regulating NF-
SIRT3 is a deacetylase found in mitochondria. In an experimental study, it was observed that mice deficient in SIRT3 exhibited significantly elevated levels of mitochondrial protein acetylation when compared to mice lacking SIRT4 and SIRT5. Therefore, SIRT3 is a crucial mitochondrial deacetylase [13]. Recent research has shown that SIRT3 regulates mitochondrial function and biosynthetic pathways through lysine deacetylation and maintains basal ATP levels by regulating the electron transport chain (ETC). Several clinical studies have also shown that SIRT3 deficiency can promote the development of metabolic syndrome, which may cause cardiovascular disease. Moreover, SIRT3 levels are reduced by 50% in the kidneys of aging mice compared with young mice. Therefore, low SIRT3 expression may affect the human lifespan. Overall, SIRT3 is protective against vascular senescence in rodents and humans [49]. SIRT3 can also protect cardiomyocytes from oxidative stress-mediated cellular damage and prevent the development of cardiac hypertrophy. SIRT3 also participates in defense against several cardiac illnesses [56]. Elevated SIRT3 levels can prevent apoptosis and oxidative stress and improve the maintenance of mitochondrial homeostasis in many patients with cardiovascular diseases. Elevated SIRT3 levels also have anti-aging, anti-fibrotic, and anti-inflammatory effects [50]. The research showed that high SIRT3 expression is associated with longer a life span in mammals. SIRT3 can also maintain cellular mitochondrial function of the kidney [57]. SIRT3 is a key regulator of renal cell mitochondrial dynamics. Furthermore, the acetylase target of SIRT3 is activated in specific conditions, such as CR [58]. SIRT3 has several deacetylase targets, including acetyl coenzyme a synthase2 (AceCS2), LKB1, ornithine transcarbamylase (OTC), and FOXO3A [34]. SIRT3 is also involved in the tricarboxylic acid cycle, redox respiratory chain, and the TCA cycle [59]. Notably, the TCA cycle is crucial in the regulation of aging and maintenance of homeostasis [3]. Overall, the acetylase activity of SIRT3 plays an important role in mitochondria by regulating multiple metabolic pathways and mitochondrial functional metabolism and resisting oxidative stress, thus stabilizing cells against aging [60]. SIRT3 also promotes DNA damage repair and maintains genome stability [10].
SIRT4 is primarily located in the matrix of mitochondria consisting of
structural domains and catalytic centers [61]. Firstly, it is involved in the
regulation of cellular metabolism [49], providing a carbon source for the citric
acid cycle by activating the glutamine cycle [59], and also inhibits
SIRT5 is a NAD-dependent protein lysine demethylase, desuccinylase, and
deglutarylase, primarily located in the mitochondrial matrix, cytoplasm, and
nucleus. SIRT5 catalyzes demethylation, desuccinylation, and
deglutarylation of mitochondrial enzymes through various metabolic pathways
related to glycolysis, fatty acid oxidation, and the urea cycle. However,
mitochondrial SIRT5 has a few targets, including carbamoyl phosphate
synthetase 1 (CPS1) [64] and succinate dehydrogenase (SDH) [65]. SIRT5
is highly expressed in the brain, heart, liver, and kidney [64]. SIRT5
can regulate amino acid catabolism and regulate ammonia production and
ammonia-induced autophagy by regulating glutamine metabolism in non-hepatocytes.
In addition, SIRT5 controls ammonia levels through interaction with the
mitochondrial GLS and desuccinylation. In addition to reducing ammonia
production, SIRT5 can also reduce autophagy and mitochondrial autophagy
[51]. Recent studies have suggested that SIRT5 can be considered a tumor
promoter or suppressor and may be associated with tumor formation. Moreover, it
may inhibit the development of neurodegenerative diseases [66]. Peroxisome
proliferator-activated receptor co-activated receptor-1
SIRT6 is a class III histone deacetylase found in the nucleus. SIRT6 exhibits deacetylase activity that can activate free histones when packaged into nucleosomes and undergo deacetylation. SIRT6 activity is nucleosome-dependent and can only participate in activation when bound to nucleosomes. SIRT6 regulates mitosis and maintains its fidelity [68]. SIRT6 has been shown to possess the ability to inhibit premature senescence and exert anti-denervation effects on endothelial cells and blood vessels. Furthermore, it has demonstrated the capacity to prevent the progression of atherosclerosis and the senescence of smooth muscle cells associated with aging [52]. SIRT6 also controls gene expression changes associated with aging by modulating nuclear factor kB [69]. Clinical studies have shown that SIRT6 deficiency can substantially increase the probability of severe metabolic disorders, genomic instability, inflammatory diseases, and oncological diseases in mice. In contrast, SIRT6 overexpression extends the lifespan of mice [70]. Notably, SIRT6 is a chromatin-associated protein that stabilizes the genome and regulates telomeric chromatin in human cells. This ensures efficient telomere replication and prevents structural abnormalities of telomeres [71]. SIRT6 also regulates the homeostasis of renal function [57]. Overall, SIRT6 mainly promotes the repair of DNA damage and maintains genomic stability [10]. Recent experiments have shown that SIRT6 overexpression increases the lifespan of male mice by approximately 16% [69].
SIRT7 is a class III histone deacetylase primarily found in the nucleus. It functions in chromatin and is involved in cellular transformation, prevention of fatty liver disease, and is also associated with tumor formation in vivo [68]. For example, SIRT6,7 may enhance the invasion and metastasis of osteosarcoma cells [72]. SIRT7 is also crucial for ribosomal DNA transcription, has acetylase activity and can regulate the transcriptional activity of RNA polymerase I [57]. In addition, SIRT7 also exhibits deacetylase, desuccinylase, and deglutaminase activity [73]. Most importantly, it is involved in a number of stress responses in the endoplasmic reticulum and reduces DNA damage, and improves cell survival under genomic stress conditions [49].
Studies have shown that AMPK interacts with the sirtuin family. Moreover, AMPK can directly or indirectly influence the sirtuin family to prolong lifespan. The findings indicate that the activation of AMPK and the sirtuin family can increase the lifespan of various organisms. AMPK can regulate lifespan and CR in worms and Drosophila. Similarly, SIRT1 is closely associated with the aging process in many organisms, including flies, C. histolytica, mice, worms, and mammals [27].
Metabolism (anabolism and catabolism) is a constant cell self-renewal that takes place in an organism, and it is associated with resistance to cellular senescence. Metabolism is a vital function in living organisms. Metabolism ensures continuous growth of cells and human life by maintaining physiological homeostasis. Therefore, metabolism is associated with cellular senescence. Anabolism involves the use of energy from adenosine triphosphate (ATP) or nicotinamide adenine dinucleotide phosphate (NADPH) to produce macromolecules in a biosynthetic process, whereas catabolism involves breaking down large components into smaller compounds that can be used directly in metabolism. Phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), mTOR, AMPK, and sirtuins are crucial to achieving homeostasis [13]. Anabolism allows the release of growth factors that promote cell division, growth, and differentiation. Growth factors belong to a family of highly effective cell surface acceptors in the receptor tyrosine kinase (RTK) family. These receptors typically promote nutrient uptake. PI3K also regulates metabolism through the interaction between RTK and growth factors, which activate the PI3K receptor. When energy is sufficient, RTK can activate PI3K. Activated PI3K can catalyze phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3), leading to the recruitment of AKT to the cell surface by PIP3, which facilitates glucose uptake via the main glucose transporter protein GLUT1. AKT is a serine/threonine-specific protein kinase that plays an important role in many cellular processes, including glucose metabolism, apoptosis, cell proliferation transcription, and cell migration. AKT is also an activator of glycolytic enzymes. AKT can also produce pyruvate, ATP, and NADPH [74]. However, the idea that AMPK and sirtuins are directly activated by affecting ATP and NADPH through the P13K/AKT pathway has not been confirmed. In addition, AKT can trigger lipid synthesis by enhancing the effective volume of acetyl coenzyme A. When under CR, ATP levels are reduced, and AMPK will be activated. Activated AMPK can inhibit mTORC1. Studies have shown that effective inhibition of mTORC1 during energy deficiency can prolong cell survival [75]. Another key protein that is activated when energy is low is the sirtuin family. An experimental animal study was conducted by Katharine et al. [76] and revealed that SIRT3 was activated during CR by comparing SIRT3 gene knock-out mice with normal mice. Activated SIRT3 was effective in reducing oxidative stress and thus delaying cellular senescence [76]. Moreover, some animal experiments have shown that a certain degree of energy restriction will delay cellular senescence. The rats in the calorie restriction experimental group lived longer than the normal group [77]. Therefore, energy restriction can delay cellular senescence by reducing oxidative stress damage. It is considered a powerful intervention to improve disease and extend lifespan in mammals [76]. In conclusion, PI3K/AKT, mTOR, AMPK, and sirtuin signaling networks can regulate metabolism and delay cellular senescence (Fig. 2).
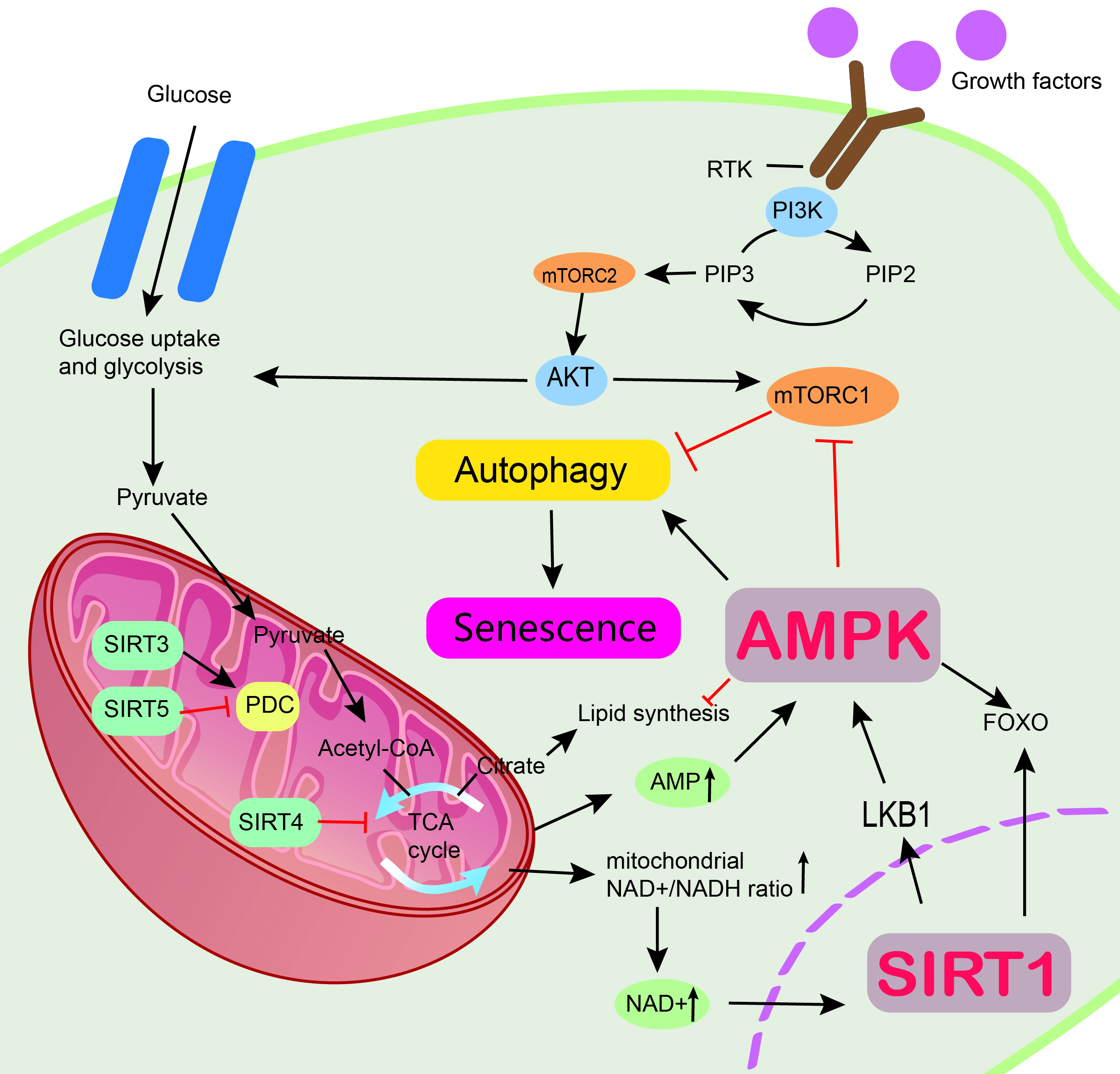 Fig. 2.
Fig. 2.Mechanism by which AMPK and sirtuin proteins regulate metabolism and resist aging. Multiple signaling pathways are involved in metabolic control, including the PI3K/AKT pathway, mTOR, AMPK, and sirtuins. Regulation of cellular senescence through the SIRT1/LKB1/AMPK pathway. AMPK, AMP-activated protein kinase; PI3K/AKT, Phosphatidylinositol 3-kinase/protein kinase B; mTOR, The Mammalian Target of Rapamycin; SIRT1, sirtuin 1; SIRT3, sirtuin 3; SIRT4, sirtuin 4; SIRT5, sirtuin 5; LKB1, liver kinase B1; FOXO, Forkhead box class O; mTORC2, mTOR complex 2; RTK, receptor tyrosine kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate.
Sirtuin SIRT1-mediated deacetylated liver kinase B1 (LKB1),
belonging to sirtuin family, is the upstream kinase of AMPK and activates both
LKB1 and AMPK [14]. When under CR, SIRT1 is activated and can
also sense intracellular NAD
SIRT1 and AMPK can mutually regulate each other’s activity through
various pathways. AMPK enhances SIRT1 activity by increasing cellular NAD
In addition to regulating various biological signaling pathways and targets, SIRT1 can also prevent and treat age-related diseases, especially cardiovascular disease, diabetes, and neurodegenerative diseases. Although SIRT1 activation can treat aging-related illness through suppression of p53, this has not been recognized in clinics. In addition, SIRT1 overexpression can prolong the life span of mice, as demonstrated in an in vivo experiment [83]. Therefore, in-depth studies targeting AMPK and SIRT1 are needed to provide therapy for aging and aging-related diseases and the development of new drugs.
The AMPK and sirtuin family inhibit cellular senescence through multiple
pathways. Some key and new clinical trials that have used AMPK and the sirtuin
family are described below (Table 3). Free fatty acid receptor 4 (FFAR4) can
activate the AMPK/SIRT3 signaling pathway, thus ameliorating renal tubular
epithelial cell senescence after acute kidney injury [84]. Some clinical trials
have found that diabetic patients have a shorter life expectancy, especially
severely obese and insulin-resistant diabetic patients. However, metformin can
treat pre-diabetic patients and also has an anti-aging effect on cells. Metformin
inhibits inflammatory response and improves cell survival. AMPK is one of the
metformin targets, indicating that metformin can inhibit cellular senescence
through the AMPK pathway. Metformin can also inhibit cellular senescence through
the mTOR signaling pathway [85]. Importantly, metformin can directly
stimulate the activation of the longevity protein STIR1, thus inhibiting
cellular senescence. Metformin can protect endothelial cells against elevated
blood glucose by indirectly stimulating the downstream targets of STIR1,
p53, and FOXO1. These findings indicate that AMPK and the
sirtuin family form a network that can regulate cellular senescence, consistent
with animal experiments. However, the effects of metformin on pathways associated
with longevity in humans are unknown [86]. A clinical phase IV trial
(NCT01765946) compared the efficacy of metformin (500 mg once daily) and placebo
in patients with pre-diabetes for two months to assess the influence of metformin
on longevity genes and inflammation in pre-diabetics. In that trial, longevity
gene expression of SIRT1, p66Shc, p53, and
mTOR was assessed in external peripheral blood mononuclear cells (PBMCs)
in vitro. Monocyte polarization was also assessed via flow cytometry
based on the expression of surface antigens (CD68, CCR2, CD163, CD206, CX3CR1) to
determine the proportion of pro-inflammatory or anti-inflammatory cells.
Inflammatory cytokines (TNF-
| Gene/protein | Study type | Allocation | Ages eligible for study | Purpose of research | Intervention | State | Phase | Numbering |
| SIRT6 | Clinical trial | Not applicable | 60 years and older | SIRT6 expression in circulating leukocytes based on the aging quality. | Different blood sample | Completed | Not applicable | NCT0156717 |
| SIRT1 | Clinical trial | Not applicable | 25 to 40 years | To prove that weight control is good for anti-aging. | Reduce calorie intake by 25% | Unknown | Not applicable | NCT01508091 |
| SIRT6 | ||||||||
| 7 sirtuins | Clinical trial | Randomized | 19 to 30 years | Effect of intermittent fasting on antioxidant gene levels, sirtuins, and markers of mitochondrial biogenesis and aging. | 400 IU vitamin D or 1000 mg vitamin C | Completed | Not applicable | NCT02132091 |
| 7 sirtuins | Clinical trial | Non-randomized | 60 years and older | Determining whether the quality of aging affects insulin sensitivity. | Insulin perfusion at 1 mU.kg |
Completed | Not applicable | NCT00951392 |
| 7 sirtuins | Observational | NR | 20 years and older | Examine the links between arterial aging with TL, as expressed in different tissues, and LTL dynamics, as expressed in the difference between TLs of muscle and leukocytes. | NR | Unknown | NR | NCT02176941 |
| AMPK | Clinical trial | Randomized | 40 to 75 years | Effects of metformin on longevity genes (SIRT1, p66Shc, p53, and mTOR) and inflammatory effects in pre-diabetic patients. | Metformin | Completed | Phase 4 | NCT01765946 |
| SIRT1 | ||||||||
| AMPK | Clinical trial | Randomized | 25 to 50 years | Effects on muscle and adipose tissue through dietary restriction and physical exercise. | Diet and exercise | Terminated | Not applicable | NCT01793896 |
| SIRT1 | ||||||||
| SIRT3 | Observational | NR | 20 years and older | The relationship between the degree of skeletal muscle senescence and aging. | NR | Completed | NR | NCT01644279 |
| SIRT1 | Observational | NR | 50 years and older | Study important factors in the pathophysiology of osteoporosis in patients with COPD. | NR | Completed | NR | NCT01067248 |
| SIRT1 | Clinical trial | Randomized | 65 years and older | The role of resveratrol in improving body function was investigated by looking at the link between changes in mitochondrial function and changes in body function. | Resveratrol 1000 or 1500 mg/day | Completed | Phase 2 | NCT02123121 |
| SIRT3 | ||||||||
| AMPK | ||||||||
| SIRT1 | Clinical trial | Randomized | 60 to 75 years | Exercise-related taurine supplementation in relation to metabolism in elderly sarcopenic obese women. | Taurine and exercise | Not yet recruiting | Not applicable | NCT05437952 |
| AMPK | Clinical trial | Randomized | 30 to 70 years | Using human leukocyte LC3 as a marker of autophagy and cellular senescence to validate the relationship between autophagy and senescence. | Metformin | Completed | Phase 3 | NCT03309007 |
| AMPK | Clinical trial | Randomized | 40 to 75 years | The effect of metformin on people without disease | Metformin | Recruiting | Phase 3 | NCT04264897 |
| AMPK | Clinical trial | Randomized | 20 to 80 years | Acute effects of high-load resistance exercise on inflammation in young and elderly people | Resistance exercise | Recruiting | Not applicable | NCT05042167 |
| SIRT1 | ||||||||
| AMPK | Clinical trial | Randomized | 65 years and older | Effects on muscle in |
Training or high-quality protein | Recruiting | Not applicable | NCT03649698 |
AMPK, AMP-activated protein kinase; SIRT1–7, Members of the sirtuin family; mTOR, The Mammalian Target of Rapamycin; TL, Telomere length; LTL, Leukocyte telomere length; COPD, Chronic obstructive pulmonary disease; NR, Not reported.
The substantial increase in life expectancy and the ongoing decline in birth
rates have resulted in a significant rise in the aging population. The process of
aging is influenced by various factors, including environmental conditions,
dietary habits, and emotional well-being. Nonetheless, cellular senescence should
be completely inhibited. AMPK and the sirtuin family can inhibit cellular
senescence. AMPK can maintain energy metabolism, increase cellular autophagy,
reduce oxidative stress, and inhibit inflammation and endoplasmic reticulum
stress. The sirtuin family has an anti-oxidation effect and can regulate cellular
metabolism, DNA damage repair, and autophagy, which are related to cellular
senescence. AMP-activated protein kinase (AMPK) and the sirtuin family can
directly or indirectly inhibit cellular senescence by regulating other
substances. For example, AMPK can extend lifespan by inhibiting the
CRTC-1/CREB signaling pathway. AMPK can also inhibit cellular senescence
by inhibiting autophagy through mTOR or increasing autophagy through
ULK1. FOXO combined with nuclear factor
kappa-light-chain-enhancer of activated B cells, NF-
XP and JH designed and supervised the study. YZ,TL, YH and WW provide ideas. YH ,YZ, TL and YL reviewed the references. YH and XP wrote the manuscript. YL, YZ, WW and TL contributed to tables and figures. YZ, YL, TL,and WW reviewing the manuscript for important intellectual content. XP, WW and JH revised the manuscript. XP acquired funding. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by National innovation and entrepreneurship training program for College Students (202010489017 to PXC), Jingzhou Science and Technology Bureau Project (2022HC78 to PXC), the College Students Innovative Entrepreneurial Training Program in Yangtze University (Yz2022297 to PXC), the Scientific Research Project of Education Department of Yangtze university (JY2020134 to PXC), Hubei Province Natural Science Foundation of China (2017CFB786 to PXC).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.