











1 Department of Nephrology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
†These authors contributed equally.
Abstract
Background: Renin-dependent hypertension with tubulointerstitial injury
remains a problem with high prevalence in the clinic. However, whether and how
renin participates in tubulointerstitial injury remains incompletely understood.
New evidence suggests that renin cleaves C3 into C3a and C3b. In the present
study, we aimed to explore the role of renin-mediated C3a/C3a receptor (C3aR)
signaling in renin-dependent hypertension-induced kidney injury and illustrate
the detailed mechanisms. Methods: C3a concentration changes in serum
from healthy volunteers incubated with recombinant renin were detected by ELISA.
C3aR expression in human tubular epithelial cells was evaluated in renal biopsy
sections from malignant arteriolonephrosclerosis and benign
arteriolonephrosclerosis patients. C3aR changes in human kidney 2 (HK2) cells
were detected after the cells were treated with human serum, renin and aliskiren.
The C3a analogue and C3aR antagonist SB290157 were used to stimulate HK2 cells to
explore the downstream signaling of C3a/C3aR activation. For in vivo
studies, two-kidney, one-clipped (2K1C) hypertensive rat model was established to
simulate renin-dependent hypertension conditions. C3a and C3aR expression was
detected in the clipped kidneys. SB290157 was injected intraperitoneally to block
C3a/C3aR signaling in 2K1C rats. Results: The results showed that renin
cleaved C3 into C3a and activated C3a/C3aR signaling in tubular epithelial cells
(TECs) from both humans and rats. In vitro results demonstrated that
C3a/C3aR activation impaired peroxisome proliferator-activated receptor alpha
(PPAR
Keywords
- hypertension-induced kidney injury
- renin
- C3a
- C3a receptor
- mitochondrial fatty acid oxidation
- profibrotic phenotype transition
Hypertension is one of the most common causes of secondary kidney diseases and a major contributor to chronic renal dysfunction [1, 2]. Overactivation of the sympathetic nervous system [3] or renal artery stenosis [4] leads to low renal blood flow and subsequently increased renin secretion from the juxtaglomerular apparatus, which result in renin-dependent hypertension. Recently, a curious relationship among the renin-angiotensin system (RAS), immune activation and tubular injury is highlighted [5]. New evidence suggests that renin can cleave C3 to produce C3a and C3b, and the application of renin inhibitors reduces renin-induced C3 cleavage and mitigates tubulointerstitial injury [6, 7]. However, the mechanisms by which renin affects tubulointerstitial injury are not fully understood.
Complement C3, as the central molecule of the complement system, has been
implicated in the pathogenesis of several chronic kidney diseases (CKDs) [8, 9, 10].
Reportedly, C3 and its cleavage fragments accumulate in the glomerular
capillaries and arterioles of the kidneys in patients with hypertension [11]. The
intensity of C3 deposition is associated with the severity of
hypertension-induced kidney injury [12, 13]. Apart from serum-derived C3
deposition, local synthesis of C3 under disease conditions also contributes to
renal damage [14]. C3a, the smaller cleavage fragment of C3, can regulate
intracellular signaling by binding to the C3a receptor (C3aR), which is located
on the cellular membrane [15]. In the kidney cortex, C3aR is mainly expressed in
tubular epithelial cells (TECs) and interstitial infiltrating immune cells such
as monocytes and macrophages [16, 17]. Evidence has revealed that C3a/C3aR
signaling plays a vital role in renal tubulointerstitial fibrosis in several CKD
models, such as unilateral ureteral obstruction model and adriamycin-induced
proteinuria model, by promoting tubular profibrotic phenotype transition and
mesenchymal transition [18, 19, 20]. In addition, C3aR inhibition has been reported to
be a promising treatment to alleviate tissue fibrosis in different disease models
[21, 22]. However, the effect of renin-induced C3a/C3aR signaling on the renal
tubulointerstitium and the effect of C3aR antagonist on renin-dependent
hypertension-induced kidney injury have not been fully elucidated. Studies
suggest that hypertension suppresses peroxisome proliferator-activated receptor
alpha (PPAR
Here, we conducted in vitro and in vivo experiments to simulate the renin-dependent hypertension-induced kidney injury microenvironment to explore how renin/C3a/C3aR signaling regulates renal tubulointerstitial injury and illustrates whether the C3aR antagonist SB290157 can protect against renin-dependent hypertension-induced kidney injury by restoring mitochondrial fatty acid oxidation (Mito FAO) and mitigating mesenchymal transition through C3a/C3aR signaling.
Serum samples collected from seven healthy volunteers were stored in aliquots at –80 °C. After a 1:10 dilution in Dulbecco’s phosphate-buffered saline (DPBS, Gibco, Waltham, MA, USA), serum samples were incubated with recombinant renin (1.2 µg/mL, BioVision, Waltham, MA, USA) or phosphate-buffered saline (PBS, Gibco) at 37 °C for 24 h. After incubation, the C3a level of the samples was detected immediately with a Complement C3a Human ELISA Kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
Immortalized human tubular epithelial cells (human kidney 2 (HK2) cells, CRL-2190, ATCC, Manassas, VA, USA) were routinely cultured in Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 (DMEM/F12, Gibco) containing 10% fetal bovine serum (FBS, Gibco) as described previously [26]. The cell lines were mycoplasma-free (Mycoplasma Detection Kit, Yeasen, Shanghai, China) and authenticated by STR identification.
HK2 cells were treated with 10% serum from healthy volunteer 7 (H7) supplemented with recombinant renin (1.2 µg/mL) or PBS at 37 °C for 24 h. Aliskiren (10 µM, MedChemExpress, Monmouth Junction, NJ, USA), a specific renin inhibitor, was preincubated with recombinant renin for 1 h before addition.
A C3a analogue, which has been reported to mimic the natural biological activity
of C3a [27, 28], was used to explore the effect of C3a/C3aR activation. HK2 cells
were treated with the C3a analogue (1 µg/mL, Sangon Biotech,
Shanghai, China) or PBS for 24 h. For C3aR inhibition, cells were pretreated with
the C3aR antagonist SB290157 (1 µM, Sigma‒Aldrich, St. Louis, MO, USA) for
1 h. HK2 cells were harvested for western blotting analysis, and the culture
medium was used to evaluate the concentration of transforming growth factor
HK2 cells after the treatment were fixed in 4% paraformaldehyde and
permeabilized in 0.2% Triton X-100, followed by incubation with 2% BSA for 1 h.
The cells were then incubated with PPAR
The fatty acid uptake probe in the Fatty Acid Uptake Assay Kit (Dojindo Laboratories, Kamimashiki-gun, Kumamoto, Japan) functions as a fatty acid analogue and shows the cellular uptake capacity of fatty acids directly. MitoBright LT Deep Red (Dojindo Laboratories) can mark the mitochondria. HK2 cells completing the treatment were stained with the fatty acid probe and subsequently with MitoBright LT Deep Red. The cells were observed by confocal microscopy (LSCM, LSM800, Zeiss, Oberkochen, Germany). The colocalization between the fatty acid probe and MitoBright LT Deep Red was measured by ImageJ with the Coloc 2 plugin and presented as Manders’ coefficients.
Three patients with malignant arterionephrosclerosis (MANS) and three patients with benign arterionephrosclerosis (BANS) were included in this study. The study protocol was approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. All of the kidney tissues were obtained from renal biopsies. Informed consent was obtained from all of the patients. Paraffin-embedded kidney sections were prepared for immunofluorescence.
Recombinant rat renin protein (active) (Abcam) and rat C3 protein (Complement
Technology, Tyler, TX, USA) were used in the experiment. Purified rat C3 protein
(100 µg/mL) was incubated with recombinant rat active renin at
concentrations of 0, 1.2, 12.5, and 25 µg/mL at 37 °C for 24 h.
After incubation, the sample was mixed with 5
Male Sprague‒Dawley (SD) rats weighing 150~170 g were purchased
from Charles River Laboratories (Beijing, China) and housed at optimal
temperature (22
The C3aR antagonist SB290157 was used to inhibit C3a/C3aR signaling activation in vivo. SB290157 was intraperitoneally injected into rats at a dose of 3 mg/kg per day for 4 weeks after the surgery. The nondrug-treated groups received an equal volume of vehicle at the same time.
The tail artery blood pressure of rats was measured at different time points (before the surgery and 2 weeks and 4 weeks after the surgery) using the BP-2000 Blood Pressure Analysis System (Visitech Systems, Apex, NC, USA). Prior to the measurement, all rats were acclimated in restraints. The blood pressure of each rat was determined by averaging 3 records.
Plasma samples were collected from rats using EDTA-coated Eppendorf tubes. The
concentration of renin in plasma was measured using a Renin Assay Kit
(Fluorometric) (Abcam). For acquiring kidney tissue homogenates, a piece of
kidney cortex tissue from each sample was weighed. We added PBS (tissue weight
(g): PBS volume (mL) = 1:9) and two beads to to each vail containing the sample
and homogenized the tissue at 60 Hz for 30 s for three times with Tissue Grinders
(Jingxin, Shanghai, China) to obtain tissue homogenates. The homogenates were
centrifuged 5000
The level of serum creatinine (Scr) was determined by using a liquid chromatography-tandem mass spectrometry (LC‒MS/MS) method according to the manufacturer’s instructions. The severity of tubular damage was assessed by two nephrologists in a blinded manner after observing whole kidney sections by hematoxylin-eosin (H&E) staining. Tubular damage was scored from 0 to 4 according to the distribution of lesions as follows: 0, no lesions; 1, less than 25%; 2, 25–50%; 3, 50%–75%; and 4, more than 75%. Renal tubulointerstitial fibrosis was quantified as the percentage of blue area per field in Masson’s trichrome-stained sections using ImageJ, and ten fields of view for each sample were used to collect the data.
The renal cortex tissue and cells were lysed in radioimmunoprecipitation assay
(RIPA) buffer (Sigma‒Aldrich) containing a protease/phosphatase inhibitor
cocktail (NCM Biotech, Jiangsu, China). The protein concentration of the samples
was measured with a bicinchoninic acid assay (BCA) protein quantitation kit
(Beyotime). Equal amounts of protein (20 µg for cells and 30 µg for
tissues) were separated via 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF)
membrane. After incubation in fast blocking buffer (EpiZyme, Shanghai, China),
the membrane was incubated with the following primary antibodies at 4 °C
overnight: anti-C3aR (1:1000, Bioss, Beijing, China), anti-C3/C3b (1:2000,
Abcam), anti-TGF
Total RNA from renal cortex tissues and cells was extracted using the FastPure
Cell/Tissue Total RNA Isolation Kit (Vazyme, Jiangsu, China). Complementary DNA
was generated using the HiScript® III 1st Strand cDNA Synthesis
Kit (+gDNA wiper) (Vazyme), and qPCR was performed using the ChamQ Universal SYBR
qPCR Master Mix (Vazyme). The specific primers used were as follows: Rat REN (F)
GATCACCATGAAGGGGGTCTCTGT (R) GTTCCTGAAGGGATTCTTTTGCAC; Rat C3aR (F)
AGGCAATGGGCTGGTGCTGT (R) CAGGAAGACACTGGCAAACAT; Rat
The renal cortex tissues were cut into 1 mm
The kidneys were fixed in 4% paraformaldehyde, embedded in paraffin and cut
into 4-µm-thick sections. After dehydration and antigen retrieval by EDTA
(pH 9.0) under high temperature and high pressure, the kidney sections were then
incubated with the following primary antibodies at 4 °C overnight: anti-E-cadherin
(E-cad) antibody (1:4000, TSA, Proteintech), anti-C3aR antibody (1:100, Bioss),
anti-CD68 antibody (1:200, Servicebio, Wuhan, China), anti-
The distribution of data were exiamined by Shapiro-Wilk test. The values for
each parameter were expressed as the mean
Serum from seven healthy volunteers was collected and incubated with recombinant
human renin or PBS. As shown in Table 1, the C3a concentration in serum incubated
with recombinant renin increased significantly compared to that in the same
samples incubated with PBS (with renin 922.16
| Patient | C3a Concentration with PBS (ng/mL) | C3a Concentration with Renin (ng/mL) | ΔC3a Concentration (ng/mL) | t-value | p-value |
| H1 | 340.78 | 444.23 | 103.45 | 2.989 | 0.024* |
| H2 | 560.23 | 758.79 | 198.56 | ||
| H3 | 644.18 | 778.64 | 134.46 | ||
| H4 | 577.3 | 851.79 | 274.49 | ||
| H5 | 835.77 | 1006.8 | 171.03 | ||
| H6 | 534.45 | 1344.69 | 810.24 | ||
| H7 | 437.96 | 1270.15 | 832.19 |
As C3aR is mainly expressed in TECs, we used serum from H7 incubated with human recombinant renin to stimulate HK2 cells and investigated the change in C3aR. As shown in Fig. 1A, treatment with serum incubated with recombinant renin significantly increased the protein level of C3aR in HK2 cells. The addition of aliskiren, a renin inhibitor, reversed the change in C3aR protein levels.
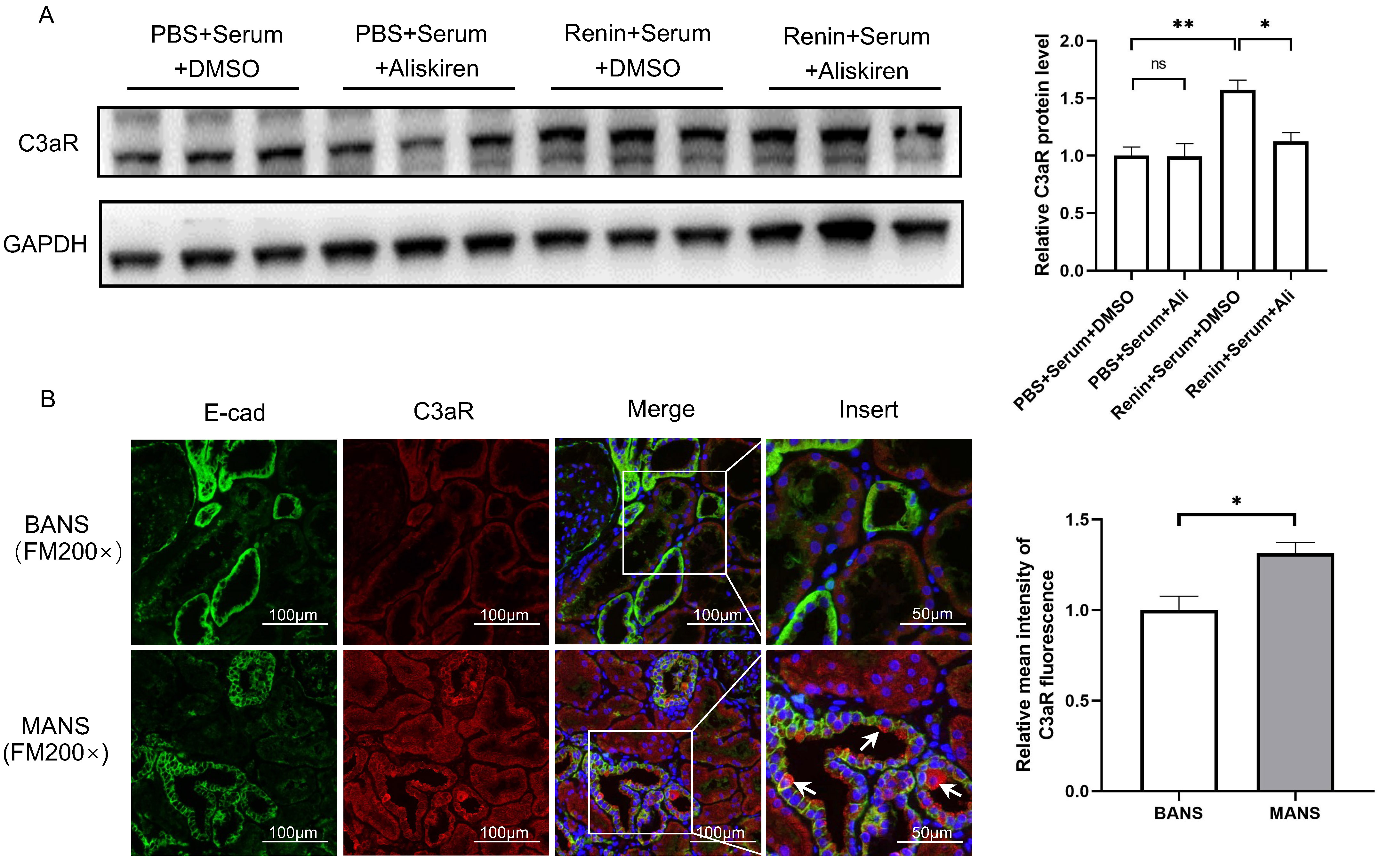 Fig. 1.
Fig. 1.Renin induces C3aR expression in renal tubular epithelial
cells. (A) Representative western blotting images and summarized data showing
the effect of renin-incubated serum and aliskiren intervention on C3aR protein
levels in HK2 cells. N = 3, by one-way ANOVA. (B) Representative
immunofluorescence staining images of C3aR and E-cad in kidney sections of
recruited patients with MANS and BANS (200
To further validate the results in human kidneys, we performed
immunofluorescence staining of C3aR in kidney samples from 6 hypertensive
patients who underwent renal biopsy. Among them, 3 were diagnosed with BANS and 3
were diagnosed with MANS. Compared to patients with BANS, the patients with MANS
were characterized by higher Scr, lower eGFR, higher renin activity and more
severe tubulointerstitial fibrosis (Table 2). The results revealed areas with
high C3aR expression in tubular epithelial cells (marked by E-cad) in patients
with MANS (Fig. 1B). The relative fluorescence intensity of C3aR (Fig. 1B), as
well as the plasma renin activity in MANS patients (with MANS 2.49
| Patients | Age (year) | Sex | Diagnosis | Disease duration | SBP (mmHg) | DBP (mmHg) | Scr (umol/L) | Proteinuria (mg/24 h) | eGFR (mL/min/1.73 m |
Renin activity (ng/mL/h) | Plasma C3 (g/L) | TIF degree |
| P1 | 33 | M | BANS | 3 y | 145 | 96 | 95 | 256 | 95.2 | 0.42 | 0.93 | + |
| P2 | 65 | M | BANS | 30 y | 143 | 79 | 140 | 1755 | 46.9 | 0.50 | 1.15 | + |
| P3 | 29 | M | BANS | 2 m | 200 | 110 | 117 | 7004 | 67.9 | 0.17 | 1.23 | ++ |
| P4 | 29 | M | MANS | 2 w | 220 | 145 | 259 | 1145 | 27.6 | 2.53 | 0.73 | +++ |
| P5 | 30 | M | MANS | 2 w | 185 | 137 | 486 | 831 | 12.8 | 3.09 | 1.40 | +++ |
| P6 | 32 | M | MANS on BANS | 3 y | 220 | 120 | 387 | 1623 | 16.6 | 1.85 | 0.83 | ++ |
M, male; SBP, systolic blood pressure; DBP, diastolic blood pressure; Scr, serum creatinine; eGFR, estimated glomerular filtration rate; MANS, malignant arteriolonephrosclerosis; BANS, benign arteriolonephrosclerosis; TIF, tubulointerstitial fibrosis degree; +, mild TIF; ++, moderate TIF; +++, severe TIF.
Although renin can cleave serum C3 into C3a, it can also catalyze angiotensinogen to generate angiotensin II, which also influences the downstream signaling of C3aR [30]. To avoid the effect of angiotensin II, we used a C3a analogue that has been reported in previous studies in the following experiments.
The C3a analogue also significantly increased the C3aR protein level in HK2
cells (Fig. 2A). Notably, the C3a analogue triggered the synthesis of
TGF
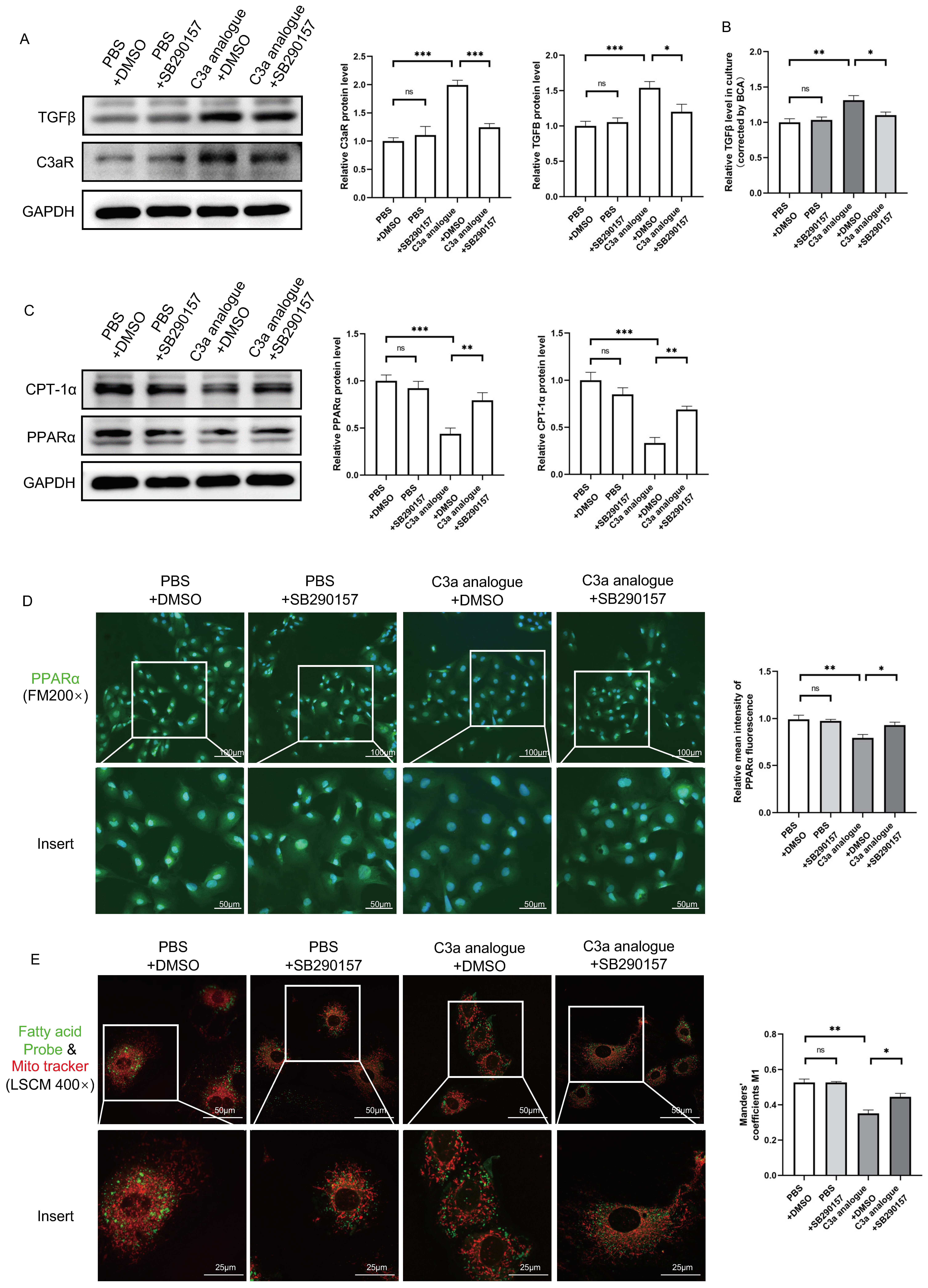 Fig. 2.
Fig. 2.The C3aR antagonist SB290157 mitigates the C3a
analogue-induced profibrotic phenotype transition and Mito FAO inhibition in HK2
cells. (A) Representative western blotting images and summarized data of C3aR
and TGF
Renin is a species-specific enzyme. No evidence has shown whether rat renin can cleave C3 in rats. Before performing the validation experiments in vivo, we carried out western blotting analysis to detect the effect of rat renin on rat C3. Interestingly, western blotting analysis showed that the level of C3 cleavage increased as the concentration of renin increased, as indicated by the increasing contents of iC3b and C3c, two C3 cleavage fragments (Fig. 3A). Slight increases in the contents of iC3b and C3c were detected when purified rat C3 protein was incubated with rat recombinant renin at 1.2 µg/mL and 12.5 µg/mL, but not statistically significant. Statistical analysis showed that renin-mediated C3 cleavage reached its highest level with 25 µg/mL renin (Fig. 3B,C). The above results demonstrated that rat renin could cleave C3.
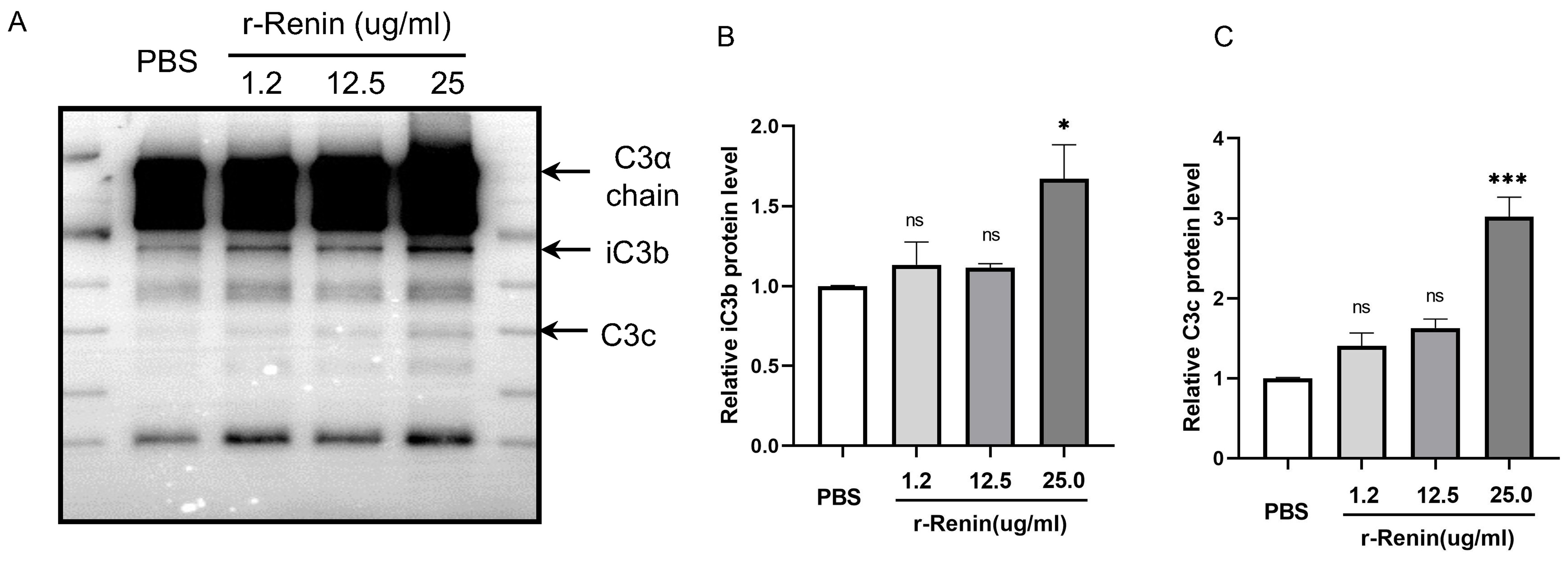 Fig. 3.
Fig. 3.Coincubation with recombinant rat renin increases the cleavage
fragments of C3. (A) Representative western blotting images, summarized data of
(B) iC3b and (C) C3c protein levels showing C3 cleavage after incubation with
recombinant rat renin at 0, 1.2, 12.5 or 25 µg/mL. r-Renin, recombinant rat
renin. N = 3,
Renin-dependent hypertension 2K1C rat model was used for in vivo
experiments. As shown in Fig. 4A, the blood pressure (BP) of 2K1C rats was
significantly elevated at 2 weeks (170
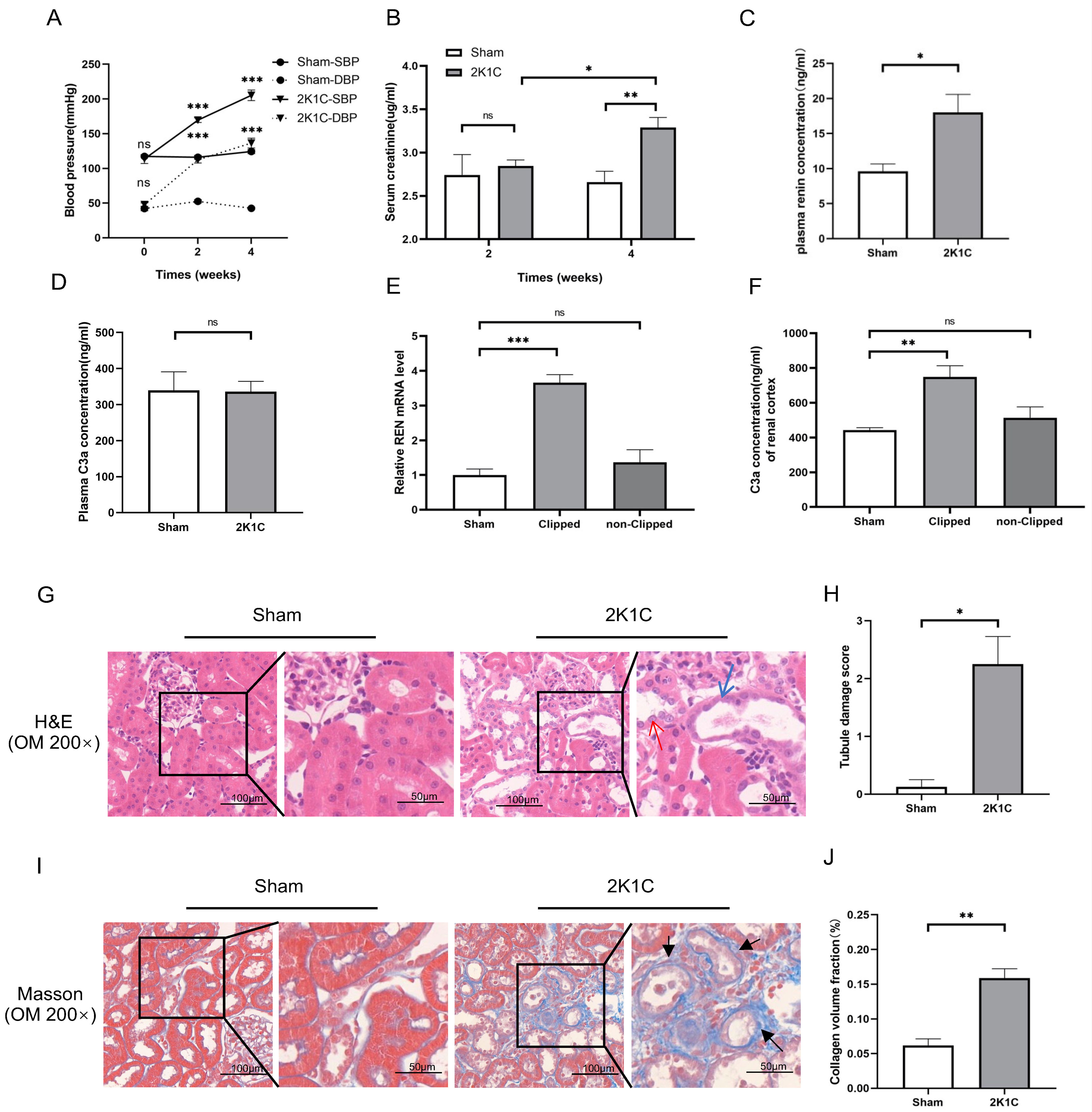 Fig. 4.
Fig. 4. Tubulointerstitial injury, renin mRNA and C3a concentration in
the clipped kidney cortex of 2K1C rats. (A) Summarized data of blood pressure,
(B) Scr levels, (C) plasma renin concentration and (D) plasma C3a concentration
in sham rats and 2K1C rats. (E) Summarized data of relative REN mRNA expression
and (F) C3a concentration in the renal cortex in sham rats and 2K1C rats. N = 4
per group, by one-way ANOVA. (G) Representative H&E staining images, (H)
summarized data of tubular damage scores, (I) representative Masson staining
images and (J) summarized data of collagen volume fraction of the kidney cortex
in sham rats and 2K1C rats (clipped kidney) (200
As shown in Fig. 4C, the plasma renin concentration of 2K1C rats increased
markedly at 4 weeks (2K1C 18.00
After 4 weeks of 2K1C, the protein levels of C3aR and
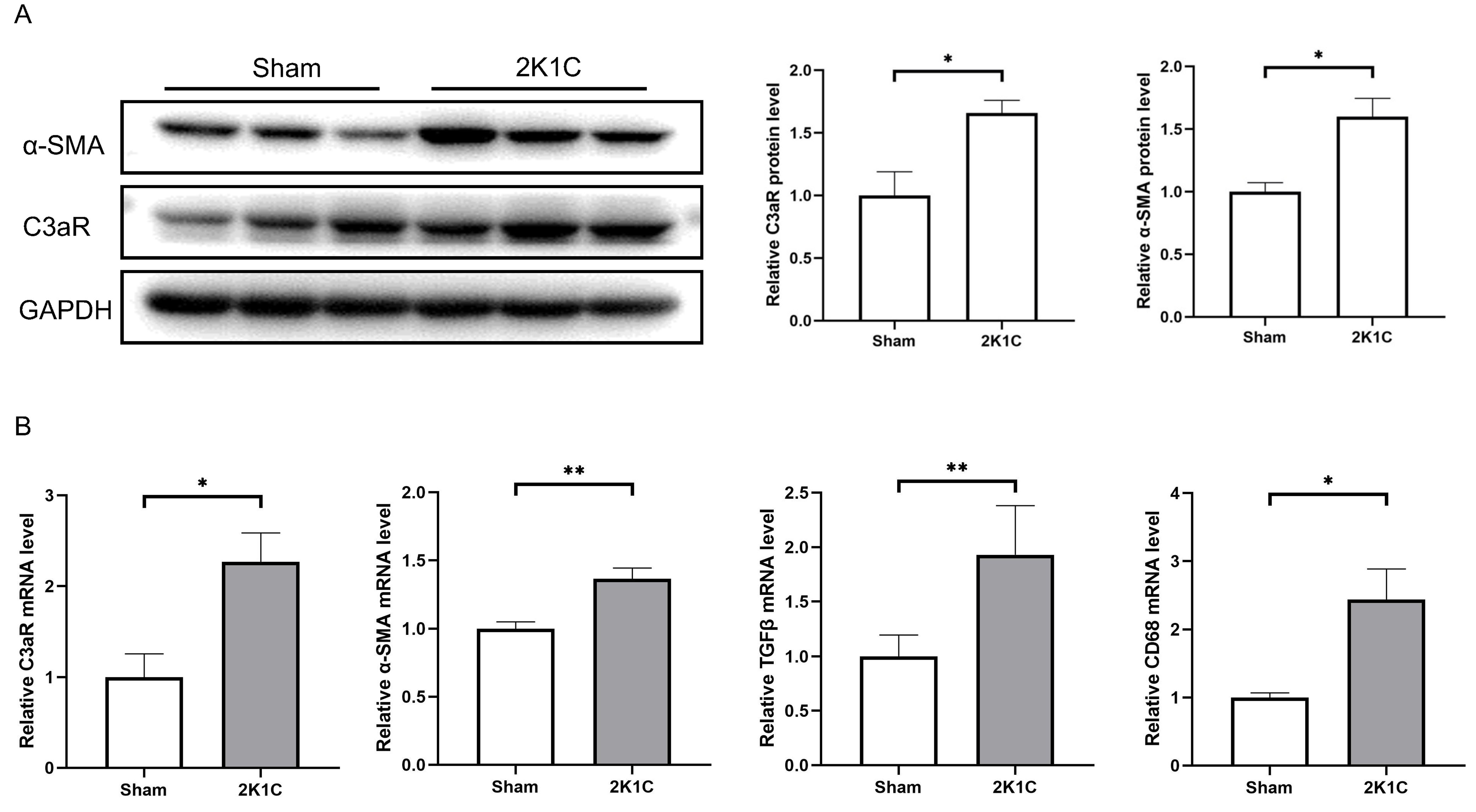 Fig. 5.
Fig. 5.C3aR and mesenchymal transition markers increase in the kidney
cortex of 2K1C rats. (A) Representative western blotting images, summarized data
of C3aR and
Treatment with the C3aR antagonist SB290157 did not influence the BP of 2K1C
rats at 4 weeks after surgery (Fig. 6A). The renin mRNA or C3a level in the
clipped kidneys of 2K1C rats was also not influenced by SB290157 (Fig. 6B,C). Of
note, tubular injury and interstitial fibrosis in the clipped kidneys of 2K1C
rats were attenuated by SB290157 treatment, as indicated by reduced Scr,
decreased tubular damage scores and reduced collagen deposition (Fig. 6D–H). The
protein levels of C3aR, TGF
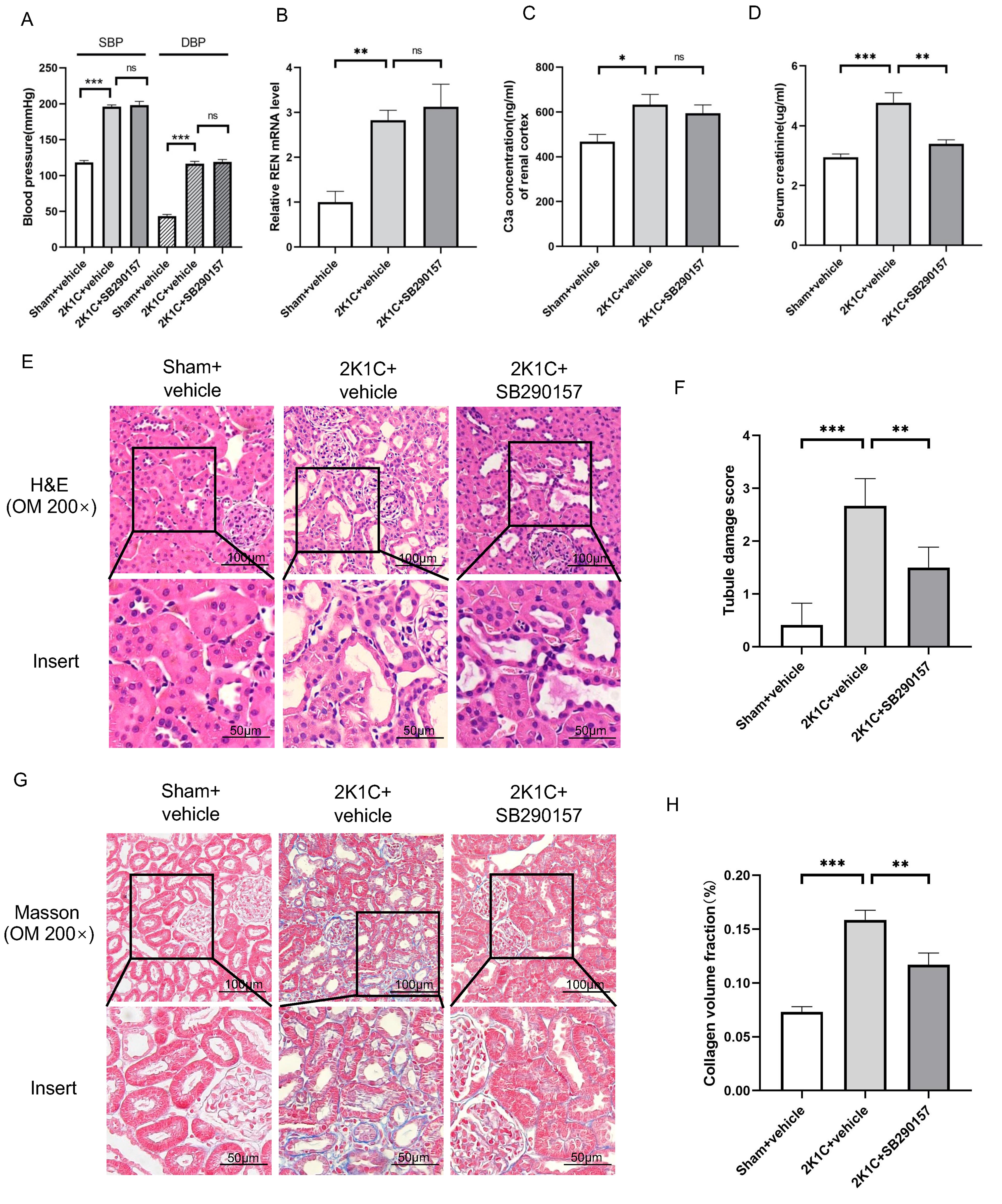 Fig. 6.
Fig. 6.The C3aR antagonist SB290157 alleviates tubulointerstitial
fibrosis in 2K1C rats. (A) Summarized blood pressure and (D) Scr levels in
sham+vehicle, 2K1C+vehicle and 2K1C+ SB290157 rats. N = 6 per group. (B)
Summarized REN mRNA levels and (C) C3a concentration in the renal cortex in
sham+vehicle, 2K1C+vehicle (clipped kidney) and 2K1C+SB290157 (clipped kidney)
rats. N = 6 per group. (E) Representative H&E staining images, (F) summarized
tubular damage scores, (G) representative Masson staining images and (H)
summarized collagen volume fraction of the renal cortex in sham+vehicle,
2K1C+vehicle (clipped kidney) and 2K1C+SB290157 (clipped kidney) rats
(200
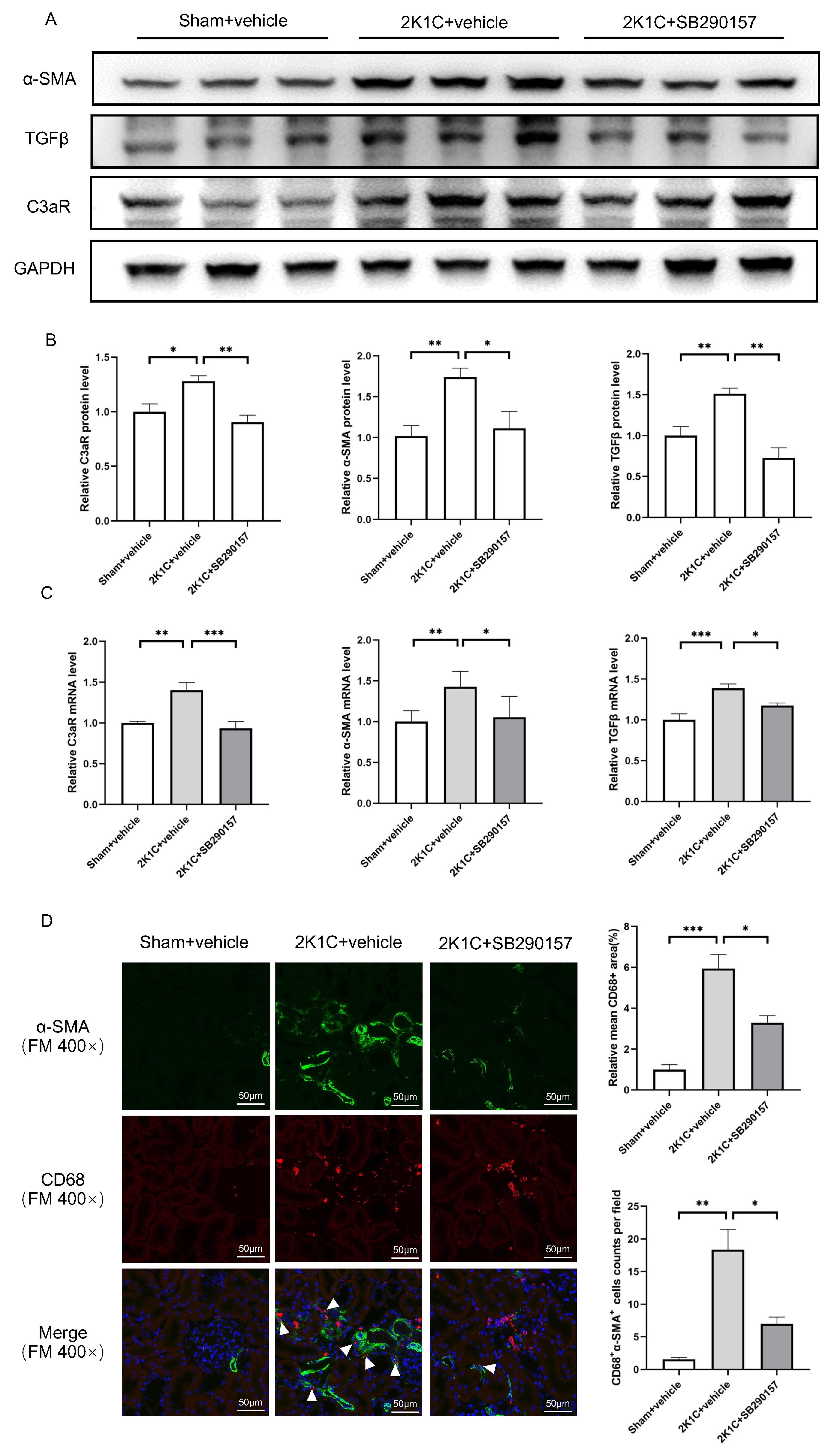 Fig. 7.
Fig. 7.The C3aR antagonist SB290157 reduces C3aR expression and
mesenchymal transition in 2K1C rats. (A) Representative western blotting images
and (B) summarized data of C3aR,
Under TEM, a higher abundance of lipid droplets was found in the proximal TECs
of the clipped kidneys of 2K1C rats. Treatment with SB290157 reduced the
accumulation of lipid droplets in the proximal TECs in 2K1C rats (Fig. 8A). The
protein levels of PPAR
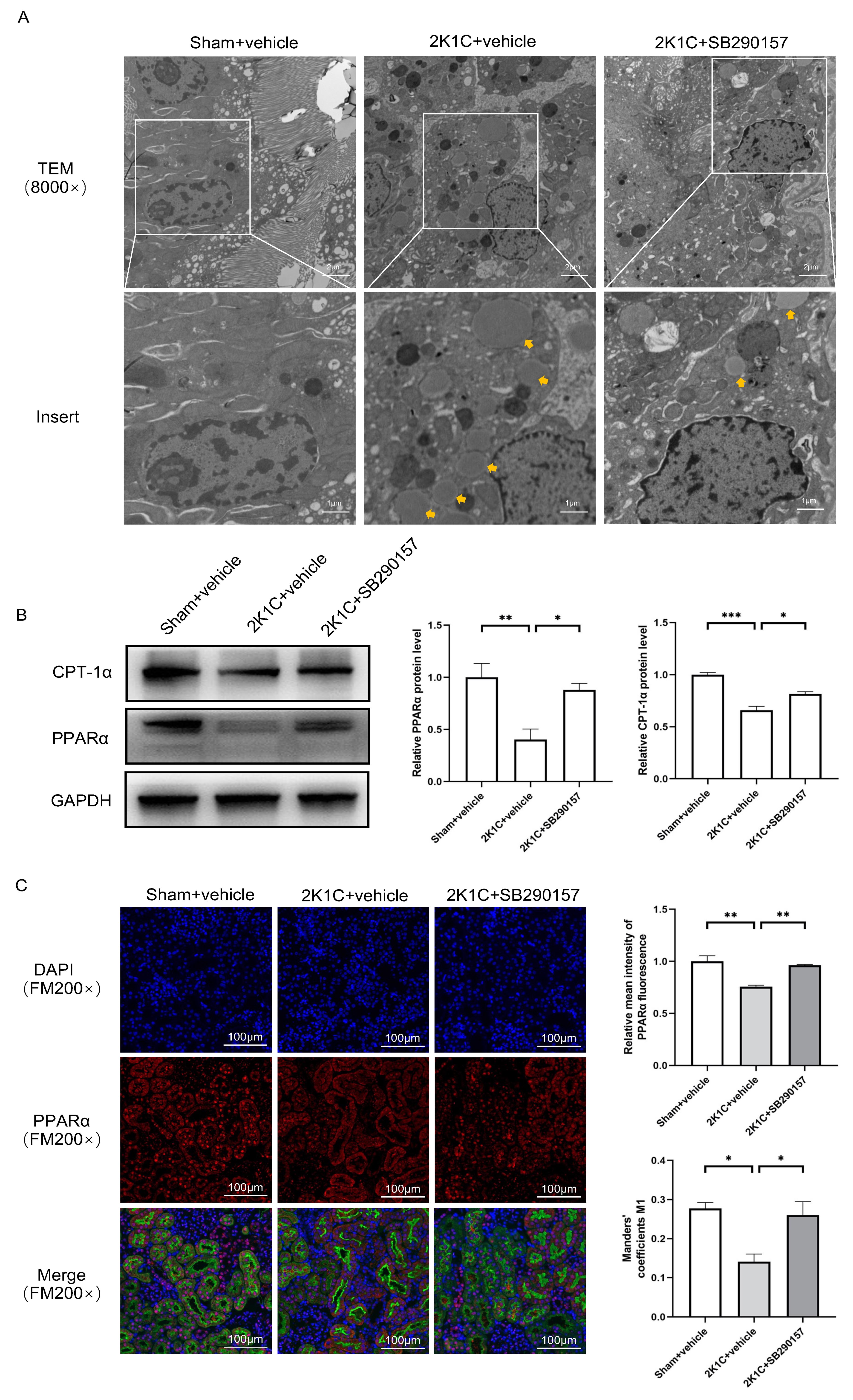 Fig. 8.
Fig. 8. The C3aR antagonist SB290157 restores
PPAR
In the clinic, renin-induced hypertension combined with tubule injury remains a
problem with a high prevalence. Here, we provide new in vivo and
in vitro evidence that targeting C3a/C3aR signaling may achieve some
benefits in the treatment of renin-dependent hypertension-induced kidney injury.
In the present study, we verified that renin cleaved C3 into C3a and activated
C3a/C3aR signaling in TECs. The activation of C3a/C3aR signaling played a vital
role in the tubular profibrotic phenotype transition by downregulating
PPAR
Few previous studies have focused on the relationship of renin with C3. Only one study has reported that human renin can cleave C3 in vitro [6]. They also reported that murine renin was unable to act on murine C3. Renin is a species-specific enzyme. In the present study, we tested the ability of renin to cleave C3 in humans and rats. Our finding that human renin could catalyze C3 cleavage in human serum is in agreement with previously reported results [6]. Notably, our results proved that recombinant rat renin could directly cleave purified rat C3. In addition, we observed that C3a levels increased synchronically with renin in the kidney cortex of clipped kidneys in 2K1C rats. Therefore, there may exist different mechanisms of renin in mice and rats. By analyzing our data and the data reported before, we find that 1.2 µg/mL recombinant human renin could achieve significant C3 cleavage while in rat 25 µg/mL recombinant renin was required, suggesting a weaker C3 cleavage effect of rat renin.
In different disease models, the location of the C3aR protein in the kidney cortex has been reported to differ from that under normal conditions. Under normal conditions, C3aR distribution is restricted to renal epithelial cells and interstitial cells [16, 17]. However, high C3aR expression in podocytes, mesangial cells and endothelial cells has been reported in patients and animals with various chronic kidney diseases [19, 31, 32]. Here, we showed increased C3aR expression in the TECs of MANS patients with high renin activity. In comparison, C3aR expression in the TECs of BANS patients was much lower. These results are in line with a previous report that more complement components, including C3a, were found in MANS patients than in BANS patients and normal controls [12]. Our results provide a novel mechanism of the RAS in renal injury by activating C3a/C3aR signaling, especially in malignant hypertensive patients with a high renin state.
The location of sites with high C3aR expression indicates the potential
involvement of C3a/C3aR signaling in renin-dependent hypertension-induced
tubulointerstitial fibrosis. It is known that TECs adopt a profibrotic phenotype
and secrete profibrotic factors, including TGF
Another important finding in our study is that renin/C3a/C3aR activation impairs
PPAR
C3a/C3aR signaling is widely involved in kidney diseases through various mechanisms [48]. In acute kidney injury, there is controversy about the role of C3a/C3aR signaling. Reportedly, C3a promotes the synthesis of inflammatory mediators and chemokines in ischemia‒reperfusion induced kidney injury [49, 50]. In Shiga toxin-associated hemolytic uremic syndrome, C3a/C3aR signaling activation induces mitochondrial dysfunction and imbalances the redox state to aggravate tubular injury [51]. However, C3a/C3aR activation in sepsis-induced kidney injury has been demonstrated to be a protective way to limit the bacterial load and renal damage [27]. In most CKD models, C3a/C3aR signaling is defined as dangerous signaling that stimulates the synthesis of proinflammatory and profibrotic cytokines in renal parachymal cells and infiltrating cells [52, 53]. In addition, C3a is reported to be an important inducer of the cellular mesenchymal transition, including that of epithelial cells and endothelial cells [19, 32]. Our study verifies the effect of C3a/C3aR activation in promoting tubulointerstitial fibrosis in hypertension-induced CKD. Inhibiting C3a/C3aR signaling is likely to be a potential therapeutic strategy in hypertensive subjects with tubulointerstitial fibrosis.
The C3aR antagonist SB290157 has been confirmed as a C3aR inhibitor in the lung [22], brain [54], heart [55] and kidney [31, 44, 56]. SB290157 treatment has proven beneficial in chronic renal fibrosis. Given the curative potential of SB290157, we performed a series of experiments to test its effect on renin-dependent hypertension-induced kidney injury. Our data are the first to demonstrate that SB290157 significantly mitigates renin-dependent hypertension-induced Mito FAO defects and tubular profibrotic phenotype transition by blocking C3a/C3aR signaling. In clinical practice, ACEIs and ARBs are frequently used to treat hypertension patients with RAS activation. However, the utilization of these drugs results in persistent renin elevation. Therefore, the blockage of C3aR may be a potent strategy for preventing tubulointerstitial injury. Of note, the inhibitor of another anaphylatoxin receptor, the C5aR inhibitor avacopan, has been approved by the FDA to treat anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis [57]. No C3aR inhibitor is currently available in the clinic. We believe that C3aR inhibitors may be a potential treatment for renin-dependent hypertension-induced kidney injury in the future.
There are a few limitations of the current study. First, our study is limited by the small sample size of patients and rats employed. And we did not directly detect the C3a protein in western blotting analysis because of the lack of a C3a-specific antibody. The levels of C3a measured in tissue homogenate may underestimate the C3a concentration in clipped kidneys due to the limitations of an ELISA from tissue homogenate, as a portion of C3a may remain within the tissue matrix. We did not use the renin inhibitor aliskiren to treat 2K1C rats since its ability to inhibit renin activity in rodents has been reported to be weak [58].
Another potential limitation of our study is that we did not verify the
specificity of SB290157 due to its agonistic effect on C5aR2 [59]. To prove the
effect of SB290157 on C3aR, an in vitro calcium assay would be a
possibility considering that SB290157 could inhibit the influx of Ca
At last, we only evaluated the role of SB290157 in 2K1C rat model to simulate renin-dependent hypertension status in vivo. For the translation of our findings to clinical applications, it is necessary to further confirm the effect of SB290157 in additional models of renin-dependent hypertension, for instance, spontaneous hypertension rats and double transgenic rats harboring both the human renin and human angiotensinogen genes (dTGR), in future studies.
The results of our study provide strong evidence that renin can cleave C3 into
C3a and activate C3a/C3aR signaling in TECs to impair
PPAR
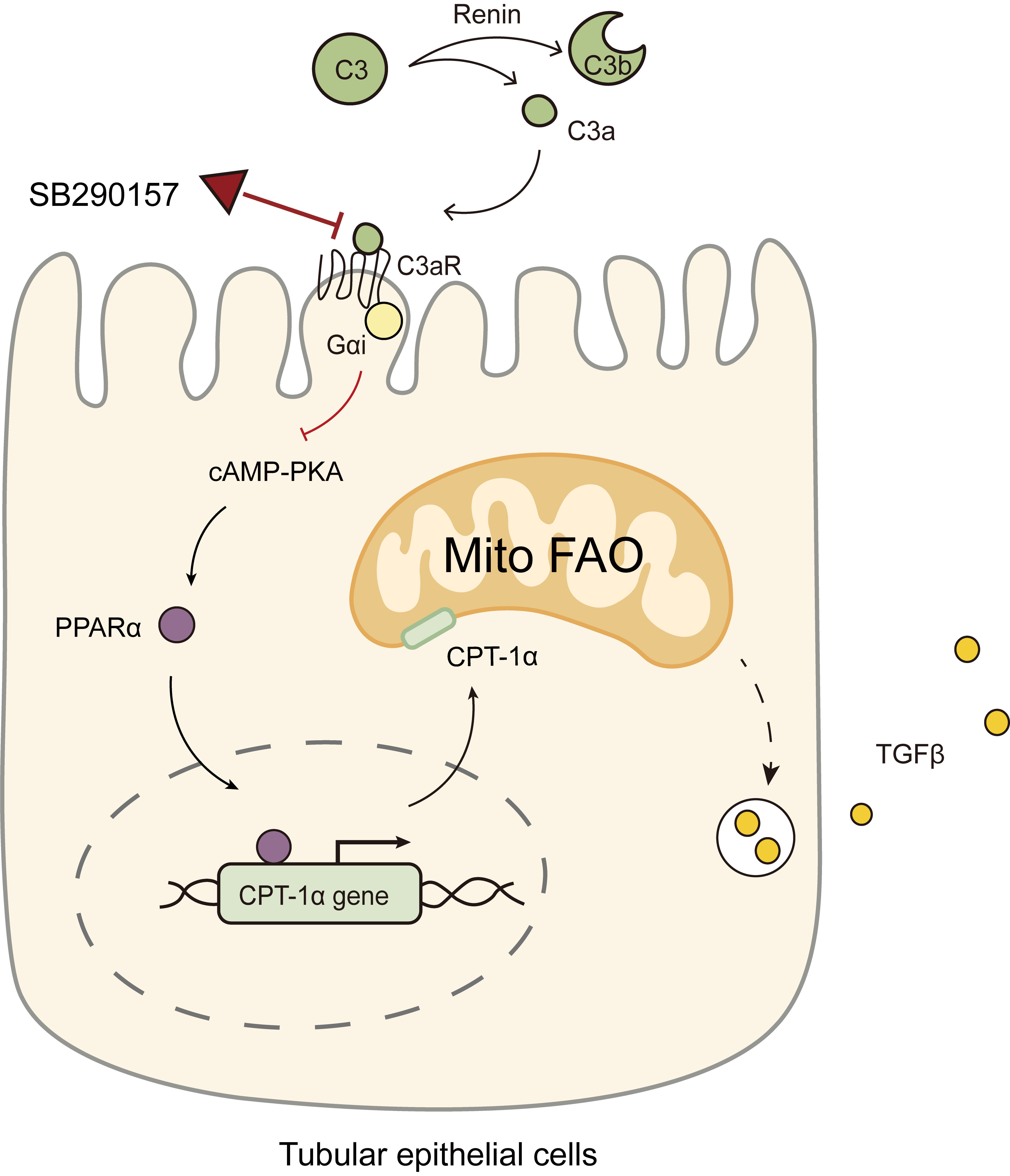 Fig. 9.
Fig. 9.
Diagram showing the C3aR antagonist SB290157 targeting C3a/C3aR
signaling to restore PPAR
ANCA, anti-neutrophil cytoplasmic antibody; AQP1, aquaporin 1; BANS, benign
arteriolonephrosclerosis; BCA, bicinchoninic acid assay; BP, blood pressure; CKD,
chronic kidney diseases; CPT-1
The datasets used and/or analyzed in the current study are available from the corresponding author on reasonable request.
CW, ZW and WZ designed the research study. CW, ZW and KJ performed the research. TX collected the clinical data. CW, ZW and HM analyzed the data. JX and XP provided help on the analysis of histological images and transmission electron microscopy images. XF made contributions to data interpretation. XF and WZ provided supervision. CW and ZW wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors approved the final version of the parper. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study protocol has been approved by the Ethics Committee of Ruijin Hospital, Shanghai Jiao Tong University School of Medicine (approval number 20220-319). Patient information was managed according to applicable data protection regulations. All participating individuals have given their written informed consent to participate in the study. Animal study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethic Committee on Animal Care of Charles River Laboratories (approval number P2021100).
We are grateful for the sacrifice of the animals in this study and thank all the participating staff of the Department of Nephrology, Ruijin Hospital, Shanghai Jiao Tong University. We thank all the patients participated in the study for their cooperation.
This study was funded by the National Natural Science Foundation of China (Grant number 81470967, 81670613 and 82270705) and the Shanghai Science and Technology Committee (Grant number 21ZR1440300).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.