
- Academic Editor
The retina, a component of the central nervous system, is composed of six distinct neuronal types and various types of glial cells. A technique for single-cell transcriptome analysis called single-cell RNA sequencing (scRNA-seq) can be employed to study the complicated dynamics of several types of retinal cells. It meticulously examines how various cell types express their genes, shedding light on all biological processes. scRNA-seq is an alternative to regular RNA-seq, which cannot identify cellular heterogeneity. Understanding retinal diseases requires research on retinal cell heterogeneity. The identification of novel cell subpopulations can provide information about disease occurrence and progression as well as the specific biological functions of particular cells. We currently have a better understanding of the interactions among the brain, the retina, and its visual pathways thanks to the use of scRNA-seq to examine retinal development and disease pathogenesis. Additionally, this technology offers fresh perspectives on the sensitivity and molecular basis of cell subtypes linked to retinal diseases. Thanks to scRNA-seq technology, we now have a better understanding of the most recent developments and difficulties in retinal development and disorders. We believe that scRNA-seq is an important tool for developing cutting-edge treatments for retinal diseases. This paper presents a systematic review of the history of sRNA-seq technology development and provides an overview of the unique subtypes of retinal cells and the specific gene markers this technology identifies.
Sight is the primary sense used by humans and other primates for orienting themselves in their environments. A tissue in the back of the eye called the retina receives light focused by the cornea and the lens of the human eye. Thereafter, the retina’s photoreceptor cells detect light to initiate the process of vision. The vertebrate eye contains photoreceptors, amacrine cells (ACs), retinal ganglion cells (RGCs), horizontal cells, and bipolar cells, all of which descend from a common progenitor pool [1]. Additionally, retinal progenitor cells (RPCs) produce Müller glia, a type of glial cell [2]. The retina’s cell types are composed of two plexiform layers and three cell body layers [3]: the outer nuclear layer has photoreceptors; the outer plexiform layer comprises photoreceptor-interneurons synapses; and the inner nuclear layer comprises the cell bodies of the visual information-transmitting bipolar, amacrine, and horizontal interneurons and the extracellular space-supporting Müller glia.
With the advancement of high-throughput sequencing technology, it is now possible to conduct large-scale studies of genomes and related elements such as chromatin structure, DNA sequences, metabolites, proteins, and RNA transcripts. For traditional high-throughput sequencing, adequate DNA samples from multiple cells are required. However, the accuracy of high-throughput sequencing is rather low, necessitating the correction of sequencing data. At the single-cell level, single-cell RNA sequencing (scRNA-seq) is high-throughput sequencing analysis of the transcriptome, genome, and epigenome [4]. scRNA-seq technology is widely employed in tumor, embryo, and stem cell research [5]. Single-cell transcription maps are used to classify tumors, and gene expression patterns are used to identify immunotherapy targets and detect cancer cell metastasis [6]. scRNA-seq has primarily been used in ophthalmology to determine cell subtypes, pathogenic gene pathways, and targeted treatments for retinal diseases (Fig. 1). scRNA-seq provides information on gene expression in individual cells and can be used to identify cell types, study gene expression patterns, and characterize cell states. Single-cell assay for transposase-accessible chromatin sequencing (scATAC-seq) measures chromatin accessibility at the single-cell level and can be used to identify regulatory regions of the genome and study changes in chromatin accessibility during development or disease. Single-nucleus RNA sequencing (snRNA-seq) measures gene expression in individual nuclei from cells that are difficult to dissociate, such as neurons or frozen tissues. SnRNA-seq can provide information on gene expression in specific cell types and has been used to study retinal development and disease. In this paper, we comprehensively review the rapid advancement of scRNA-seq technology and present challenges and concerns regarding retinal development and disease.
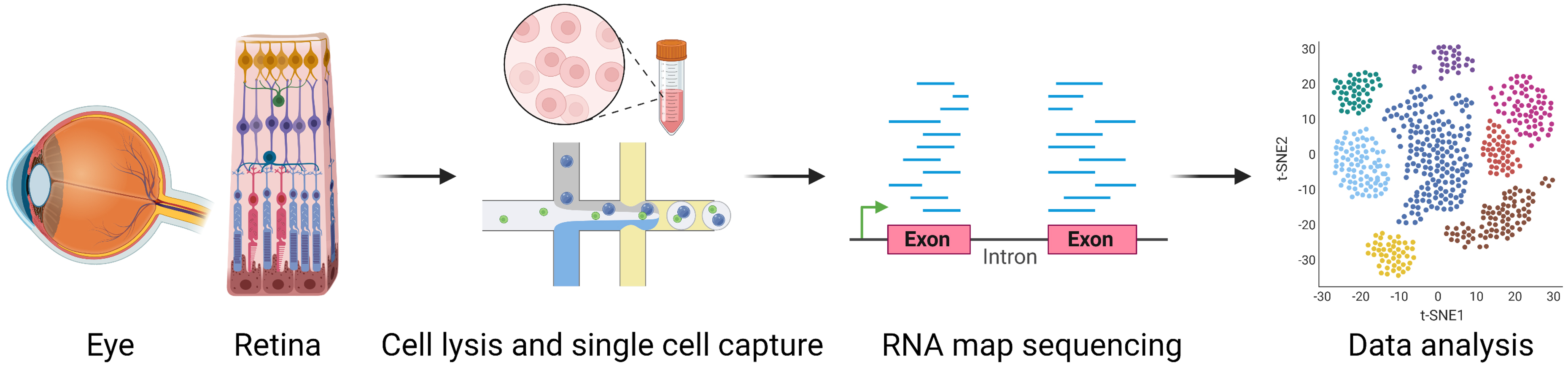 Fig. 1.
Fig. 1.General workflow of single-cell RNA sequencing. Single-cell suspensions made from harvested organs and tissues are prepared. The best technique is chosen for single-cell capture. RNA is reverse transcribed into cDNA after cell lysis, and cDNA is then amplified by PCR to create a cDNA library for sequencing. The resulting sequence reads are aligned to specific genes and assigned to cells using cell-specific barcodes incorporated into the cDNA by the primers used for reverse transcription. Finally, a detailed analysis of the collected data is performed. Figure was created with BioRender. cDNA, complementary DNA; PCR, polymerase chain reaction.
The development of the human retina follows a similar pattern to that of other mammals, but with some important differences [7]. During the first stage, the optic vesicle invaginates to form the optic cup, which is the precursor to the retina. The second stage involves the differentiation of RPCs into different cell types including photoreceptors, horizontal cells, bipolar cells, ACs, ganglion cells, and Müller glia. This stage occurs during the first trimester of pregnancy. The third stage is characterized by the migration of retinal cells to their final positions within the retina. During this stage, cells differentiate into specific subtypes and migrate radially to form the layered structure of the retina. This stage occurs during the second trimester of pregnancy. The fourth stage involves the formation of synapses and the establishment of functional connections between retinal cells. This stage is critical for proper functioning of the retina and occurs during the third trimester of pregnancy, continuing into the postnatal period. The final stage is characterized by the refinement and maturation of neuronal connections. This includes the pruning of excess synapses and the establishment of functional circuits between different retinal cell types. This stage occurs during the first few months after birth. Overall, the development of the human retina is a complex and highly orchestrated process that spans several months of prenatal development and continues into the postnatal period. Understanding the stages of retinal development is important for identifying potential causes of developmental disorders and developing new treatments for retinal diseases [8].
According to the neural retina and retinal pigment epithelium (RPE) transcriptome landscape before 12 weeks of gestation, RGCs appear in humans at 5 weeks and peak at 8 weeks of gestation, whereas horizontal cells are detected at 7 weeks and peak at 9 weeks of gestation [9]. Rod and cone photoreceptor cells reach their peak at 17 weeks, followed by ACs. Müller glia and bipolar cells are the last to be detected. Transcriptome analysis has revealed the timing of the appearance and peak expression of various retinal cells, and the identification of receptor-ligand pairs offers insights into cell-cell interactions. Receptor-ligand pairs of RPCs and photoreceptor cells, including FLT1/NRP1/NRP2 and VEGFA, NGFR and TTR, and PTPRZ1 and PTN/MDK, were identified using CellPhoneDB, which offers insights into the interactions between RPE and photoreceptor cells. Several neuroepithelial progenitors are found in the developing embryonic eye’s optic vesicle, where they differentiate to form the RPE, optic stalk, and neural retina among other optic cup domains with distinct functions [10]. Several structurally and metabolically important genes, including collagen, type IX, alpha 1 (Col9a1) and brain-type creatine kinase, show altered expression in these cells just before regionalization of the optic cup.
scRNA-seq profiling has further elucidated the expression patterns of key genes involved in retinal development and revealed the regulatory networks governing RPC competence, neurogenesis, and cell fate specification. Lu et al. [11] examined 118,555 retinal cells across 16 stages of human retinal development and four stages of distinct maturation of retinal organoids (RtOgs), from early neurogenesis to adulthood, to create a comprehensive scRNA-seq profile. Along with early-stage neurogenic RPCs, atonal homolog 7 (ATOH7) is also expressed in late-stage neurogenic immature photoreceptors, bipolar-photoreceptor precursors, RPCs, ACs, and horizontal cell precursors, enhancing cone genesis in the developing human retina. Neural progenitor cells can generate numerous subtypes of neurons and glia and undergo typical stage-dependent transitions during neurogenesis [12]. Clark et al. [13] discovered significant variations between primary and neurogenic progenitors as well as early and late stages by scRNA-seq, which was used to reconstruct developmental trajectories of various retinal cell fates and determine the genes and gene networks that affect RPC competence, retinal neurogenesis, and cell fate specification during development. Additionally, the authors discovered that nuclear factor I (NFI) factors regulate late-born cell type generation and cell cycle exit.
Furthermore, investigating the transcription factor profiles of the developing
human retina, particularly the activated transcription factors, can provide
valuable information on human retinal development. It is challenging to ascertain
the roles of transcription factors in maturation of the human retina, and the
significance of these transcription factors can be inferred from analyses of
their activities. Orthodenticle homeobox 2 (OTX2) is an essential transcription
factor for the proper development and function of the retina. ScATAC-seq and
scRNA-seq can provide an unprecedented view of the developing human retina,
identifying crucial transcription factors associated with particular fates and
the sequence of transcription factor cascades that characterize the primary types
of retinal cells. OTX2, which is essential for the formation of photoreceptor and
bipolar cells, is upregulated by a sizable subset of RPCs during retinal
development [14]. According to scRNA-seq, basic helix-loop-helix (bHLH) factors
Achaete-scute-like 1 (Ascl1) and neurogenin2 (Neurog2) may regulate the
development of Otx2
Transcription factor AP-2 delta and some other transcription factors necessary for RGC development are expressed in the developing human retina as early as 5 weeks [17]. Furthermore, one cut homeobox 1 (ONECUT1) and ONECUT2 are involved in the development of the human retina’s horizontal cells [18]. Through alterations in accessible chromatin that accompany cell-state changes, scATAC-seq characterizes putative gene regulatory networks [19]. Zhang et al. [20] studied the functions of ATOH7 and Neurog2, which are expressed early in the development of vertebrate retinal epithelium, in human RGC development using three-dimensional (3D) RtOgs made from embryonic stem cells (ESCs). Moreover, both ATOH7 and Neurog2 exert positive autoregulation and increase the levels of bHLH factors expressed by differentiating neuroblasts and exiting RPCs while suppressing important bHLH factors linked to pre-neurogenic states.
The human retina is a complicated sensory tissue that recognizes light photons, translates their photic patterns into neurochemical information, and then registers that information as vision in the brain [21]. The mammalian retina is an intricate tissue consisting of various interconnected neuronal and glial cells, predominantly photoreceptors. RNA leakage from damaged cells occurs due to photoreceptors’ structural fragility, which exposes them to mechanical and enzymatic dissociation. The dissociation of retinal samples presents significant challenges in scRNA-seq processing, particularly for comparisons. It is difficult to accurately identify different cell types because the ambient RNA in cell suspensions can be integrated into scRNA-seq microfluidic droplets, contaminating other cell types. In particular, rod-dominant retina scRNA-seq results can be considerably impacted by such contamination. In addition to preventing inaccurate cell clustering, contamination should be minimized for samples with highly expressed genes, such as rhodopsin in rod cells. Fadl et al. [22] modified the retinal dissociation protocol by using low temperature during digestion and adding anti-oxidants (D-alpha-tocopheryl acetate, catalase, and superoxide dismutase) to the digestion and inactivation solutions, thereby lowering papain enzymatic activity, reducing mechanical cell damage, and reducing oxidative stress. This protocol effectively dissociated the retina and was optimized to reduce cell death while maintaining cell morphology, allowing a higher gene detection rate per cell. Additionally, the authors created a bioinformatics pipeline for scRNA-seq that was optimized to remove low-quality retina sample cells and lessen the technical noise caused by ambient RNA contamination.
Individual photoreceptor cells (rods and three cone classes) in the adult retina transform light into an electrical signal, and the bipolar cells receive this electrical signal before passing it to the RGCs [23]. The populations of various cell types differ greatly depending on where they are in the retina, which further complicates retina dissociation. Lukowski et al. [24] used 20,090 single cells from three healthy donors to create a transcriptome atlas of the human neural retina. Cells from various eye regions including the iris, cornea, choroid, sclera, retina, and RPE were extracted for scRNA-seq. A total of 18 clusters were identified by this study, including those for cone photoreceptors (GNGT2, GUCA1C, and ARR3), retinal astrocytes (GFAP), rod photoreceptors (RHO, CNGA1, and PDE6A), Müller glia (RLBP1/CRALBP), bipolar cells (VSX2 and OTX2), RGCs (ONECUT1, ONECUT2), and microglia (HLA-DRA, HLADPB1, and HLA-DPA1). These data recorded the molecular profiles of both putatively early-degenerating rod photoreceptors and healthy photoreceptors. Longer post-mortem periods showed the loss of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) expression, indicating a new role for MALAT1 in rod photoreceptor aging. According to the RNA velocities of all ocular cell types, COL9A1-high and pigmented ciliary body cells may have been derived from a putative stem cell type. Human RPE cells can be divided into two clusters: peripheral RPE (expressing crystallin alpha B [CRYAB]) and macular RPE (highly expressing inhibitor of DNA-binding 3) [25]. CRYAB demonstrates greater specificity in peripheral RPE cells than insulin-like growth factor-binding protein 5, suggesting that CRYAB may serve as a novel marker gene for peripheral RPE.
Fovea centralis, a small depression measuring 0.6–0.7 mm in diameter, is found in the retina of humans and other primates [26]. It lacks vascular components, bipolar cells, rods, and ganglion cells, and is predominately comprised of cones with distinct morphologies and Müller cells. The enlarged inner and outer segments and relatively long axons that make up Henle’s nerve fiber layer serve as distinguishing characteristics between foveal and extrafoveal cones [27]. Peripheral cones have short exterior segments and teardrop-shaped “cone” inner segments. It would be very interesting to characterize the molecular variations between the central and peripheral retina. A better knowledge of the regional differences across cell types may be valuable in clinical settings. Several human retinal disorders can either selectively protect or harm foveal cones. Even in patients with known genetic etiologies, gene modification can avert or allow foveal cone damage. Voigt et al. [28] used scRNA-seq on the foveal and peripheral retinas of three individuals to catalog cell type-specific transcripts in the human retina and uncover region-specific changes in these cell types. There were 17 cell clusters representing the most common retinal cell populations. Each retina was punched with a 2 mm fovea-centered and 4 mm periphery punch. A total of 8217 cells (4639 peripheral cells and 3578 foveal cells) met the criteria for quality control and filtering. Local gene expression analysis is possible for samples that are greatly enriched with foveal cells by using 2 mm fovea-centered punches. Cone photoreceptor cells and glial cells produce unique cell clusters of foveal and peripheral cells. The beta-carotene oxygenase 2 gene is more highly expressed in peripheral than foveal cones. Yan et al. [29] dissected the entire retina and retrieved 84,982 transcriptomes of high quality (29,246 from the peripheral retina; 55,736 from the fovea). With the aid of a dissecting microscope, the foveal samples were observed to have a diameter of about 1.5 mm and were centered on the foveal pit. The authors found 58 different cell types among the six classes, namely, non-neuronal cells, amacrine, horizontal, photoreceptor, retinal ganglion, and bipolar, and the ratio of the horizontal cells in the fovea was nearly four times higher (7.3:1) compared to the 1.9:1 ratio of the peripheral retina. The ratio of GABAergic to glycinergic AC types was higher in the fovea compared to the periphery (1.8:1 and 1.1:1, respectively). Furthermore, only the OFF cone bipolar RGC type was enriched in the fovea, whereas five of the eight rare RGC types (clusters 7, 8, 9, 10, and 12) were more prevalent in the peripheral retina than in the fovea. Additionally, Yi et al. [30] dissected foveal and peripheral cells and collected 19,520 single-cell transcriptomic profiles, yielding 38,558 from six healthy human samples. RGR, HTR1, DIO2, and CYP26A1 are abundant in foveal Müller glia cells. The cellular specificity of peripheral and foveal cones may be controlled by a thyroid hormone signal that is delivered locally by the foveal Müller glial cells. Voigt et al. [31] employed paired parafoveal and foveal retinal cells from four human donors, comprising 28,637 parafoveal and 5856 foveal cells, in a complementary scRNA-seq experiment to assess gene expression. The authors discovered that the cone:rod:RGC ratio in a 4 mm parafoveal punch was 1:6.7:1.5 and 1:0.5:0.3 in a 1 mm foveal punch. Müller cells are predominant in both the fovea and parafovea. Müller glia is the only other cell type observed in the foveola. Foveal cones express more transcription coactivators, including POUF2A1, YBX1, and LBH, compared to the parafovea [32].
ACs, a diverse group of interneurons, regulate photoreceptor input to the RGCs, making every RGC type selectively sensitive to distinct visual cues that are relayed. Yan et al. [33] profiled more than 32,000 ACs from male and female mice using high-throughput scRNA-seq and computational techniques and identified 63 AC types. The authors investigated the transcriptomic relationships between the various AC types and discovered transcription factors, such as transcription factor 4 and Meis homeobox 2, which are expressed by the majority of glycinergic and GABAergic types individually or by several closely related AC types.
Müller glia, the retina’s primary glial cells, perform various functions such as neurotransmitter recycling, structural support, potassium siphoning, osmotic regulation, and bicarbonate transport [34]. NFI gene products promote gliogenesis, which is required for Müller glia differentiation in the developing rodent retina [35]. At E5, RPCs do not significantly express NFIA, NFIB, NFIC, or NFIX, and all cell types exhibit low levels of NFIC and NFIX expression during retinal development. NFIA and NFIB, on the other hand, are expressed by a few immature Müller glia and relatively few E8 progenitors at E8, E12, and E15. Additionally, NFIA and NFIB are found in fewer differentiating amacrine, photoreceptor, bipolar, and ganglion cells at later developmental stages (E12 and E15). The complement system and Müller glia serve to defend the neuroretina. Although the underlying mechanism is still unknown, neurons tightly control activation of the microglia under normal physiological conditions [36]. Retinal microglia are found in the inner and outer plexiform layers as well as the ganglion cell layer. Using published scRNA-seq data sets Liu et al. [37] discovered that a variety of retinal neurons express immune regulatory molecules such as cluster of differentiation 200 (CD200), CD47, C-X3-C motif chemokine ligand 1, transforming growth factor, and CD59a (complement inhibitor). Ascl1 overexpression can induce Müller glia in the adult mouse retina to regenerate functional neurons because different types of retinal neurons are produced by Ascl1:Atoh1 [38].
Gene expression in retinal cells can vary depending on their location within the tissue, with the macula/fovea region being particularly unique [39]. The macula is a small, central region of the retina that is responsible for high acuity vision and contains a high density of cone photoreceptors. By contrast, the peripheral retina contains a higher proportion of rod photoreceptors and is responsible for detecting movement and low light levels. The unique cellular composition of the macula/fovea, with a high density of cones and absence of blood vessels and other retinal layers, presents challenges for studying gene expression in this region. However, recent advances in single-cell sequencing technologies, such as scRNA-seq and snRNA-seq, have allowed researchers to investigate gene expression patterns in individual cells from the macula and fovea with greater precision. It is also worth noting that while the macula is a prominent feature in humans and primates, it is either absent or present in a different form in other species such as rodents. This highlights the importance of considering species-specific differences when studying retinal biology and disease.
Age-related macular degeneration (AMD) is a leading cause of irreversible blindness worldwide [40]. The compositional changes of Müller glia, microglia, astrocytes, endothelium, and rods in the macula are more prominent during the progression from standard to progressive AMD. Differentially expressed genes (DEGs) between normal and advanced AMD are enriched in antigen presentation, tissue remodeling, complement and coagulation pathways, and other signaling pathways such as repressor activator protein 1, Toll-like receptor, nucleotide oligomerization domain-like receptor, and phosphoinositide 3-kinase/Akt [41]. These results have paved the way for future investigations into the molecular underpinnings of retinal illness and the development of new treatment targets by proving the ability of scRNA-seq to infer cell-type-specific gene expression and cell-type compositional alterations in intact bulk tissue. Microglial activation, monocyte infiltration, and monocyte differentiation into macrophages can all take place simultaneously and rapidly within 48 h of the start of photoreceptor degeneration [42]. All immune subgroups have different levels of C-C motif chemokine ligand 2 (CCL2) expression, indicating that C-C motif chemokine receptor 2 (CCR2) is rapidly degraded and downregulated after extravasation into the retinal parenchyma. During retinal degeneration, CCL2-CCR2 signaling is essential for attracting circulating immune cells [43].
The RPE connects the choroid and neural retina in the human eye. AMD, one of the pathologies of RPE degradation that causes blindness, is a multifactorial genetic disease, and its pathogenesis has been widely linked to RPE and choroid dysfunction [44]. Following the evaluation of total RPE/choroid preparations, RPE-specific expression profiles and large choroidal cell populations were discovered [45]. Choroidal endothelial cells are critical for AMD pathogenesis despite representing a small portion of the RPE/choroidal cell population; therefore, a second scRNA-seq experiment was carried out using magnetically separated choroidal endothelial cells. The regulator of the cell cycle gene (RGCC) is highly and specifically expressed in the choriocapillaris. AMD donors have substantially upregulated RGCC, which activates complement and induces endothelial cell apoptosis. Finally, retinal degeneration transcriptomes show altered splicing, including increased intron retention and abnormal alternative splicing. The recessive gene, CWC27 spliceosome-associated cyclophilin, has been linked to both syndromic and non-syndromic autosomal recessive retinal degeneration [46].
Diabetic retinopathy (DR) is a prevalent diabetes-related complication and a
major cause of vision loss and blindness [47]. When 80 post-mortem human retinal
specimens from 43 patients with varying stages of DR were examined by RNA-seq,
the results showed that most differentially expressed transcripts were associated
with late-stage DR and pathways including Hippo and Gap junction signaling [48].
Additionally, the authors discovered transcripts with considerable overlap with
sphingolipid and cyclic guanosine monophosphate-protein kinase G signaling and
progressive alterations throughout disease stages using scRNA-seq. It is
difficult to analyze DR pathology due to the intricate cytoarchitecture of the
retina. scRNA-seq of retinal tissues from Akimba
(Ins2
Another typical diabetes-related complication, proliferative diabetic retinopathy (PDR), is a significant contributor to severe vision loss and blindness [54]. It is characterized by development of the tractional retinal detachment and vitreous hemorrhage-causing epiretinal fibrovascular membrane (FVM), which develops between the retina and posterior hyaloid of the vitreous [55]. After surgically harvesting PDR-FVMs, Hu et al. [56] conducted scRNA-seq to generate an intricate cell atlas of FVM and discovered eight different cellular compositions, with microglia being the predominant cell type. Ligand-receptor interactions in profibrotic microglia with upregulation of cytokines in the PDR vitreous suggest that several signaling pathways, such as interferon gamma receptor 1, CD44, and CCR5, are involved in the activation of microglia within the PDR microenvironment.
In autoimmune retinopathy (AIR), anti-retinal antibodies destroy retinal cells, causing blindness [57]. AIR etiology includes paraneoplastic and non-paraneoplastic diseases. AIR diagnosis is aided by antiretinal antibodies. However, the specificity of this diagnostic assay is inhibited because unaffected individuals might also need to circulate antiretinal antibodies. Voigt et al. [58] described the AIR-attributed clinical progression of progressive photoreceptor degeneration in a 70-year-old patient. Additionally, to explore how various retinal cells react to photoreceptor degeneration, researchers used scRNA-seq on peripheral retinal and paired foveal samples from a 70-year-old patient and four unaffected healthy patients. A total of 23,429 cells were extracted, of which 7189 were from a patient with AIR. This study provides evidence that glial cells have a distinct transcriptome in the context of human retinal degeneration and constitutes a complementary clinical and molecular analysis of a case of progressive retinal disease.
Glaucoma is linked to distinctive optic nerve damage and certain types of visual field loss, which primarily involves the loss of RGCs [59]. Gelatinase activity is associated with human and animal diseases and retinal pathophysiology. Todd et al. [60] conducted scRNA-seq of N-methyl-d-aspartate (NMDA)-damaged mouse retina and found that Müller glia express Slc1a3, Lhx2, Vim, Sox9, and Rlbp1 differentially and at higher levels than all other cell types, and that reactive microglia defend retinal neurons by using IL-1 and other macroglia. A subclass of matrix metalloproteinases (MMPs) called gelatinases breaks down the extracellular matrix to control cell migration and intercellular signaling. Campbell et al. [61] identified the different cell types that produce gelatinases and other MMPs in the avian retina using scRNA-seq libraries of normal and NMDA-damaged retinas. MMP-2 was observed in oligodendrocytes and non-astrocytic inner retinal glia cells, but MMP-9 was very weakly expressed in non-astrocytic inner retinal glia cells, Müller glia, and amacrine. Additionally, MMP-2 could reprogram Müller glia to develop into Müller glia progenitor cells.
Retinitis pigmentosa (RP) is a group of genetic disorders that cause degeneration of photoreceptor cells in the retina, leading to vision loss and eventual blindness. scRNA-seq technology has revolutionized our understanding of the molecular mechanisms underlying RP. Several studies have used scRNA-seq to identify different cell types in the retina and their gene expression patterns. Peng et al. [62] performed scRNA-seq on the retina of mice with RP and found that Müller glia cells and microglia cells were activated and upregulated the expression of inflammatory genes. Similarly, Liu et al. [63] identified different cell types in the human retina and found that Müller glia cells and RPE cells exhibited gene expression changes in RP patients compared to healthy controls. Furthermore, scRNA-seq has been used to study the molecular mechanisms underlying RP. Menon et al. [64] identified a novel mechanism for the activation of autophagy in RP, where autophagy-related genes were upregulated in photoreceptor cells, leading to their degeneration. Lukowski et al. [65] demonstrated that mitochondrial dysfunction is a key driver of RP progression. They found that photoreceptor cells in RP patients had reduced mitochondrial respiration and increased oxidative stress, leading to cell death. Finally, scRNA-seq has also been used to identify potential therapeutic targets for RP. The solute carrier family 16 member 8 (SLC16A8) gene, which was upregulated in Müller glia cells in RP patients. Inhibition of SLC16A8 has been shown to improve photoreceptor survival in mouse models of RP. The SLC7A11 gene as a potential therapeutic target for RP, as its inhibition reduced oxidative stress and improved photoreceptor survival in RP mouse models. Therefore, scRNA-seq has provided valuable insights into the pathogenesis of RP, including the identification of different cell types in the retina, the molecular mechanisms underlying RP, and potential therapeutic targets. These findings have the potential to inform the development of novel therapies for RP.
Adeno-associated viruses (AAVs) are the preferred viral-vector candidates for therapeutic gene delivery in gene therapy [66]. AAV engineering has produced vectors with a greater capacity to deliver therapeutic genes, and newly engineered AAV vectors are being increasingly used in clinical trials [67]. However, due to interspecies variations, it has been difficult to anticipate the translational potential of gene therapies developed in animal models. Quantitative comparison of AAVs is difficult, even though the choice of vector is crucial, especially in large animals. Öztürk et al. [68] developed a scRNA-seq AAV engineering (scAAVengr) pipeline for the rapid quantitative in vivo comparison of transgene expression from newly engineered AAV capsid variants across all different cell types in tissue in parallel and the same animals. The authors were able to quantitatively assess the clinical potential of numerous lead candidates across all retinal cell types in the foveal and peripheral retina using the scAAVengr pipeline in large animal models with eyes similar to those of humans. The new human-DNA MiniPromoter Ple345 created by Simpson et al. [69], was identified by scRNA-seq to exhibit specific and robust expression in the RGCs of the nonhuman primate rhesus macaque retina when combined with intravitreal delivery in rAAV9. Using the scRNA-seq AAV engineering pipeline, Xi et al. [70] evaluated wild-type and engineered AAV capsids in human retinal explants, and measured the effectiveness of 18 AAV serotypes in ex vivo cultures of the human retina and found that K91, K912, and 7m8 were the most effective serotypes.
Globally, 285 million people are estimated to be visually impaired, with retinal disorders accounting for about 26% of blindness cases. The loss of photoreceptors results from numerous inherited and age-related retinal dystrophies. At present, there is no treatment to stop retinal degeneration; therefore, cell replacement has evolved as a prerequisite for therapeutic transplantations. The creation of human embryonic stem cells (hESCs) in 1998 and human-induced pluripotent stem cells (hiPSCs) in 2007 led to a breakthrough in the treatment of retinal disease. Both cell types can be expanded endlessly in vitro and used to produce photoreceptors and RPEs. Creating different types of neurons from RPCs in a spatiotemporal-specific manner is necessary for developing the mammalian retina. RPC proliferation, cell-fate commitment, and particular neuronal differentiation are required for RPC progression during retinogenesis.
Organoid technologies derived from pluripotent stem cells have created new opportunities for preclinical basic science research, drug discovery, and organ transplantation therapy [71]. RtOgs, which can be produced from hESCs or iPSCs, are self-organized tissues that mimic in vivo retinal development [72]. Single-cell RNA research has revealed that RtOg cellular makeup and lamination at various developmental stages mirror those found in vivo [73]. According to the scRNA-seq analysis of RtOgs from two distinct developmental stages (D57 and D171), RPCs (51%), RGCs (21%), transition phase 1 cells (12%), and photoreceptor progenitor cells (7%) comprised the majority of the young age group (D57); and bipolar cells (21%), photoreceptor cells (20%), and RPCs (24%) were seen in D171, with rods and cones comprising 25% and 55% of the photoreceptors [74]. Potential models for retinal degeneration have been identified as human pluripotent stem cell (hPSC)-derived RtOgs [75]. Sridhar et al. [76] evaluated how well hPSC-derived RtOgs perform compared to the fetal retina and demonstrated that despite the relative disorganization of the inner retina, RtOgs accurately recapitulate the timing of retinogenesis of the fetal retina, with cellular composition and gene expression comparable across a wide range of developmental stages. Following the development of protocols to generate 3D RtOgs from hESCs/hiPSCs, it was found that numerous retinal cell types are generated along with structures resembling the developing eye and laminated retina [77]. Mao et al. [78] dissected the temporal development of RPCs in the early stages of human retinogenesis by performing scRNA-seq on cells isolated from 3D RtOgs derived from hESCs. Chromatin remodeling and genes involved in Notch and Wnt signaling pathways were dynamically regulated during RPC commitment, and ASCL1 was co-expressed with cyclin D1, a G1 phase cell cycle regulator, in a cell cycle-independent manner, which plays a role in promoting early retinal neurogenesis by temporally controlled overexpression. Despite these rapid developments, it has not yet been fully realized how to precisely define and characterize the cells that develop within these organoids over time [79]. High-throughput integrated fluidic circuits, with a capacity of 800 capture sites, were used in this study for scRNA-seq to evaluate hESC-derived RtOgs throughout differentiation [80]. Collin et al. [81] differentiated an hESC (H9) cell line into RtOgs and used the Fluidigm C1 Single-Cell mRNA-Seq HT IFC to dissociate and partition the samples into single cells, which were then processed for scRNA-seq at 60, 90, and 200 days. This study demonstrated the viability and capability of scRNA-seq to analyze the inherent complexity of RtOgs and the orderly emergence of important retinal cell types therein, which recapitulates the order of retinal development.
The scRNA-seq technique has facilitated the in-depth study of retinal disorders, including the identification of eye cell type-specific markers; the classification of retinal cell types and subtypes at the transcriptome level; and the discovery of novel, uncommon types, and subtypes. Furthermore, it has shed light on the differences between individual cells, shown how the retina develops at various developmental stages, shown the molecular link between disease states and cellular responses, and identified particular cell types linked to various retinal diseases. However, using single-cell sequencing technology has its challenges. (1) The resulting single-cell suspension must accurately represent the cell composition of the original tissue to be considered representative. Given the difficulty in obtaining human eye samples and the possibility of animal differences affecting the cell type ratio, this could be difficult. (2) MicroRNA and other regulatory RNAs are not typically captured by scRNA-seq because it only detects polyadenylated RNA. The last issue is the difficulty of effectively separating individual cells from biological samples and analyzing vast amounts of sequencing data. A powerful detection method to identify the gene regulatory networks during cell development and differentiation can be produced by the close coupling of scRNA-seq and bioinformatics technology. (3) During tissue digestion and processing, immune cells such as microglia can become activated and may skew the gene expression profiles of cells. Thus, it is important to carefully consider the potential effects of immune cell activation when interpreting scRNA-seq data. Furthermore, techniques such as fluorescence-activated cell sorting or magnetic-activated cell sorting can be used to isolate specific cell populations and minimize potential immune cell activation during tissue processing. (4) Endothelial cells are indeed an important component of the retinal vasculature and play a critical role in retinal function and disease. Unfortunately, they constitute a relatively minor population within retinal tissue and are often lost during tissue processing. To address this, researchers have developed methods to enrich for retinal endothelial cells, such as using magnetic beads coated with endothelial cell-specific antibodies. These enrichment techniques can improve the sensitivity and accuracy of scRNA-seq analysis for endothelial cells in the retina. It is important to carefully consider the potential effects of tissue processing and cell isolation methods when designing scRNA-seq experiments in retinal biology. By addressing these challenges and optimizing protocols, researchers can obtain more accurate and comprehensive insights into the gene expression profiles of retinal cells, including immune cells and endothelial cells. Single-cell transcriptome sequencing technology is constantly improving, it is anticipated that the application of scRNA-seq will change from describing tissue heterogeneity to figuring out the mechanism onset and progression, which may be quickly incorporated into research on eye-related diseases in the coming years, such as assessing gene expression changes during stem or precursor cell differentiation into eye tissue; determining the mechanisms causing immune rejection after intraocular stem cell transplantation; and evaluating the pathogenesis, specific treatment, and prognostic assessment of various ocular tumors. Future work should focus on making scRNA-seq a more comprehensive, high-throughput, affordable, and simple-to-use tool for clinical research.
YZ, HL, SX and DL—Substantial contributions to the conception and design of the work; the acquisition, analysis, and interpretation of data for the work. SX and DL—Drafting the work or reviewing it critically for important intellectual content. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.