
- Academic Editor
†These authors contributed equally.
Background: Ferroptosis is a form of iron-dependent regulated cell
death, and prior work has highlighted the potential utility of
ferroptosis-inducing agents as tools to treat heart failure (HF). To date,
however, no detailed examinations of the prognostic utility of
ferroptosis-related genes (FRGs) in HF have been conducted.
Methods: We used established genomic identification of FRGs for
total samples in the gene expression omnibus (GEO) database, screened for differentially expressed FRGs,
performed protein-protein interaction analysis and functional analysis of HF
immune microenvironment subtypes. Subsequently, we applied tools to calculate
immune cell infiltration, compare immune cell, immune response genomic and
HLA gene differences between subtypes, and perform candidate drug
identification. Finally, preliminary in vivo validation of the screened
central genes was performed in animal models. Results: FRGs
were compared between samples from HF and healthy control donors, revealing 62 of
these genes to be differentially expressed as a function of HF status. HF
patient-derived tissues exhibited significant changes in the expression of
HLA genes, increase immune cell infiltration, and higher levels of other
immune-related genes within the associated immune microenvironment. These FRGs
were then leveraged to establish two different immune-related subtypes of HF
based on clustering analysis results, after which these subtypes were
characterized in further detail. Functional enrichment analyses revealed the
identified differentially expressed genes to be enriched in key immune-related
pathways including the primary immunodeficiency, natural killer cell-mediated
cytotoxicity, Fc
Heart failure (HF) remains a major threat to global public health, affecting approximately 64.3 million individuals and necessitating consistent and systematic management to ensure optimal patient outcomes [1]. Measurements of key circulating biomarkers have been shown to be strongly correlated with the clinical prognosis of HF patients [2], and international guidelines have recommended the use of a range of biomarkers as guides to gauge HF patient outcomes including N-terminal pro-B type natriuretic peptide (NT-proBNP) and high-sensitivity troponin T (hs-cTNT) [3]. Traditional biomarker assays are inexpensive and straightforward, enabling the routine evaluation of patients with the goal of cost-effectively identifying individuals at high risk without the need for invasive procedures or expensive instrumentation, thereby allowing these individuals to receive further personalized care [4]. The establishment of a more detailed understanding of key biomarkers associated with HF may represent a means of more rationally and effectively facilitating patient risk stratification.
Ferroptosis is a form of regulated cell death that differs from apoptotic or necrotic processes [5], occurring due to the iron-mediated accumulation of lipid peroxides within affected cells [6]. The induction of ferroptosis holds promise as a therapeutic strategy in certain diseases, including in HF patients that have responded poorly to more traditional forms of clinical management [7, 8, 9]. Prior work has revealed a range of genes that regulate ferroptosis in positive or negative manners [10, 11], but the relationships between these different genes and the prognosis of HF patients has yet to be clarified. Efforts to more clearly define the molecular etiology of ferroptosis-related genes (FRGs) in the pathogenesis of HF may thus aid in clarifying the heterogeneous phenotypes observed among HF patients, thereby revealing new opportunities to provide affected individuals with more efficacious care.
For the present study, a series of analyses of gene expression profiles and microarray-derived data were used to compare transcriptomic differences between HF patients and healthy controls with the goal of defining key FRGs that are differentially expressed as a function of HF status and to define hub biomarkers of HF-related immunoregulation. Together this research effort will advance current understanding of the molecular factors that drive HF at the systems biology level.
Microarray data used for this study included the GSE57338 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57338), which included 313 total samples (177 diseased samples, 136 control samples). FRGs were identified using established gene sets including PMID: 33867820 (n = 103), PMID: 33767582 (n = 386), PMID: 32760210 (n = 60), and the FerrDb database (n = 369), yielding 449 total FRGs.
The 449 identified FRGs were used to generate a protein-protein interaction network (https://string-db.org), with the MCC algorithm in the CytoHubba v0.1 (New York City, NY, USA) plug-in being used to select hub genes from this network within the Cytoscape application for further downstream analyses.
Functional enrichment analyses for different HF immune-related subtypes were
assessed based on enrichment scores for particular gene sets in individual
samples as determined with a non-parametric unsupervised gene set variation
analysis (GSVA) approach with the R GSVA v1.38 package (Bioconductor, Boston, MA, USA), which enables the
transformation of gene expression data from individual genes in a characteristic
expression matrix. Differential expression analyses were then performed with the
R limma package (Bioconductor, Boston, MA, USA), with a false discovery rate (FDR)
There are several tools to calculate immune cell infiltration, such as CIBERSORT, XCELL, SSGSEA, TIMER, etc. To avoid the limitations of the database and to improve the accuracy of the data analysis results, we applied here at least three tools to calculate immune cell infiltration, comparing immune cells between subtypes, immune response gene sets, and HLA gene differences.
Hub modules of interest were introduced into the DGIdb database to facilitate the identification of candidate gene-drug interactions, with the ggplot2 v3.3.4 R package (Hadley Wickham, London, UK) being used to generate a Sankey map with geom alluvium as a means of highlighting the associations between hub genes and drug candidates.
Primary antibodies specific for HDAC1, LNPEP, PSMA1 and PSMA6 were from ABclonal
(A19571, A11959, A3460, and A2188), while antibodies specific for
C57BL/6 mice (specific pathogen-free 8-week-old males, 20–25 g; Experimental
Animal Center of Nantong University) were housed under standard conditions (21
Over 80% of the mice in both surgical groups survived, with comparable rates in both groups. Surviving mice underwent echocardiographic assessment 8 weeks after surgery, followed by further experimentally appropriate examination.
Following isoflurane-mediated anesthetization, mice were fixed in the supine position on an operating table. The skin of the anterior thoracic region was then prepared, coated with coupling agent, and a high-resolution small animal ultrasound imaging system (Vevo 2100, Visual Sonics, Canada) was used to conduct echocardiographic analyses of these animals using the FMS-250 probe (depth: 2.0–2.5 cm, frequency: 13–24 MHz). Under M-mode ultrasonographic visualization of cardiac structures in these animals, parameters measured using the left ventricular (LV) short-axis and parasternal LV long-axis views included LV internal diastolic diameter (LVIDd), LV internal diameter end-systole (LVIDs), and interventricular septal dimension in diastole (IVSd). LV systolic function-related indices were then computed based upon these values over an average of 3–5 cardiac cycles, including ejection fraction (EF) and fractional shortening (FS).
Mouse hearts were collected and fixed in 4% paraformaldehyde for 48 hours and embedded in paraffin. Next, the tissue was cut into 5 mm cross-sectional sections. The sections were stained with hematoxylin-eosin (HE) to observe the histopathological changes in the myocardium, WGA staining to measure the cross-sectional area of cardiomyocytes, and Masson staining to evaluate the extent of interstitial fibrosis in the mouse myocardium.
After collection, cardiac tissue samples were fixed for 10min with 1% glutaraldehyde, followed by further fixation for 1 h at room temperature using 3% glutaraldehyde. Samples were then further fixed for an additional 1 h at room temperature using 1% osmium tetroxide in 0.1 mol/L dimethylarsinate buffer, followed by embedding in resit. Polymerized ultrathin sections were then stained using uranyl acetate and lead nitrate, followed by TEM imaging to assess mitochondrial morphology and transverse muscle arrangement.
Cells were treated as above, and a bicinchoninic acid (BCA) kit was used to
detect protein concentrations in sample lysates. A ferrous ion colorimetric test
kit (E-BC-K773-M, Elabscience, Wuhan, China) was then used to detect iron ion levels based on provided directions.
Briefly, the provided probe bound to ferrous ions released by cells, with the
resultant absorbance at 593 nm being quantified to measure intracellular Fe
After cells had been harvested and lysed, a BCA kit was used to measure protein concentrations. MDA levels in these cells were assessed using an MDA assay kit, with absorbance at 530 nm ultimately being measured. Similarly, GSH levels in these cells were analyzed based on the directions provided with a micro-reduced GSH assay kit. Absorbance was assessed at 405 nm, and GSH levels were established based on a standard curve.
RNA was isolated from cardiac tissue samples and cardiomyocytes with Trizol
(Servicebio, Wuhan, China). HiScript II Q RT SuperMix (Vazyme, Nanjing, China)
was used to prepare cDNA, after which ChamQ Universal SYBR qPCR Master Mix
(Vazyme, Nanjing, China) and a fluorescent qPCR instrument (Bio-rad; CFX, CA, USA)
were used for qPCR analyses. Primers used for this study (RiboBio, Guangzhou,
China) had the following sequences: GAPDH: (F: CCTCGTCCCGTAGACAAAATG, R:
TGAGGTCAATGAAGGGGTCGT); HDAC1: (F: TCATAAATACGGAGAGTACTTCCCA, R:
CGTCTCGCAGTGGGTAGTTCAC); LNPEP: (F: CAAACCAGTCAGCAGAACTCATC, R:
TAGAGGAATAATGGCAGTGGGAAG); PSMA1: (F: CGGGTCTAACTGCTGATGCC, R:
CATATCGCTGTGTTGGGATCTG); PSMA6: (F: GCAGAGATTGACGCTCACCTT, R:
GGCCTCTATGATGTCAGTTTGTTG). GAPDH was used as a control to normalize gene
expression, with relative expression being measured using the
2
A protein extraction reagent was used to isolate proteins from tissue or cell samples, after which a BCA protein analysis kit (Servicebio, Wuhan, China) was used to measure protein concentrations. Proteins were then separated via 8% SDS-PAGE (Servicebio, Wuhan, China), transferred to polyvinylidene difluoride (PVDF) membranes, and blots were then blocked for 2 h at room temperature with 5% non-fat milk prior to incubation overnight with appropriate primary antibodies at 4 °C. Blots were rinsed with TBST three times, then probed with secondary antibodies for 2 h. Chemiluminescent reagents were then used to detect protein bands, followed by quantification using ImageJ V1.8.0 (National Institutes of Health, Bethesda, MD, USA).
Data are reported as means
To gain initial insight into the relationships between ferroptosis and HF, a comprehensive set of 449 FRGs with corresponding genomic location information was established (Fig. 1A). Protein-protein interaction analyses were also conducted for these FRGs (Fig. 1B). The relationship between these FRGs was then examined by comparing all samples in the selected microarray dataset to samples from HF patients (Fig. 1C). Differential FRG expression between healthy control individuals and HF patients was then compared using established cutoff criteria, revealing 62 differentially expressed FRGs of which 42 and 20 were respectively upregulated and downregulated in HF samples (Fig. 1D–F, Supplementary Table 1).
 Fig. 1.
Fig. 1.Ferroptosis-related genes (FRGs) landscape in heart failure. (A) Genomic position configuration of 449 FRGs. (B) Protein-protein interactions of 449 FRGs. (C) Differential FRG expression correlations (Expression correlation analysis in all samples (top) and in disease samples (bottom), scatter plots showing expression of positively correlated FRGs in different samples). (D) volcano plot showing up-or down-regulation of FRGs in disease compared to control. (E) Box plot showing significant differences in genes in the samples. (F) Heat map showing the expression of FRGs in all samples.
The immunological microenvironment of HF-related patient samples was next
characterized. Analyses of the HLA expression profiles in these samples revealed
significant downregulation of most HLA genes in HF samples relative to healthy
tissues (Fig. 2A). Correlations between these HLA genes and the 62 identified
differentially expressed FRGs were also examined (Fig. 2B). Immune infiltration
analyses performed through three computational approaches revealed higher levels
of B cell and CD8
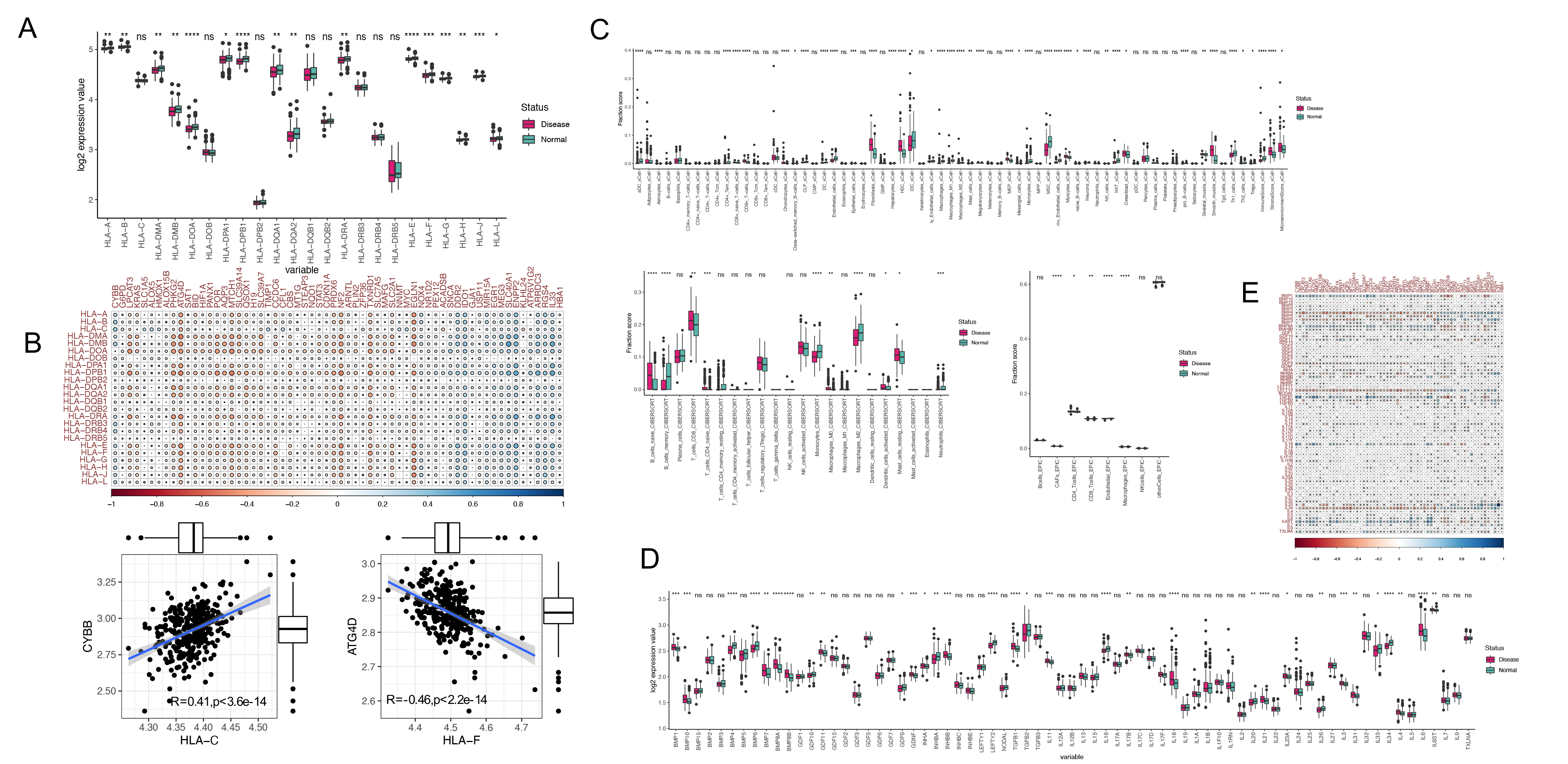 Fig. 2.
Fig. 2.The role of FRGs in the immune regulation in heart
failure. (A) Expression levels of HLA-related genes in disease and control
samples. (B) Correlation analysis of HLA gene expression with FRGs in disease.
(C) Immune cell infiltration analysis by XCell, CIBERSORT, and EPIC methods. (D)
Expression levels of immune-associated genes in disease and healthy samples. (E)
Correlation analysis of immune-associated genes and FRGs in disease samples (ns,
not significant, *:
A consensus clustering strategy was next used to group HF patient samples based on the 62 identified HF-associated FRGs (Fig. 3A). Samples were separated into two groups based on the fact that the strongest intra-group and weakest inter-group connections were evident at a clustering variable (k) value of 2 in generated CDF curves (Fig. 3B,C). A subsequent principal component analysis (PCA) indicated that these two HF subtypes were distinct from one another based on gene expression profiles (Fig. 3D). Differences in the 62 HF-associated FRGs were also compared between these two subtypes of HF (Fig. 3E,F).
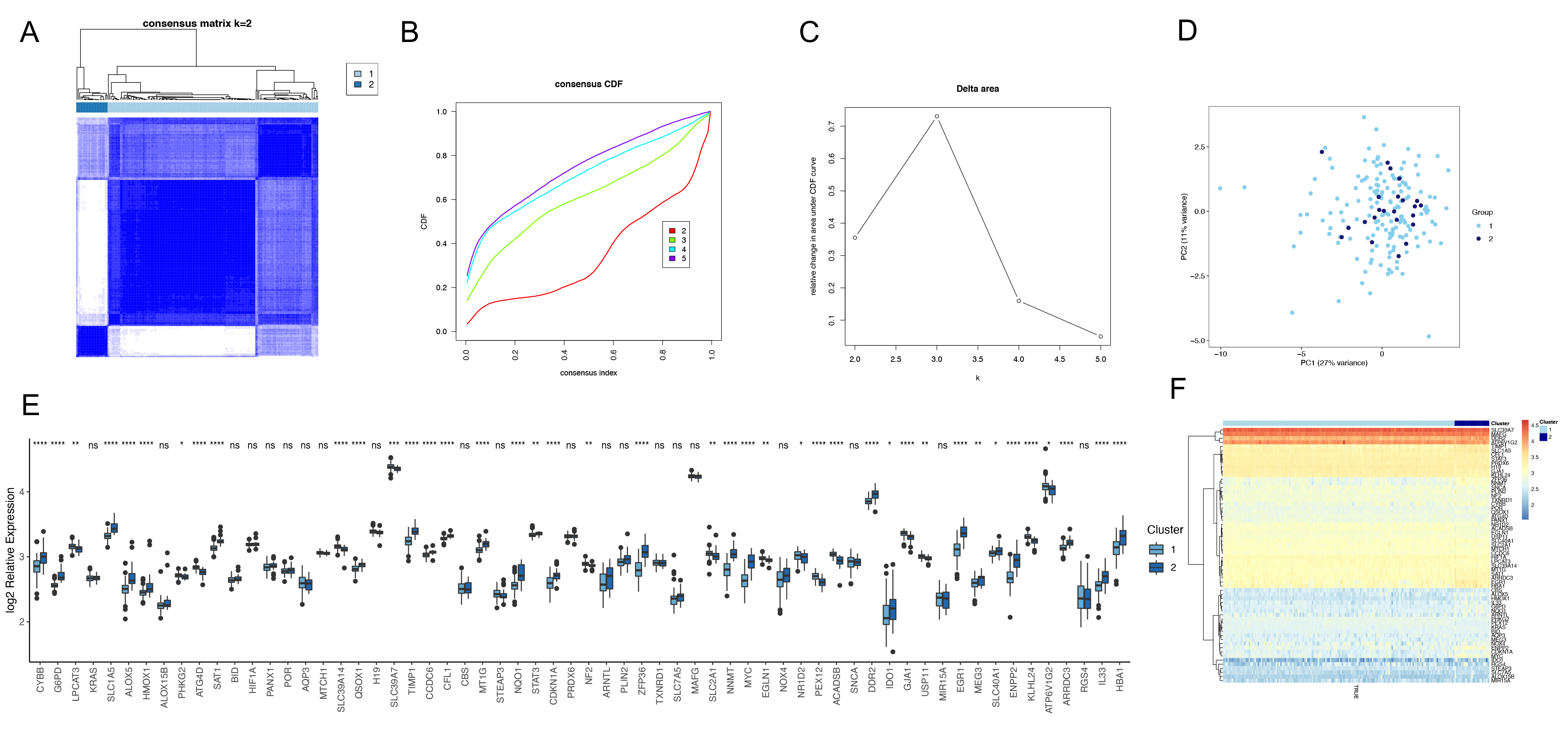 Fig. 3.
Fig. 3.Identification of FRGs-related molecular subtypes in
heart failure. (A) Heat map showing the results of consistent cluster analysis
for subtype identification. (B) The CDF value of consensus index. (C) Relative
change in area under CDF curve for k = 2–5. (D) Principal component analysis
showing the clustering of each disease sample according to subgroup groupings.
(E) Box plot showing the distribution of FRGs expression according to subgroups,
with the x-axis representing FRGs and the y-axis the logarithm of relative
expression. (F) Heat map showing the expression of FRGs according to subgroups.
(ns, not significant, *:
To clarify the biological differences between these HF patient clusters, a GSVA
enrichment analysis was conducted for each of these subtypes. KEGG pathway
enrichment analyses revealed that relative to samples in Cluster 1, those in
Cluster 2 were more closely associated with immune-associated pathways including
the primary immunodeficiency, natural killer cell-mediated cytotoxicity,
Fc
 Fig. 4.
Fig. 4.Functional analysis of different molecular subtypes. (A) KEGG. (B) Gene Ontology.
The immunological microenvironment associated with these established HF patient subtypes was next explored by using three algorithms to assess immune cell infiltration in these patient samples. This approach revealed increased immune cell infiltration in samples from Subtype 2 relative to Subtype 1 (Fig. 5A). Moreover, Subtype 2 samples exhibited significant increases in the expression of immune-related genes including IL-6, IL-26, IL-18, TGFB1, and TGFB3 (Fig. 5B). These results highlight key differences in immune cell infiltration among these ferroptosis-associated HF patient subtypes, underscoring the close interplay between ferroptotic processes and the HF-related immune microenvironment.
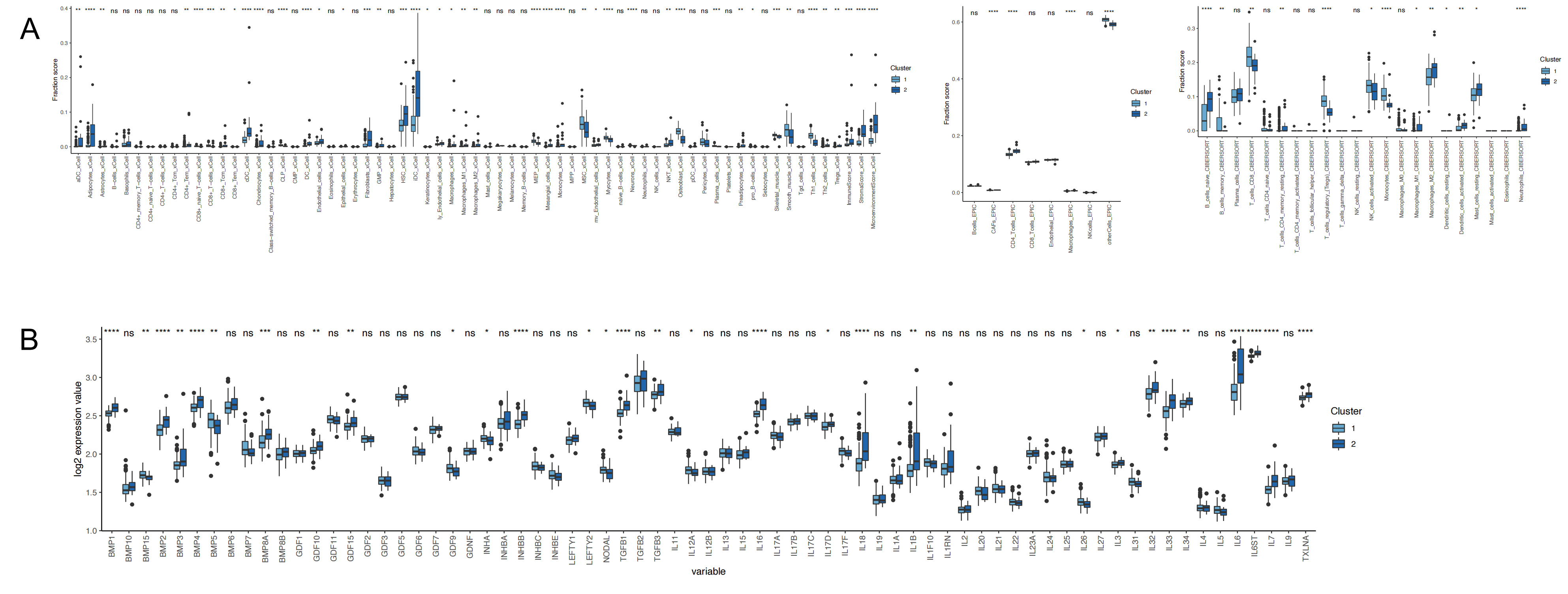 Fig. 5.
Fig. 5.Immune microenvironment of different molecular
subtypes. (A) Analysis of immune infiltrating cells between subtypes. (B)
Differences in expression of immune response genes between subtypes, ns: not
significant, *:
A co-expression network was next developed based on the identified
differentially expressed genes (DEGs) and HF patient cluster phenotypes in an
effort to define gene modules significantly associated with these clusters.
Differential gene expression analyses were initially performed among these
clusters with established significant thresholds, revealing genes and functions
that were distinct among these clusters (Fig. 6A–C). Interactions between these
DEGs were then examined through the construction of a co-expression network (Fig. 6D,E). The DynamicTreeCut method revealed three total co-expression modules (Fig. 6F), and the modules that were most highly correlated were filtered based on
cluster information. For example, the blue module from Cluster 2 was most
strongly correlated with the turquoise module from Cluster 1 (Fig. 6E). Given the
large numbers of genes in these modules, they were further assessed to identify
hub genes of interest by calculating module feature vectors and their
associations with genes to define module membership (MM) and gene significance
(GS). In total, 111 hub genes in Cluster1 and 32 hub genes in Cluster2 were
identified based on established criteria (GS
 Fig. 6.
Fig. 6.Weighted Coexpression Network Construction and Identification of Hub genes. (A) Volcano plot showing the differential genes between clusters. (B,C) Functional analysis between two clusters. (D) Clustering of samples and removal of outliers. (E) Analysis of network topology for various soft-thresholding powers. (F) Cluster dendrogram of genes in all patients. Each branch in the figure represents one gene, and each color represents one coexpression module. (G) Correlation between the gene module and clusters. (H) The scatter plot of Gene significance vs. Module membership in the turquoise co-expression module. (I) The scatter plot of Gene significance vs. Module membership in the blue co-expression module.
Next, DGIdb was utilized to identify candidate drugs and compounds with the potential to treat HF based on the hub genes identified above. Accordingly, gene-drug interaction information was used to establish a protein-protein interaction network, with hub nodes in this network then being leveraged to determine candidate drugs of interest (Fig. 7A,B). In total, 27 drug candidates associated with 4 hub genes (HDAC1, LNPEP, PSMA1, and PSMA6) were identified (Fig. 7C). Of these compounds, 21 were predicted to interact with HDAC1, including abexinostat, an-9, apicidin, belinostat, cudc-101, and dacinostat.
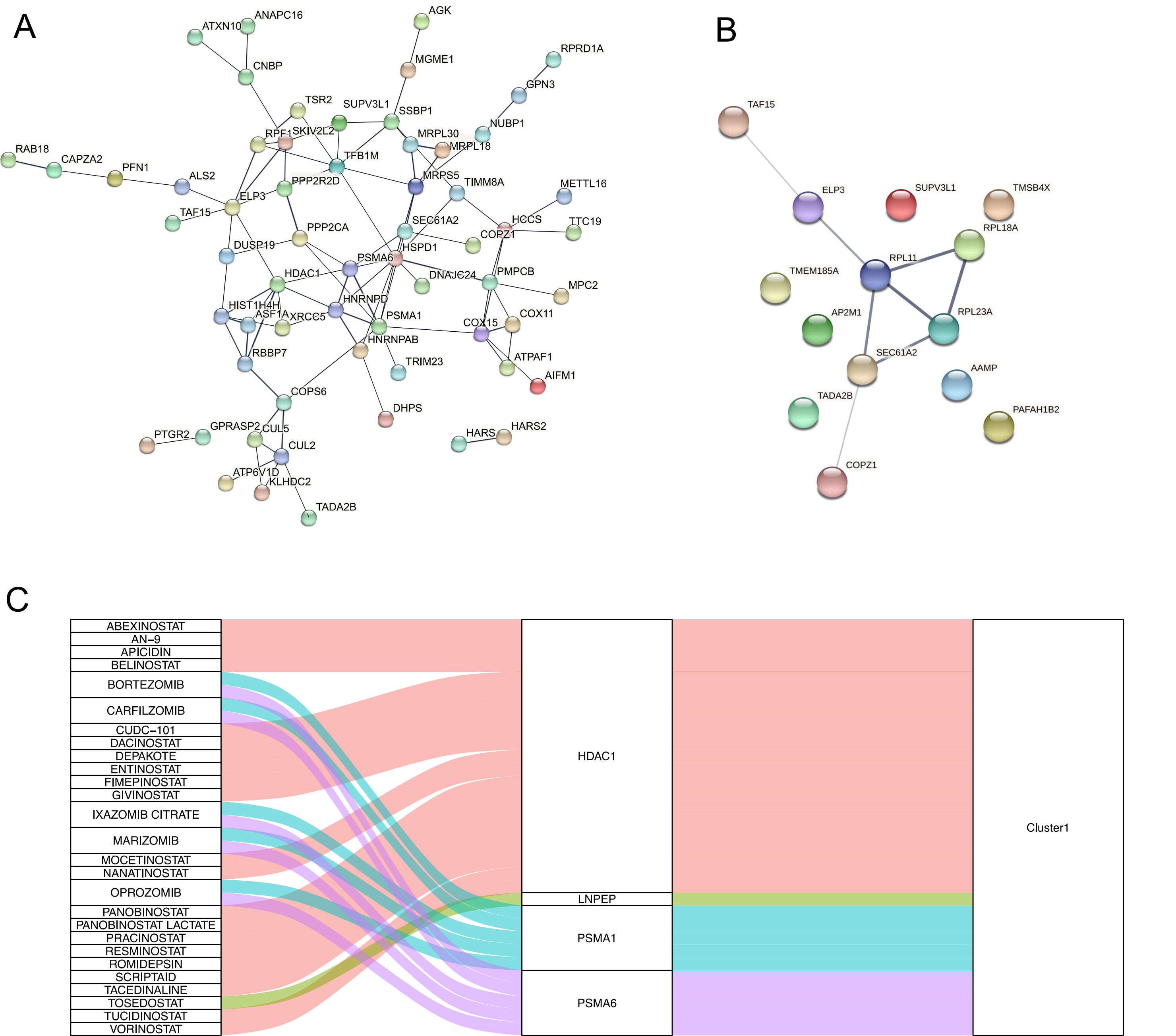 Fig. 7.
Fig. 7.Relationships between core genes and potential drugs. (A) Protein–protein interaction (PPI) network diagram of Cluster1 hub gene. (B) PPI network diagram of Cluster2 hub gene. (C) Sankey diagram showing the potential drugs corresponding to each cluster hub core gene.
To confirm the in vivo relevance of the above findings, an in vivo model of HF was established by performing TAC surgery on C57BL/6 mice, with echocardiographic analyses of these animals being performed at 8 weeks post-surgery (Fig. 8A). These analyses revealed that LV EF and LV FS were significantly reduced in mice in the TAC group relative to the sham control group (Fig. 8B), while the IVSd, LVIDd, and LVIDs were increased (Fig. 8C). Heart volume was also increased significantly in mice in the TAC surgery group (Fig. 8D). HE staining results also showed myocardial hypertrophy in the TAC group of mice, with a large number of disorganized cardiomyocytes, significantly increased cytoplasm, and extensive inflammatory cell infiltration (Fig. 8D). Meanwhile, WGA staining demonstrated a significantly larger ventricular cross-sectional area in TAC mice (Fig. 8E). Subsequently, we assessed myocardial interstitial and perivascular fibrosis in ventricular tissue in Masson-stained sections. As expected, TAC surgery promoted the progression of cardiac fibrosis (Fig. 8F). These results confirmed successful HF model establishment.
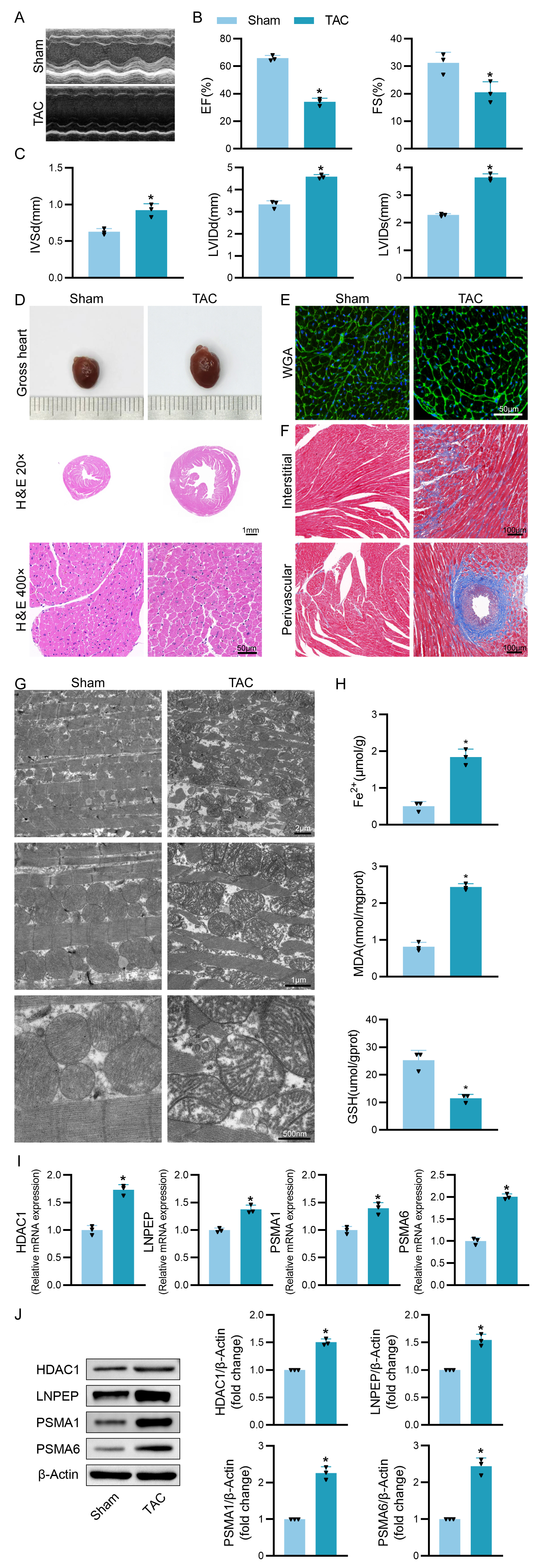 Fig. 8.
Fig. 8.Validation of the four hub genes expressions in animal models.
(A–C) Mice in the indicated groups underwent M-mode echocardiographic imaging to
assess left ventricle parameters in the long axis of the left parasternal
sternum. Analyzed parameters included ejection fraction (EF), fractional
shortening (FS), left ventricular internal diastolic diameter (LVIDd), left
ventricular internal diameter end-systole (LVIDs), and interventricular septal
dimension in diastole (IVSd). (D) Representative of gross cardiac morphology and
H&E-stained cross-sections (20
Based on the successful establishment of this model, we observed the changes of mitochondria in cardiomyocytes of TAC mice using transmission electron microscopy. The results showed that compared with the Sham group, the mitochondria in the cardiomyocytes of mice after TAC were apparently smaller, disordered in arrangement, with indistinct cristae and ruptured outer membranes (Fig. 8G). Meanwhile, we measured the ferrous ion content and the expression levels of ferroptosis -related factors MDA and GSH, and the results showed that the mice in the TAC group had obviously higher ferrous ion content and significantly increased MDA levels, while GSH levels were significantly lower (Fig. 8H), which further confirmed the close association between HF and ferroptosis.
Subsequent qPCR analysis showed that mRNA expression of HDAC1, LNPEP, PSMA1 and PSMA6 differed significantly between these two groups of mice (Fig. 8I). Western blot analysis also showed differences in the expression of these four genes at the protein level, which was consistent with the results of the bioinformatics analysis described above (Fig. 8J). All of these evidences support a possible relationship between immune microenvironment-related ferroptosis and HF.
Cardiovascular diseases (CVDs) are the leading cause of death worldwide, and heart failure (HF) is the end stage of various CVDs with poor prognosis [1, 2]. There is a considerable amount of evidence to support that MicroRNAs and phytochemicals have emerged as attractive therapeutic directions in CVDs [12, 13], but therapeutic approaches for HF still need to be further explored. Ferroptosis is a recently defined and increasingly important type of regulated cell death that occurs in an iron-dependent manner and influences a range of pathogenic conditions including HF and other types of cardiovascular disease such as arrhythmia, diabetic cardiomyopathy, myocardial ischemia-reperfusion injury, sepsis-related cardiomyopathy, and atherosclerosis [14, 15]. Investigating the underlying mechanisms involved in regulating ferroptosis in cardiomyocytes may be an effective therapeutic approach for the treatment of HF.
The goal of treatment efforts for HF patients is the improvement of ventricular function and associated symptoms in an effort to ameliorate long-term morbidity and mortality [16]. Many factors have been used to guide HF diagnosis, and echocardiographic parameters and BNP levels are among the best-studied biomarkers in this context [17]. The advent of new high-throughput sequencing technologies and other forms of so-called “big data”, however, has provided new opportunities to define key molecular correlated for HF development. The immune microenvironment is thought to play a pathological role in the onset of HF [18]. Environmental risk factors can increase the production of reactive oxygen species caused by oxidative stress, which in turn induces ferroptosis in cells [19], and the close relationship between the immune microenvironment and ferroptosis has been demonstrated in many fields, including oncology. As such, this study was developed to explore correlations between ferroptotic cell death and HF in an effort to provide a new foundation for the immunotherapeutic treatment of patients suffering from this condition.
Here, possible relationships between HF and ferroptosis were explored by
comparisons of HF patient and control tissue samples, ultimately leading to the
identification of 62 FRGs that were differentially expressed in HF samples (41
upregulated, 20 downregulated). Analyses of the immune microenvironment in these
samples revealed the significant downregulation of most HLA genes in HF patient
samples with corresponding increases in B cell and CD8
Using DGIdb, candidate drugs capable of treating HF that targeted the HDAC1, LNPEP, PSMA1, and PSMA6 hub genes were also identified, offering promising tools that may aid in the future clinical management of affected patients. Several previous studies have shown that HDAC1, LNPEP, PSMA1 and PSMA6 are closely associated with ferroptosis and heart failure. Zhang et al. [20] found that Yixinshu capsules improve cardiac hypertrophy by modulating the RB/HDAC1/GATA4 signaling pathway based on proteomics and mass spectrometry image analysis. Meanwhile, it has been indicated that CaMKII can exacerbate the progression of heart failure by activating class I HDAC [21]. Overexpression of PI16 reduced nuclear levels of HDAC1 after angiotensin II treatment, and overexpression of PI16 prevented cardiac hypertrophy and fibrosis by inhibiting stress-induced CF activation [22]. In addition, Huang et al. [23] identified ferroptosis in glomerular intrinsic cells of diabetic nephropathy patients with a potential key candidate gene, ALOX15, which may be regulated by HDAC1 and E3 ligase-related regulation. Emerging evidence suggests that differentially expressed protein and WGCNA analysis identified 18 upregulated proteins in non-ischemic dilated cardiomyopathy (DCM), including PSMA3 and PSMA6 [24].
To confirm the in vivo relevance of the above findings, an in vivo model of HF was established by performing TAC surgery on C57BL/6 mice. Our results revealed that LVEF and LVFS were significantly reduced in mice in the TAC group relative to the sham control group, while the IVSd, LVIDd, and LVIDs were increased. Heart volume was also increased significantly in mice in the TAC surgery group. The same was demonstrated for HE and WGA staining, while Masson staining verified significant fibrosis of cardiomyocytes in mice with heart failure. These results confirmed successful HF model establishment. Numerous studies have shown that ferroptosis plays an important role in the development of HF [4]. Excess intracellular iron can trigger ferroptosis by increasing reactive oxygen species levels through the Fenton reaction, promoting lipid peroxidation, and producing oxidation products that are highly toxic to cells, such as malondialdehyde (MDA). Secondly, it has been shown that excess intracellular free iron can enter mitochondria and promote mitochondrial oxidative stress, leading to cardiac mitochondrial dysfunction and GSH depletion. Consistent with these findings, we detected typical cardiomyocyte ferroptosis phenotypes such as elevated MDA and ferrous ion concentrations, decreased GSH expression and smaller mitochondrial volume and mitochondrial cristae crinkling in TAC-induced heart failure mice.
Subsequent qPCR and western blot analysis revealed that HDAC1, LNPEP, PSMA1and PSMA6 expression differed significantly between these two groups of mice in line with the above results from bioinformatics analyses, again supporting a possible relationship between ferroptosis and HF. It also provides a rationale for its use as a potential heart failure-related biomarker, which will likely be a therapeutic strategy to stop the progression of heart failure in the future. This study also has some limitations. It simply verified the changes in gene expression. However, their specific functional validation and mechanism of action still need further study.
In summary, the present study offers new foundational insights regarding the relationship between ferroptosis and HF. To our knowledge, these analyses represent a new direction to investigate the link between the immune microenvironment and ferroptosis in heart failure patients, and the resulting data will drive future efforts to better treat this debilitating disease and improve patient survival and quality of life.
The importance of ferroptosis as a regulator of the immune microenvironment associated with HF was confirmed by bioinformatics analysis between ferroptosis and the immune microenvironment. Four related central genes, HDAC1, LNPEP, PSMA1 and PSMA6, were identified to provide new strategies for individualized treatment of HF.
The original data supporting the conclusions of this paper will be provided by the authors without reservation.
JZ, JS and PX performed the main experiments. GW, CW and JL contributed to collection and assembly of data. HS and WY performed statistical analysis of all data. QL contributed to conception, supervision, administration, and validation of this project. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors reviewed the manuscript.
All experiments involving animals were approved by the Animal Ethics Committee of Nantong University. (Permit Number: S20220310-010).
Not applicable.
This work was supported by the grants from the Nantong Science and Technology Project (No. JC2021140).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.