1. Introduction
Podocytes are terminally differentiated epithelial cells located on the medial
side of the Bowman’s capsule [1]. The nucleus of a podocyte is located in the
center of the cell, which has a large cell body. Foot processes (FPs) are
irregular protrusions extending from the cell body [2]. The basal area of the FP
adheres to the glomerular basement membrane (GBM). In addition, adhesion
molecules and connexins exist between individual FPs, thereby forming a porous
structure known as the slit diaphragm (SD) [3]. The unique morphological and
structural characteristics of podocytes are essential for the maintenance of the
glomerular filtration barrier function. In a variety of pathologic conditions,
the structure and morphology of podocytes are affected, and the glomerular
filtration barrier is impaired, leading to the occurrence of podocyte-related
diseases [4].
Acting as an important second messenger for cells, calcium (Ca)
participates in and regulates various cellular activities, including cell
apoptosis and proliferation [5]. Cellular and extracellular Ca are in
dynamic balance, and calcium-sensing receptors (CaSRs) can sense and regulate
changes in Ca concentrations [6]. At the same time, the strict regulation
of intracellular Ca is inseparable from the expression and activity of
various channel proteins. Ca signals transmitted by channel proteins are
essential for maintaining podocyte function [7]. Ca signaling is required
for podocyte morphogenesis and FP formation [8]. Normal Ca signaling can
maintain the structure of podocytes by stabilizing cytoskeletal proteins [9, 10].
In addition, downstream signaling mediated by Ca can regulate the
autophagy balance in podocytes and participate in cell survival [11]. The
function of podocytes is critically dependent on the processing of intracellular
Ca signaling.
In the pathological states of the podocyte, enhanced Ca signaling in
podocytes is observed [12]. Changes in Ca transport and signaling are
related to the occurrence and development of various podocyte-related diseases
[9, 13, 14]. Further understanding of the status of Ca signaling in
podocytes could provide promising insights for the treatment of podocyte-related
diseases. This review summarizes the current studies on Ca sensing,
Ca channels, and the Ca-signaling pathway in podocytes, as well as
the status and targets of the Ca-signaling pathway in podocyte-related
diseases.
2. Ca Sensing and Transport in Podocytes
2.1 Ca Sensing
In mammals, Ca homeostasis is mainly mediated by CaSRs [6]. In 2008,
Piecha et al. [15] first described the presence of CaSRs in podocytes.
The podocyte-specific CaSR knockout model suggested that the CaSR represented an
important receptor in the maintenance of glomerular filtration barrier function
[16].
A CaSR is a G-protein-coupled receptor (GPCR), which encodes a gene that is
located on chromosome 3 in humans. CaSRs can be activated by type I and type II
agonists. Type I agonists include divalent ions, such as Ca and magnesium,
-amyloid peptide, and polycation, which directly stimulate the CaSR.
Type II agonists are a class of regulatory substances, including spermidine,
spermine, aromatic amino acid residues, extracellular pH, and ionic strength. In
the presence of extracellular Ca, type II agonists increase the Ca
affinity of the CaSR by allostery [6]. The CaSR activates the downstream
signaling cascade through coupling with heterotrimeric guanine nucleotide-binding
proteins (G proteins), such as the Gi/o, G12/13, and Gq/11 families [17]. An
increase in Ca stimulates the activity of Gq/11 and phospholipase C (PLC),
inducing the accumulation of inositol 1,4,5 triphosphates (IP3). Subsequently,
IP3 attaches to the endoplasmic reticulum (ER), via the IP3 receptor, and induces
Ca influx via the storage manipulation channel located on the plasma
membrane, which results in Ca being released from the intracellular stores
[18]. In podocytes, the activated CaSR can promote TRPC6-dependent Ca
entry [19].
The CaSR-mediated Ca balance of podocytes, which is key for podocyte
normal function, has gradually become a research focus.
2.2 Ca Channels
The transport of Ca in podocytes is associated with multiple channel
proteins (Table 1, Ref. [20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32]). TRPCs, which belong to
the transient receptor potential (TRP) superfamily, are the most widely studied
channel family in podocytes [5]. There are seven isoforms of the TRPC proteins,
including TRPV1-7. TRPCs are “non-selective” cation channels that can promote
Ca influx into the cells [33]. Studies demonstrated that TRPC1, TRPC3,
TRPC4, TRPC5, and TRPC6 are all expressed in podocytes [7, 20, 21, 22], although only
TPRC 3, TRPC5, and TRPC6 were shown to contribute to Ca entry into the
podocyte [23]. In podocytes, TRPCs can be activated by a variety of
upstream signals, whereas Ca stimulates the downstream Ca-dependent
signaling cascade [34, 35, 36]. Depending on their location in the cell, TRPCs are
involved in different cellular physiological processes. In organelles such as the
ER, Golgi, and lysosomes, their activation also promotes Ca influx [24].
Table 1.Subcellular localization and function of Ca channels in
podocytes.
| Calcium channels |
Subcellular localization |
Function |
Reference |
| TRPCs |
TRPC1 |
Unclear |
Promotes Ca influx |
[20] |
| TRPC4 |
| TRPC3 |
Plasma membrane and organelle membranes, such as ER, Golgi, lysosomes, etc. |
Promotes Ca influx |
[21, 24, 31] |
| TRPC5 |
[22, 24, 32] |
| TRPC6 |
[22, 23, 24] |
| SOCs |
STIM1 |
ER |
Sensor for luminal Ca concentration in ER |
[25, 26, 27] |
| ORAI1 |
Plasma membrane |
Promotes an influx of Ca into the cytoplasm |
| RyR 2 |
ER |
Calcium release from ER |
[28] |
| VDAC 1 |
Mitochondria |
Transports Ca into the mitochondria |
[29] |
| Cav2.2 |
Plasma membrane |
Promotes Ca influx |
[30] |
Abbreviations: TRPCs, transient receptor potential canonical channels; SOCs, store-operated calcium channels; STIM1, stromal-interacting molecule 1; ORAI1, Ca-release-activated Ca channel 1; RyR 2, type 2 ryanodine receptor; VDAC 1, voltage-dependent anion channel 1; Cav2.2, N-type calcium channel; ER, endoplasmic reticulum.
SOCs in the plasma membrane are triggered and activated by the depletion of
Ca from the ER. SOCs consist of a single-channel transmembrane protein,
STIM1, in the ER, and a small plasma membrane protein, ORAI1 [25, 26]. When
Ca is depleted in the ER, STIM1 is activated and aggregates in the
vicinity of ER-plasma membrane junctions. STIM1 activates ORAI1, resulting in an
influx of Ca into the cytoplasm [27]. The expressions of STIM1 and ORAI1
were observed in mouse podocytes [37]. In addition, overexpression of STIM1 and
ORAI1 increased the Ca influx in podocytes [37].
Increased intracellular Ca is a burden on organelles that may cause
dysfunction [38]. RyR2 in the ER, as well as VDAC1 in the mitochondria, mediate
Ca transport in the organelles [28, 29]. Researchers have mentioned the
role of the RyR2/Ca-release channel in ER–Ca balance [28]. In
podocytes, VDAC1 controls the Ca transport into and out of the
mitochondria, which is essential for mitochondrial function [29]. The N-type
calcium channel, Cav2.2, in podocytes, as a Ca transport channel, may be
associated with the activation of the local renin-angiotensin system (RAS) [30].
These Ca transport channels are important components of cells and
organelles. Due to the different pathophysiological statuses, activity changes in
these channels cause an imbalance in cellular Ca homeostasis, which may
relate to the pathogenesis of some podocyte-related diseases [39]. Thus, using
calcium channels as therapeutic targets has the potential for podocyte-related
diseases.
3. Role of Ca Signaling in Podocyte Cell Biology and Physiology
Ca signaling is necessary for multiple pathophysiological functions of
podocytes. Recent studies have shown that Ca signaling is involved in the
morphology, architecture, and survival of podocytes. We now describe in detail
the roles of Ca signaling in different biological processes involving
podocytes.
3.1 Podocyte Morphology
Podocytes are essential for the maintenance of the glomerular filtration
barrier. Studies have shown that mutations in TRPC6 associated with nephrotic
syndrome have led to the disruption of the filtration barrier through elevated
cytoplasmic Ca in podocytes [40, 41]. The distribution of TRPC6 channels
changes as podocyte differentiation matures [42]. This suggests that Ca
signaling may play a critical role in filtration-barrier formation. A study used
transgenic zebrafish larvae and human kidney organoids to explore the role of
Ca signaling during podocyte development [8]. They found that Ca
signaling was active during podocyte differentiation, resulting from the
triggering and release of intracellular Ca. This Ca influx may be
mediated through PLCg/IP3, leading to podocyte-foot-process differentiation and
glomerular-filtration-barrier formation. The role of Ca signaling
in podocyte morphogenesis cannot be ignored and deserves more attention.
3.2 Podocyte Architecture
The tight and complex interactions between the slit diaphragm, focal adhesion,
and the actin cytoskeleton are essential for maintaining the normal structure and
function of podocytes [43]. Slit diaphragm proteins, focal adhesion protein
complexes, and actin cytoskeleton proteins play important roles in the precise
organization and regulatory role of the actin cytoskeleton. Ca signaling
can have an impact on podocyte structure by affecting cytoskeletal proteins.
Podocin and nephrin are known to be critical SD proteins in maintaining podocyte
structural stability [44]. Lu et al. [10] elucidated the role of TRPC6
in high glucose-induced podocin reduction. In their study, TRPC6 knockdown
resulted in a diminution of the decreased podocin in podocytes, and a high level
of glucose decreased podocin through activating TRPC6. This suggested that
podocin can be mediated by TRPC6, the activation of which results in an increase
in intracellular Ca. However, the exact mechanism of action between TRPC6
and podocin remains unclear. Ca is known to regulate protein degradation
through ubiquitination [45], which could be an interesting focus for future
research.
Bhargava et al. [9] reported that IgG from kidney transplant recipients
with transplant glomerulopathy activated the calmodulin kinase IV
(CaMK4)/glycogen synthase kinase-3 (GSK3) pathway.
Phosphorylated GSK 3 stabilized nephrin transcriptional repressor SNAIL,
which caused a reduction in nephrin expression [9]. Their group also demonstrated
that a reduction in nephrin expression, induced by IgG from lupus nephritis, was
associated with the activation of CaMK4 [46]. Although Ca channels were
not mentioned in these studies, IgG, which has been produced in the pathological
state, has been shown to trigger TRPCs in several studies [47, 48]. This may also
be the mechanism through which IgG activates CaMK4 in kidney diseases.
Several signaling pathways, including CaMK4, play essential roles in actin
remodeling after an increase in Ca. Synaptopodin, an actin-binding
protein, regulates the integrity and motility of podocytes through the Rho family
of small GTPases (including Rac1, Cdc42, and RhoA) [49]. The pCaMK4
phosphorylates scaffold protein 14-3-3, which leads to the release and
degradation of synaptic foot proteins [50], by enhancing Rac1 expression and
decreasing RhoA expression [50, 51]. Rac1 promotes cell motility by forming
lamellipodia, and RhoA inhibits cell motility by promoting the formation of
contractile actin [52]. Insulin triggers store-operated Ca entry, which
leads to a decrease in synaptopodin via the Ca/calcineurin pathway [9, 10, 53].
Talin-1 is a podocyte cytoskeleton-associated anchor protein and a specific
target of calpain [54]. Studies have shown that aberrant Ca signaling can
be transmitted through the TRPC to activate downstream calpain and calcineurin,
which then, exert effects on the actin cytoskeleton by reducing Talin-1
expression [43, 55]. Farmer et al. [55] reported that TRPC6 can interact
with calpain 1 and calpain 2 and maintain calpain localization at the membrane.
This physical interaction is significant for cytoskeletal stability in podocytes,
where the actin cytoskeleton forms a highly dynamic contractile state, which is
critical for the integrity of the foot processes in podocytes [56]. The CaSR is
directly connected to the cytoskeleton by binding to the thin filament protein A.
Oh et al. [57] showed that CaSR activation increased the actin density
and content in podocytes, and reduced the degradation of the actin cytoskeleton
caused by PAN.
The biological aspects of Ca signaling in podocytes cannot be ignored.
Multiple pathological states cause podocyte structural changes via Ca
imbalance; thus, by regulating structure-related proteins, Ca signaling
also influences the mobility and stability of podocytes.
3.3 Podocyte Survival
The survival of podocytes is a complex physiological process associated with
multiple factors, such as the integrity of the cytoskeleton and expression of
various pro-survival genes, and autophagy [58, 59, 60]. The stability of the
cytoskeleton underlies cell survival. Ca signaling can affect podocyte
survival by mediating the impairment of the cytoskeleton in podocytes. Recent
studies have shown that Ca signaling also regulates the expression of
multiple survival factors. Oh et al. [57] reported that the CaSR
mediates the survival of podocytes through the MAPK pathway. The transcription
factor, the cAMP-response element-binding protein (CREB) is known to be a
pro-survival gene activator, while BAD and Bcl-xl are also pro-survival factors
[57]. When the CaSR was activated, the expression of phosphorylated CREB, BAD,
and Bcl-xl increased. Hence, an activated CaSR mediates the phosphorylation of
the p90 RSK through ERK1/2, ultimately leading to the phosphorylation of CREB,
the pro-survival gene activator [57].
In addition to the CaSR, the relationship between downstream signaling mediated
by Ca channels and cell survival should not be ignored. Autophagy is
essential for the homeostasis and survival of podocytes. Calpain in podocytes can
be activated by Ca/calcineurin [54]. Bensaada et al. [61] found
that the inhibition of calpain can maintain autophagy in podocytes during
hypertension. The TRPC6 pathway also mediates autophagy in podocytes [62].
Ma et al. [11] reported that AngII can activate TRPC 6 and cause an
increase in Ca influx, which results in the activation of
calmodulin-dependent protein kinase- (CaMKK), which, when
activated, promotes the formation of activated protein kinase (AMPK) and
autophagy in podocytes. AngII-induced abnormal autophagy and damage in the
podocyte can be alleviated following the inhibition of TRPC6-mediated Ca
influx or downstream Ca-signaling pathways, such as calcineurin [11, 63].
Stabilization of Ca channels in multiple organelles is also essential for
maintaining podocyte homeostasis. The mitochondrial Ca unidirectional
transporter (MCU) maintains mitochondrial Ca homeostasis to ensure its
normal function [64]. RyR2 in the ER is involved in ER–Ca leakage as well
as ER stress [28]. The trigger of SOCE in the ER also has a major impact on
podocyte homeostasis [65].
4. Ca Signaling in Podocyte-Related Diseases
As critical components of the glomerular filtration barrier, podocytes can
modulate glomerular filtration function. Podocyte dysfunction is central to the
pathophysiology of many common glomerular diseases, including diabetic kidney
disease, glomerulonephritis, and genetic forms of nephrotic syndrome (NS) [66].
By intravital imaging of podocyte Ca, one study suggested that the
conduction of Ca signaling was a key pathogenic mechanism in various
podocyte-related diseases [12]. Ca signaling is involved in various
podocyte-related kidney diseases, including focal segmental glomerulosclerosis,
diabetic kidney disease, lupus nephritis, transplant glomerulopathy, and
hypertensive renal injury. Below we describe the mechanisms of Ca
signaling in various diseases.
4.1 Focal Segmental Glomerulosclerosis (FSGS)
FSGS is an important cause of end-stage renal failure (ESRD), characterized by
podocyte loss and dysfunction. Most patients show steroid-resistant nephrotic
syndrome (SRNS).
Several studies have shown that genetic FSGS is associated with mutations in the
TRPC6 gene [39, 67, 68, 69] (Fig. 1). Among the mutated genotypes, TRPC6 always showed
a gained functional phenotype involving Ca-mediated podocyte pathogenic
mechanisms [13, 70]. The mutant form of TRPC6 that causes FSGS also increases
intracellular Ca. Multiple studies have shown that downstream Ca
signaling participates in podocyte damage in FSGS after the delayed inactivation
of TRPC6. Ca activates calcineurin [71] and CaMK4 [50], resulting in a
decrease in nephrin and synaptopodin expressions, which are critical for podocyte
structure. The gain-of-function TRPC6 mutant led to the phosphorylation of ERK1/2
in podocytes, which is involved in the regulation of cell proliferation and
survival [72]. However, the pathogenic role of TRPC6 activation may be through
activating NFAT-dependent transcription [73]. As substrates of calcineurin,
activation of NFATc transcription factors leads to podocyte dysfunction, and
ultimately, to glomerulosclerosis [73].
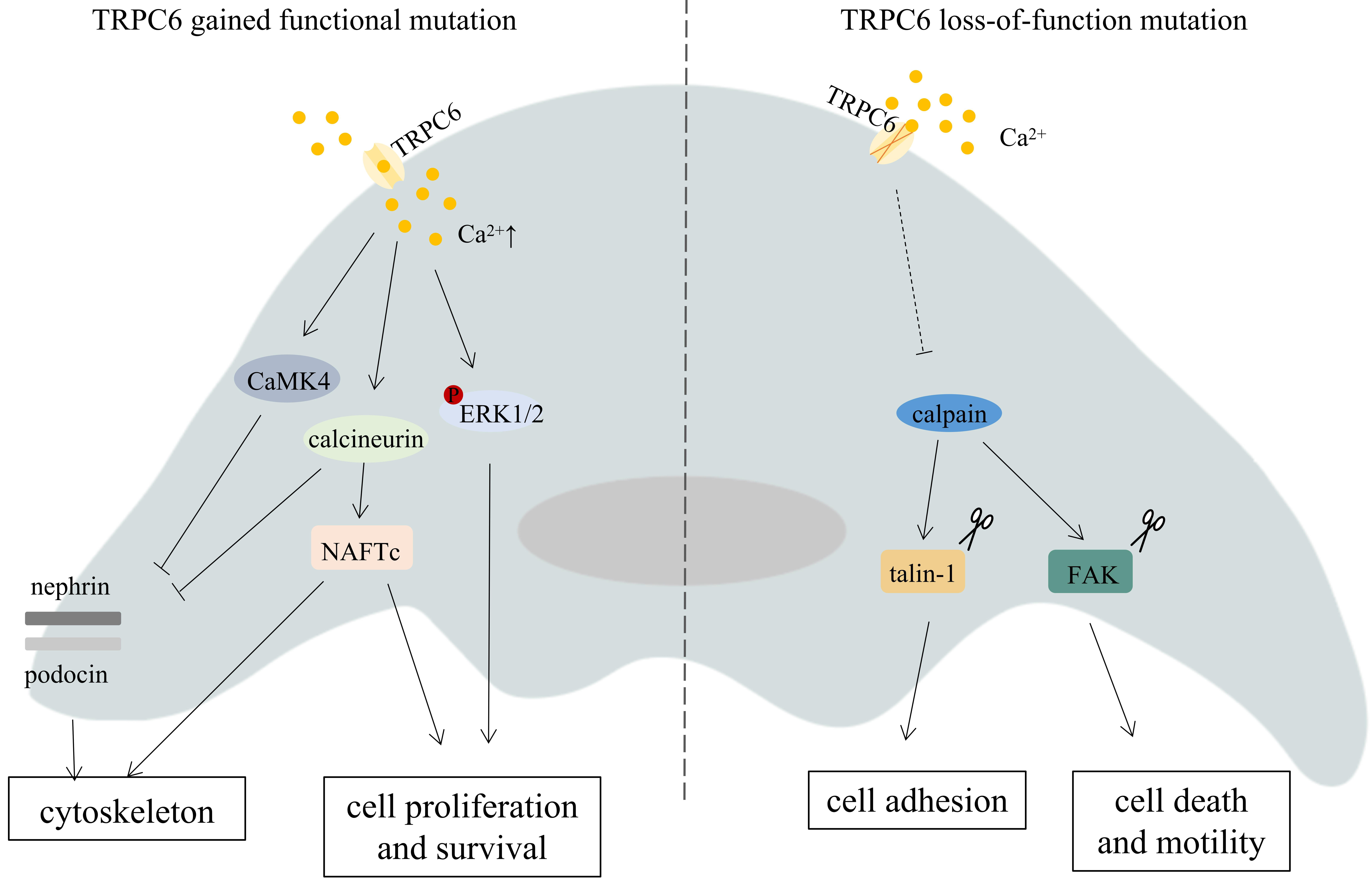 Fig. 1.
Fig. 1.
Calcium signaling in focal segmental glomerulosclerosis.
The loss-of-function TRPC6 mutation has been shown to be associated with the
pathogenesis of FSGS, although the mechanism is currently unclear [67].
Farmer et al. [55] reported that TRPC6 podocytes showed
decreased motility, stronger adhesion, and an altered actin cytoskeleton. FAK
cleavage is involved in cell death and motility, whereas talin-1 cleavage is
associated with cell-adhesion breakdown [74]. Calpain activity was lost and
appeared to be mislocalized after the knockdown of TRPC6. In addition, cleavage
of the calpain targets, talin-1 and FAK, was reduced [55], which may account for
the pathogenesis of the TRPC6 loss-of-function mutation.
In addition to the functional changes of TRPC6 and downstream pathways
contributing to podocyte damage in FSGS, RyR2 may also play a role in the
pathogenesis of FSGS. In the NS/FSGS mouse model, the RyR2 channels in the ER of
podocytes are phosphorylated, which leads to Ca depletion and stress in
the ER, resulting in podocyte damage [28].
4.2 Diabetic Kidney Disease (DKD)
An increase in Ca-channel activity was observed in the diabetic kidney
disease model [14]; the knockdown of Ca channels in Akita mice promoted
insulin resistance and aggravated glomerular injury [75]. These results highlight
the importance of the roles Ca channels perform in the pathophysiological
state of DKD.
As a metabolic-related kidney disease, the underlying pathogenesis of DKD,
especially in the dysfunction of mitochondria, is gradually gaining attention
[76]. Several studies have shown that Ca signaling participates in the
development of podocyte injury in DKD by regulating mitochondrial function (Fig. 2). Yu et al. [77] reported that TRPC6 mediated high-glucose-induced
mitochondrial fission in DKD podocyte injury. TRPC6 mediates mitochondrial
fission by Ca-activated calpain1 [77]. Tao et al. [78] found that
an increase in SOCE in podocytes was induced by high glucose and angiotensin II
(AngII), thereby mediating impaired mitochondrial membrane potential (MMP), ATP
and mitochondrial superoxide production, and mitochondrial respiratory damage in
DKD. Disruption of Ca homeostasis in mitochondria leads to increased
oxidative stress and cell apoptosis [79]. The connection between the
“mitochondria-associated membrane” (MAM) and the ER can maintain Ca
homeostasis in the mitochondria [80]. Wei et al. [81] reported that the
activation of TRPV1 attenuated mitochondrial dysfunction in podocytes caused by
hyperglycemia. Transient Ca influx, mediated by TRPV1, reduced the
transcription of Fundc1, a key molecule involved in MAM formation, by activating
5 AMP-activated protein kinase (AMPK). Therefore, the activation of TRPV1
reduced MAM formation and the transport of Ca from the ER to the
mitochondria [81]. When the ER releases Ca through the inositol
1,4,5,-triphosphate receptor (IP3R), Ca enters the mitochondrial membrane
space via VDAC1 and finally enters the mitochondrial matrix through the
mitochondrial Ca unidirectional transport protein (MCU) [82]. High glucose
and AngII induced enhanced effects of IP3R-Grp75-VDAC1-MCU, which increased
Ca in the mitochondria as well as increased active caspase-3 [64]. This
process participates in podocyte apoptosis in DKD.
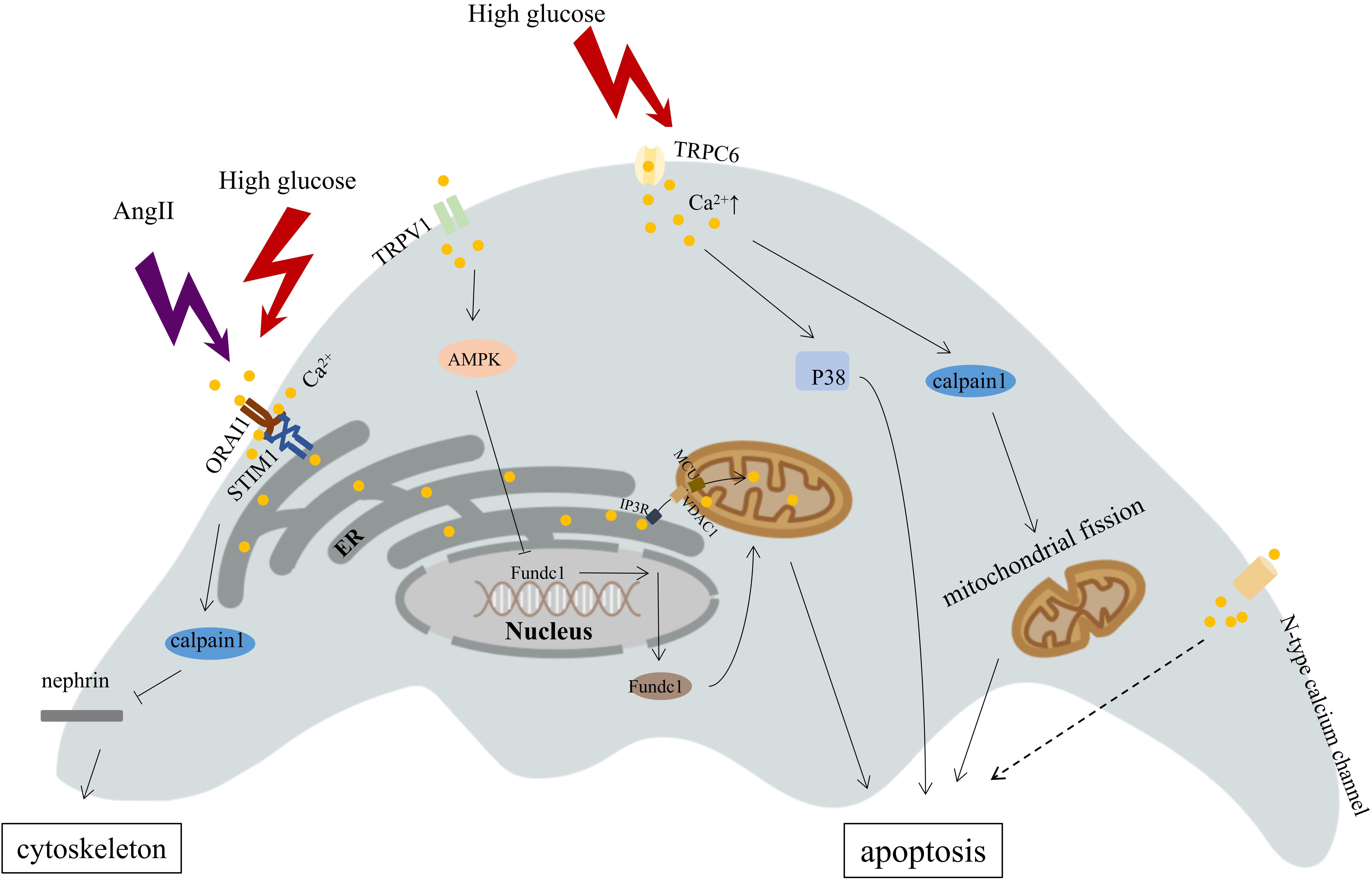 Fig. 2.
Fig. 2.
Calcium signaling in diabetic kidney disease.
High glucose-induced apoptosis is one of the key factors contributing to
podocyte injury in DKD [83]. Upregulation of Ca/calcineurin (CaN)
signaling promotes podocyte apoptosis, which is a key mechanism in podocyte
injury in DKD [84]. Podocyte apoptosis may also relate to the increased
expression of p38, caused by TRPC6 knockout in Akita mice [75]. Tao et
al. [85] also showed that a high glucose-induced increase of SOCE led to
decreased expression of the nephrin protein by promoting calpain activation. In
addition to the above Ca channels, the N-type calcium channel is also
involved in proteinuria production due to podocyte damage in DKD [86].
4.3 Lupus Nephritis (LN)
LN, as a chronic immune-complex-mediated renal lesion, is mainly characterized
by renal glomerular impairment [87]. Abnormal IgG can be transported and
deposited in podocytes via the neonatal Fc receptor (FcRn), causing
podocytopathies [88]. These recent studies have indicated that the
Ca/CaMK4 pathway plays an essential role in IgG-mediated podocyte injury
(Fig. 3).
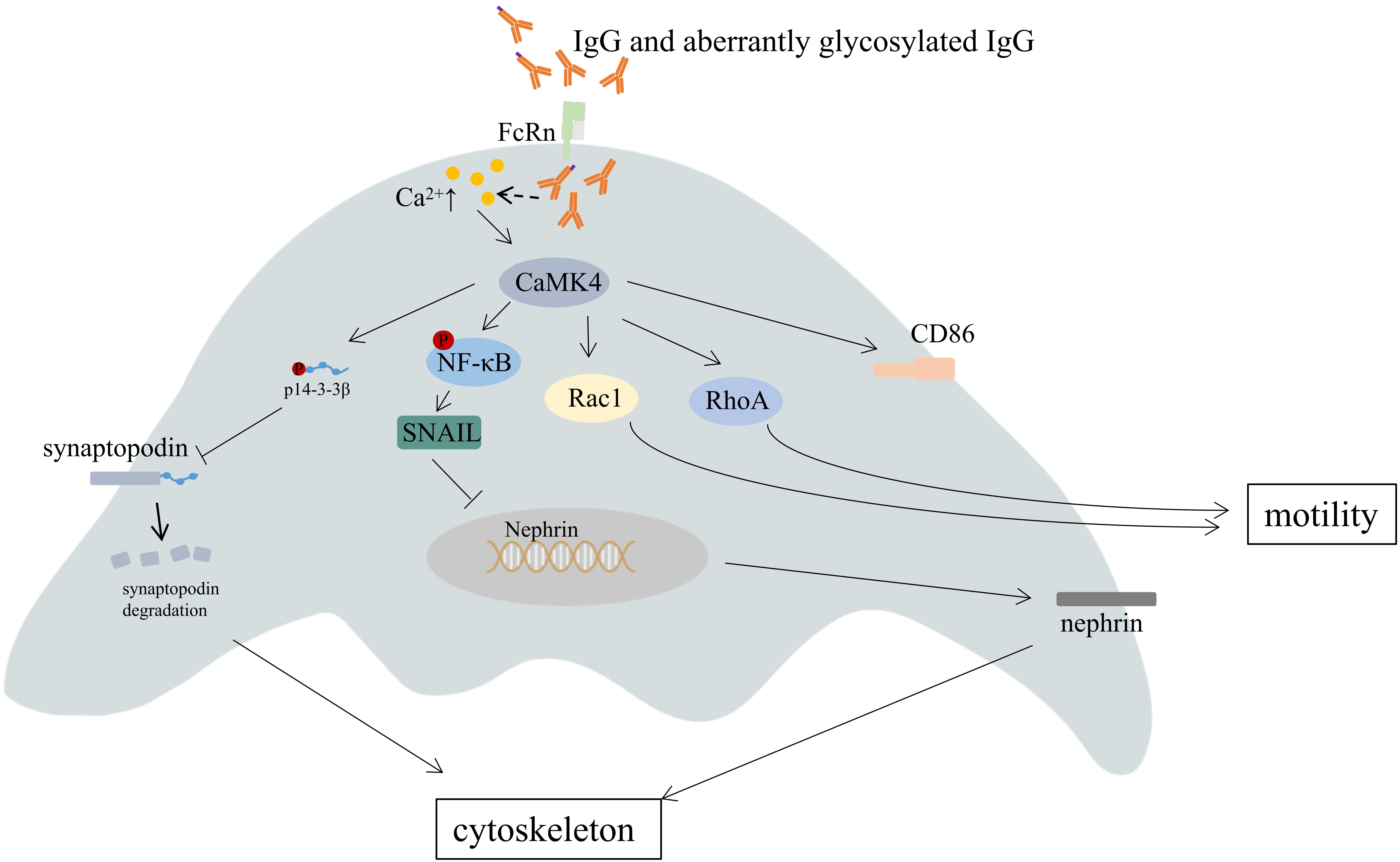 Fig. 3.
Fig. 3.
Calcium signaling in lupus nephritis.
The expression of Ca/CaMK4 is significantly elevated in podocytes of LN
patients and lupus-prone mice [46]. Ichinose et al. [88] showed that
pathogenic IgG upregulated CaMK4 and altered the expression of some genes
associated with podocyte injury, including CD86 costimulation and
skeleton-related genes, such as nephrin and synaptopodin. Bhargava et
al. [46] also reported that CaMK4 upregulation, mediated by aberrantly
glycosylated IgG, can phosphorylate NF-B, which decreases the
expression of nephrin by upregulating SNAIL. In addition, CaMK4 can also modulate
podocyte motility in LN through the regulation of Rac1 and RhoA, while can also
regulate the degradation of synaptopodin in LN via the phosphorylated scaffold
protein 14-3-3 [50]. These studies suggest that Ca/CaMK4 may be a
valuable target for the treatment of podocyte injury in LN.
4.4 Transplant Glomerulopathy (TG)
Chronic antibody-mediated immune rejection, ultimately, leads to renal graft
failure. TG is the main histological feature. Podocyte shedding was closely
associated with the occurrence of proteinuria and eGFR decrease in TG [89]. Both
TG and LN are thought to be the result of antibody-mediated damage.
Bhargava et al. [9] reported that N-glycosylated IgG in TG patients can
also enter podocytes via FcRn. N-glycosylated IgG activates CaMK4, which
phosphorylates GSK3. Phosphorylated GSK3 reduces nephrin
expression, resulting in the regulation of podocyte migration and the
rearrangement of the actin cytoskeleton [9] (Fig. 4).
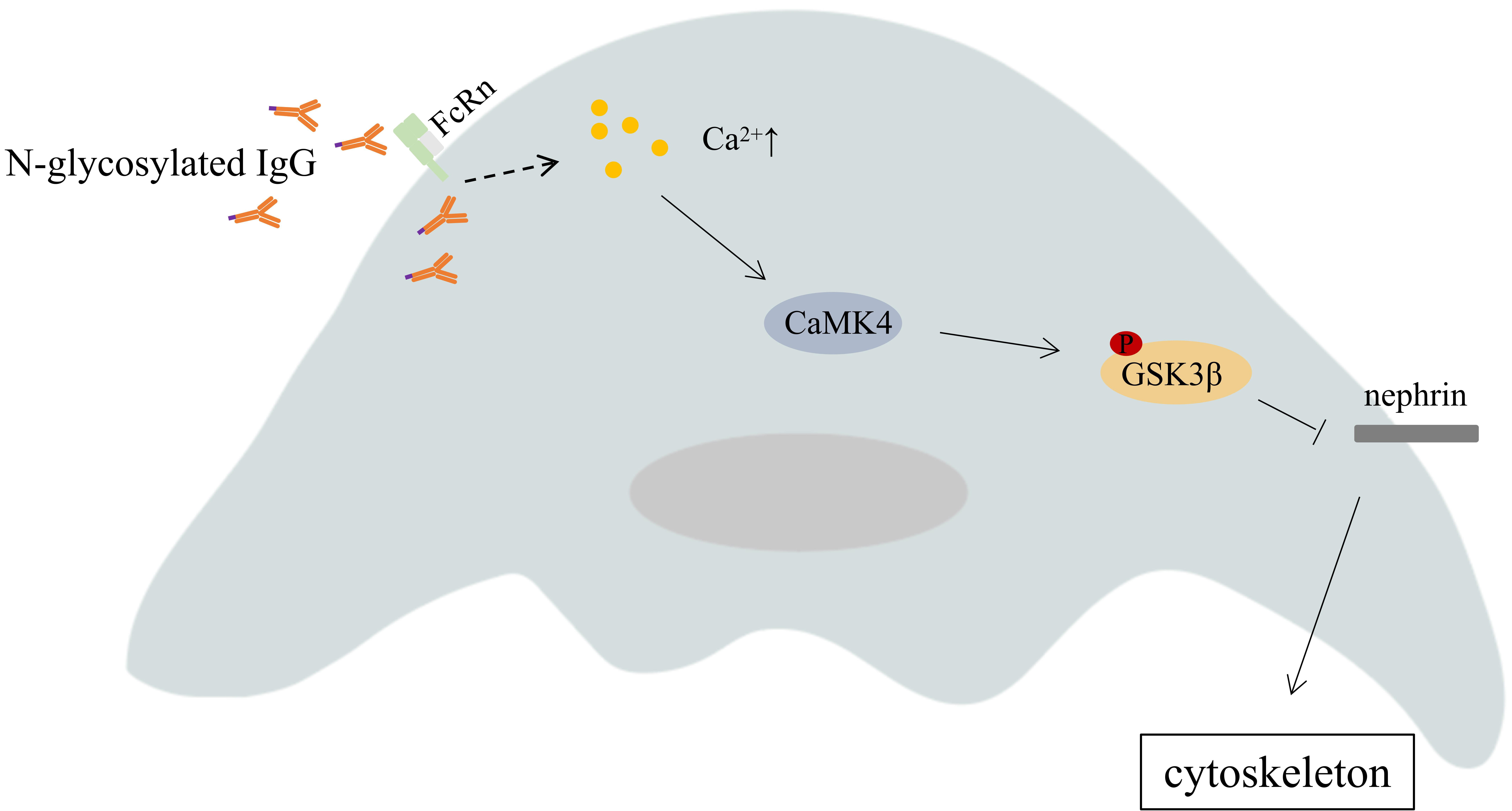 Fig. 4.
Fig. 4.
Calcium signaling in transplant glomerulopathy.
4.5 Hypertensive Renal Injury
Hypertension damages podocytes due to increased pressure in the glomerular
capillaries, leading to the loss of podocytes [90]. The activity of RAS is
enhanced in hypertensive patients [91]. AngII-induced autophagy in the podocytes
of spontaneously hypertensive rats was explored by Ma et al. [11]. They
found that the activation of TRPC6 can be stimulated by AngII, thereby resulting
in Ca influx and CaMKK-AMPK pathway activation, which leads to
podocyte autophagy. N-type calcium channel is also involved in AngII-induced
podocyte injury [92], which may be related to the production of ROS caused by
AngII [93]. Golosova et al. [94] showed that -opioid receptors
(ORs)/TRPC6 signaling is activated in hypertensive rats and human
podocytes, while pathological Ca signaling leads to actin cytoskeleton
rearrangement. These results suggest a critical role for Ca signaling in
hypertensive podocyte injury (Fig. 5).
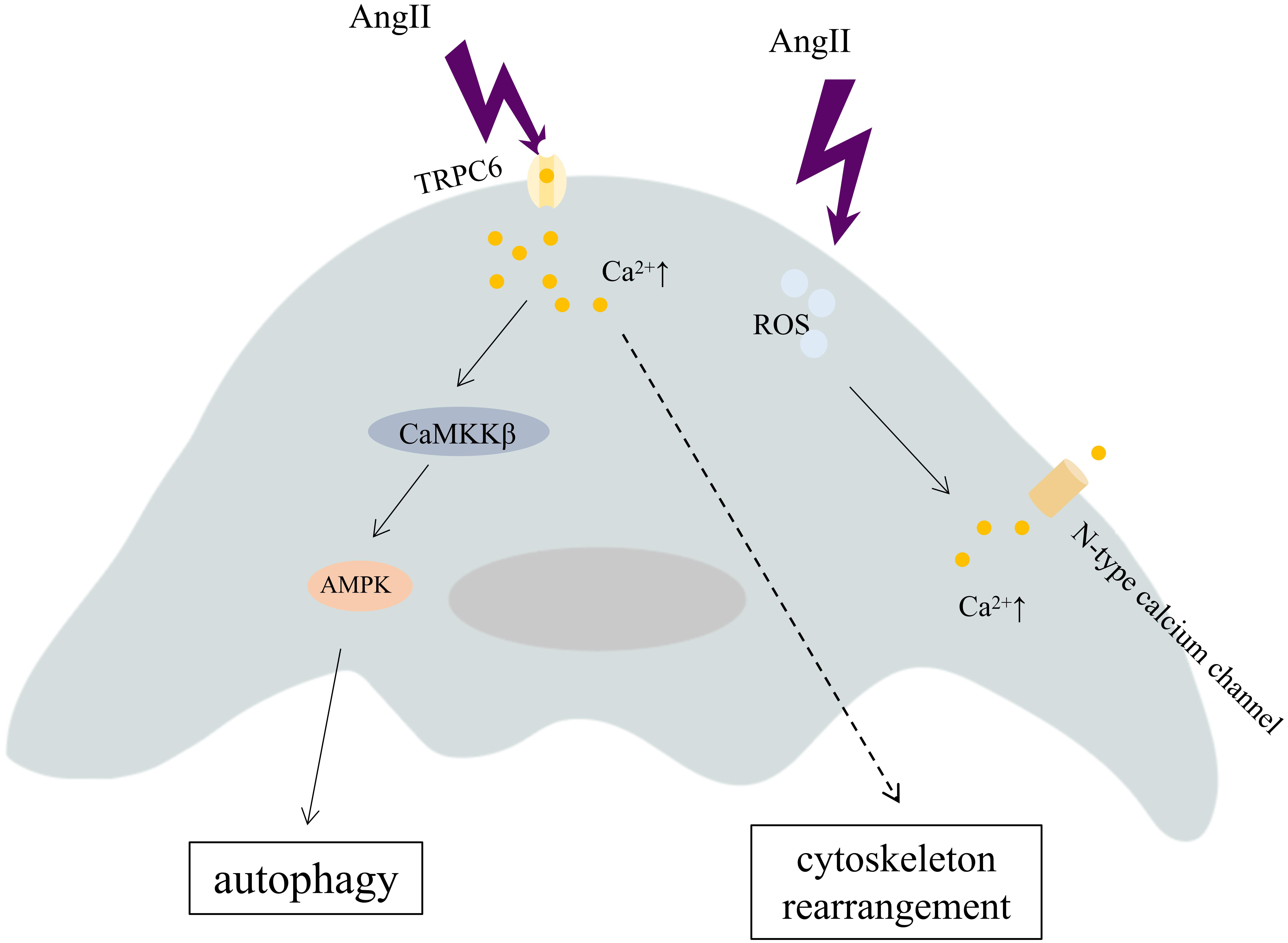 Fig. 5.
Fig. 5.
Calcium signaling in hypertensive renal injury.
Ca channels and Ca-signaling pathways are closely related to the
pathogenesis and progression of podocyte-related diseases. Activation of
Ca channels, such as TRPC6, RyR2, SOCs, and VDAC1, has been reported to
contribute to podocyte-related diseases. Ca/CaMK4 was found to be involved
in podocyte injury in LN and TG, and the activation of TRPV1 attenuated
mitochondrial dysfunction in podocytes in DKD. These results suggest that
Ca channels and Ca signaling are promising targets for the
treatment of podocyte-related diseases.
5. Targets of Ca-Signaling Pathways in Podocytes
The implementation of Ca-signaling pathways as promising therapeutic
targets is the focus of researchers; thus, we summarized the current
Ca-signaling targets associated with podocytes in Table 2 (Ref. [11, 16, 28, 57, 71, 86, 93, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110].
Table 2.Drugs and inhibitors targeting Ca signaling pathways in
podocytes.
| Drugs and inhibitors |
Targets |
Mechanism |
Disease |
Reference |
| R-568 |
CaSR |
Activates the sensitivity of CaSR to calcium ions |
Glomerulosclerosis |
[57] |
| Cinacalcet |
NS |
[16] |
| Atractylodis rhizoma water extract (ARE) |
TRPC 6 |
Inhibits the activation of TRPC 6/p-CaMK4 signaling |
Fructose-induced kidney disease |
[95, 96] |
| Astragaloside IV (AS-IV) |
Inhibits the activation of TRPC 6 |
DKD |
[97] |
| Tetrandrine |
Inhibit TRPC 6 expression and downstream RhoA/Rock and calpain 1 signaling pathway |
NS |
[98, 99, 100] |
| Taurine (2-aminoethenonic acid) |
Suppresses TRPC 6 expression by upregulating H2S synthesis |
DKD |
[101] |
| Semi-synthetic compound larixyl carbamate (LC) |
Inhibits TRPC6 activity |
|
[102] |
| Qian Yang Yu Yin (QYYY) granule |
Inhibits TRPC6/CaMKK pathway |
Hypertensive kidney injury |
[11] |
| Metformin |
Inhibits TRPC 6 expression through AMPK1 activation |
DKD |
[103] |
| Inhibits the ZEB 2/TRPC 6 pathway |
CKD |
[104] |
| Liraglutide |
Reduces TRPC6 expression |
DKD |
[105] |
| Sildenafil (Viagra) |
Increases cGMP levels by inhibiting PDE 5 and cGMP, to reduce TRPC 6 expression |
DKD |
[106] |
| Riociguat |
Activates sGC, causing an increase in cGMP production and inhibiting the TRPC 6 pathway |
Adriamycin-induced nephropathy |
[107] |
| FK506 |
Reduces TRPC 6 expression |
DKD |
[110] |
| K201 |
RyR 2 |
Blocks RyR 2-Ser2808 phosphorylation |
NS |
[28] |
| MANF |
Restores defective RyR 2 function |
| Cilnidipine |
Cav 2.2 |
Inhibits N-calcium channels |
DKD |
[86, 93] |
| microRNA-30 family members |
TRPC, calcineurin, NFATC3 |
Inhibits TRPC 6, PPP3CA and PPP3CB, PPP3R1 and NFATC3 |
FSGS |
[71] |
| CNI |
calcineurin |
Inhibits the calcineurin/NFAT/Angptl4 pathway |
NS |
[108, 109] |
Abbreviations: CaSR, calcium-sensing receptor; NS, nephrotic syndrome; DKD, Diabetic Kidney Disease; FK506, tacrolimus; MANF, mesencephalic astrocyte-derived neurotrophic factor; RyR 2, type 2 ryanodine receptor; Cav 2.2, N-type calcium channel; FSGS, Focal Segmental Glomerulosclerosis; TRPC, transient receptor potential canonical; NFATC3, Nuclear factor of activated T cell transcription factors; PPP3CA/B, protein phosphatase 3 catalytic subunit alpha/beta; CNI, calcineurin inhibitors; NFAT, Nuclear factor of activated T cell.
5.1 CaSR
The specific knockdown of CaSR in podocytes disrupted the actin cytoskeleton and
reduced cell attachment and migration speed, resulting in proteinuria.
Calcimimetics allosterically activate the sensitivity of the CaSRs to Ca.
Oh et al. [57] found that R-568, a calcimimetic that activates the CaSR
signal, stabilizes the podocyte cytoskeleton and enhances cell survival, thereby
reducing aminonucleoside (PAN)-induced glomerulosclerosis. In addition to R-568,
the calcimimetic cinacalcet has also been used in children with nephrotic
syndrome to relieve proteinuria [16]. Huang et al. [111] proposed that
the CaSR has important molecular associations in the pathophysiology of primary
membranous nephropathy. Activation of CaSRs may cause podocyte damage through
Ca imbalance. Yadav et al. [112] suggested that the Ca
imbalance caused by CaSR activation may also be one of the possible mechanisms of
renal dysfunction in sepsis. These results suggest that CaSR is also a potential
therapeutic target in primary membranous nephropathy and cardio–renal syndrome,
which still needs further exploration.
5.2 Ca Channels
Therapies targeting Ca channels in podocytes are receiving increased
attention.
The TRPC6 channel acts as the most important link in podocyte Ca
signaling. Multiple herbal extracts and synthetics can act to treat podocyte
injury by inhibiting the activity of TRPC6 and its downstream pathways. Studies have shown that atractylodin and Atractylodis rhizoma water
extract (ARE) components can prevent fructose-induced glomerular damage.
Atractylodin inhibits the activation of TRPC6/p-CaMK4 signaling in podocyte
hypermotility under fructose conditions [95]. ARE also has a therapeutic effect
by downregulating the TRPC6/Ca/CaMK4 pathway resulting from reducing
hydrogen peroxide (HO) and malondialdehyde (MDA) levels in the
glomeruli [96]. As Astragaloside IV (AS-IV), an active
ingredient isolated from Astragalus membranaceous (Fisch). Bge,
inhibited Ca channel activity by targeting TRPC6.
AS-IV can reduce the Ca influx caused by palmitic acid, thereby alleviating the apoptosis of podocytes [97]. Tetrandrine, the main active ingredient isolated from the Chinese
herbal radix Stephania tetrandra, has been used in a Chinese
traditional medicine preparation called Fangji Huangqi Tang for the
treatment of NS [98]. Studies have indicated that tetrandine can protect
podocytes from intracellular Ca influx by inhibiting TRPC6 overexpression
and downstream RhoA/Rock signaling [99], or the calpain 1 signaling pathway
[100]. Taurine (2-aminoethenonic acid) is a semi-essential amino acid,
synthesized from cysteine, which inhibits high glucose-induced podocyte damage
[101]. Taurine increases cystathionine--lyase (CSE) expression and
suppresses TRPC6 expression by upregulating H2S synthesis, thereby alleviating
the pathogenic effects of high glucose. Researchers composed a semi-synthetic
compound larixyl carbamate (LC), extracted from larixol, as an inhibitor of TRPC
6 channel activity [102]. In addition to synthetic chemicals, Qian Yang
Yu Yin (QYYY) granule, which is widely used in hypertensive kidney injury, has
been shown to protect podocytes by inhibiting the TRPC6/CaMKK pathway
[11]. The inhibitory effect of metformin on the TRPC6 pathway has also been
demonstrated. Szrejder et al. [103] showed that metformin inhibited
high-glucose-induced TRPC6 expression in podocytes by activating AMPK1,
thereby protecting the podocyte cytoskeleton. Metformin also abrogates
hypoxia-induced podocyte injury by inhibiting the ZEB2/TRPC6 pathway [104].
Liraglutide, a GLP1 receptor agonist, reduced TRPC6 expression in the kidneys of
Type 1 diabetic rats, thereby preventing the progression of diabetic kidney
disease [105]. Previous drugs used in non-renal diseases were also revived by
researchers. Sonneveld et al. [106] found that sildenafil (Viagra)
increased cyclic guanosine phosphate (cGMP) levels by inhibiting
phosphodiesterase type 5 (PDE5)- and cGMP-reduced TRPC6 expression and activity,
to prevent the progression of glomerular damage. Hart et al. [107] also
found that riociguat, through activating soluble guanylate cyclase (sGC), led to
an increase in cGMP production, thereby inhibiting the TRPC6 pathway and podocyte
damage caused by TRPC6-mediated Ca influx. Wang et al. [113]
reported that an 1-AR agonist (phenylephrine hydrochloride) could
participate in the loss of the cytoskeletal structure by inducing a
TRPC6-dependent increase in intracellular Ca in human podocytes. This
provides a possible rationale for the inhibition of TRPC6 by the 1-AR
inhibitor (silodosin) [114], to improve podocyte injury.
The Ca-release channel RyR2 on the ER of the podocyte is phosphorylated
in the NS/FSGS mouse model, causing an imbalance in intracellular Ca
homeostasis and ER stress. Park et al. [28] proposed that a novel
compound, K201, blocked RyR2-Ser2808 phosphorylation and that a brain
astrocyte-derived neurotrophic factor (MANF) restored the defective RyR2
function.
Albuminuria and ultrafiltration were significantly improved after specific
blockade of the -1 subunit of the N-type calcium channel Ca 2.2
in diabetic mice [86]. This suggests that N-type calcium channels are a future
therapeutic target for podocyte injury. Cilnidipine is an
L/N-type dihydropyridine calcium channel blocker (CCB), which
can pharmacologically inhibit the N- type calcium channels
[115]. Cilnidipine reduced the urinary protein/creatinine ratio in
hypertensive patients with chronic kidney disease more effectively than
amlodipine (L-type CCB) [116, 117]. A subsequent study
suggested that it may reduce the AngII-induced damage through the inhibition of
N-type calcium channels in podocytes [93].
5.3 Ca/Calcineurin Signaling
Presently, studies targeting Ca/calcineurin are of great interest to
researchers. Wu et al. [71] proposed that microRNA-30 family members
could inhibit the PAN-induced increase in Ca/calcineurin signaling in
podocytes by targeting the inhibition of TRPC6 (Ca channel), PPP3CA and
PPP3CB (calcineurin -catalytic subunit members [118]), PPP3R1
(calcineurin -regulatory subunit member [118]), and NFATC3
(transcription factor). The antiproteinuric effect and podocyte protection of
calcineurin inhibitors (CNI) have been confirmed [108]. Cyclosporine (CsA) and
tacrolimus (FK506) are currently the most widely used calcineurin inhibitors
[119]. CNI can ameliorate PAN-induced podocyte damage and apoptosis by inhibiting
the CaN/NFAT/Angptl4 pathway [109]. Calcineurin can dephosphorylate synaptopodin,
be degraded by CatL, and alter the morphology of the podocyte cytoskeleton. CNI
destabilizes the podocyte cytoskeleton by targeting calcineurin [120]. In
addition, one study showed that FK506 could reduce TRPC6 expression of the
podocyte in DKD [110]. The specific mechanism for the beneficial effect of CNI on
podocyte injury requires further exploration.
6. Conclusions
Ca in podocytes can be stimulated by different signals and transmitted to
multiple downstream signaling pathways. Podocytes sense intracellular Ca
concentrations through the CaSR, generating responses that regulate the balance
of Ca, which is essential for the function of the glomerular filtration
barrier. Similarly, the downstream signaling pathways mediated by the activity of
Ca channels are involved in various biological functions, such as the
morphogenesis, structure, and survival of podocytes. In addition to the TRPCs
widely studied in podocytes, recent studies have also suggested the importance of
N-type calcium channels for Ca transport in podocytes. RyR2, VDAC1, and
SOCs are also important for maintaining organelle Ca homeostasis.
Increased Ca influx and Ca signaling activation in various
podocyte-related diseases can achieve cell communications. FSGS, as one of the
diseases most commonly manifested as SRNS, is associated with mutations in
various Ca-signaling-pathway-related genes. Therapies targeting Ca
signaling have broad application prospects in patients with FSGS and also provide
a novel option for therapy in patients with SRNS. Podocytopathy in DKD is widely
recognized, and some progress has been made in the study of Ca signaling
in podocyte damage that has been induced by high levels of glucose and AngII.
However, the effect of focusing Ca-signaling therapy on podocyte damage in
DKD still needs further exploration. Abnormal IgG-antibody-mediated podocyte
injury in LN and TG also gradually attracted the interest of researchers to the
role of Ca; however, it too requires additional exploration. Ca
signaling targets may also be used as treatment strategies for immune-related
kidney injuries. Currently, the therapeutic strategies targeting the
Ca-signaling pathway focus on channel proteins, yet with the research
progress on CaSRs and downstream signaling of Ca, they will soon be
researched as potential therapeutic targets.
Author Contributions
YCT and LJY selected the topic. YCT wrote the paper. LJY provided guidance to
the paper. LJY, YCT, HPS, LLS, QQL, LF, and MR participated in the discussion,
revision, and correction of the manuscript. All authors contributed to editorial
changes in the manuscript. All authors read and approved the final manuscript.
All authors have participated sufficiently in the work to take public
responsibility for appropriate portions of the content and agreed to be
accountable for all aspects of the work in ensuring that questions related to its
accuracy or integrity.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research was supported by the National Natural Science Foundation of China
(No.81974102).
Conflict of Interest
The authors declare no conflict of interest.






