
 , Haribaskar Ramachandran 1,†, Andrea Rossi 1,*
, Haribaskar Ramachandran 1,†, Andrea Rossi 1,*
1 Genome Engineering and Model Development lab (GEMD), IUF-Leibniz Research Institute for Environmental Medicine, 40225 Düsseldorf, Germany
†These authors contributed equally.
Academic Editor: Said El Shamieh
Abstract
Genomic mutations are the driving force of biological diversity but they are also the cause of a plethora of human diseases ranging from heritable disorders to neurological pathologies and cancer. For most genetic disorders, there is no curative treatment available to date. The demand for precise, preferably patient-specific, treatment regimen offering cure is naturally high. Genome editing by Zinc Finger Nucleases (ZFNs), Transcription Activator-Like Effector Nucleases (TALENs), and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas enables targeted manipulation of genomes, thereby offering the opportunity to treat such diseases. While ethical and regulatory guidelines need to be developed and considered, the prospect of genome editing for curative treatment is certainly exciting. Here, we review the current state of therapeutics based on genome editing techniques. We highlight recent breakthroughs, describe clinical trials employing genome editing-based medicine, discuss the benefits and pitfalls, and take a look into the future of genome editing.
Keywords
- genome editing
- CRISPR/Cas
- ZFN
- TALEN
- prime editing
- clinical application
The term genomic mutation denotes an alteration in the nucleic acid sequence of an organism’s genome [1]. Irrespective of whether changes occur in coding or non-coding regions, mutagenesis can lead to silent/synonymous, loss-of-function, or gain-of-function mutations [2, 3, 4]. The latter two are the actual main generators of functional diversity, and they will be sampled by natural selection, the driving force of evolution [2, 3, 4, 5, 6]. Thus, nature has a forward genetic screening strategy.
On the other hand, and from a human-centric point of view, these mutations can lead to genetic disorders [7]. Hitherto, up to 8000 monogenic diseases [8], i.e., diseases that arise due to mutations in a single gene, and several genetic mutations associated with cancer [9] have been classified. In order to offer a permanent cure to such disorders, targeting and/or replacing mutated genes using reverse genetics constitutes an attractive therapeutic approach. Conventional gene therapy involves the complete replacement of defective genes or gene products with a wild type gene using viral vectors [10, 11]. Initial attempts employing ex vivo retrovirally modified cells were met with limited success, were accompanied by severe side effects, and resulted in death of one patient due to a strong immune response [12, 13, 14, 15]. Currently, about 20 conventional gene therapy products are approved by the US Food and Drug Administration (FDA) [11]. Major points of concern include potential severe immune reactions in response to treatment, insufficient clarification of long-term adverse side effects, and potential risk of oncogenesis, mainly due to virus-dependent gene transfer [14, 16, 17]. Targeting the mutated part of a gene directly and specifically could avoid the risk of insertional oncogenesis associated with traditional gene therapy. A targeted approach would allow more precise treatment and potentially result in a lower rate of adverse events.
Exploiting genetic alterations has a long history in humankind, but the development of modern genetics by Gregor Mendel and others in the 19th and 20th century marked a major step in this regard [18, 19, 20]. The discovery of restriction enzymes during the 1970s and the discovery of meganucleases, which induce DNA double strand breaks (DSBs) at specific genomic sites, opened the doors to targeted DNA modifications in test tubes and cells [21, 22, 23].
Such targeted DNA modifications were, and still are, mainly based on the induction of DSBs by endonuclease activity followed by their resolution through cellular DNA repair [24]. DNA repair events are mainly controlled by two different pathways: non-homologous end joining (NHEJ) or homology-directed repair (HDR) [25]. NHEJ is an error-prone pathway that results in the generation of small nucleotide insertion and deletion (indel) mutations (indels), and it is widely exploited to generate gene knockout (KO) models [26]. The HDR pathway is largely restricted to the G2/S phase of the cell cycle and it is employed for the generation of knock-in (KI) models through the co-delivery of a donor template [27, 28].
Currently, three major methods make use of genomic targeting combined with endonuclease activity to mediate gene KOs and KIs:
(1) Zinc Finger Nucleases (ZFNs) represent the first developed modular genome editing technique. ZFNs are engineered by fusing a DNA-binding domain of zinc finger proteins with a non-specific catalytic domain of the FokI endonuclease [29, 30];
(2) The DNA-binding protein Transcription Activator-Like (TAL) Effector, combined with FokI termed TAL effector nuclease (TALEN) represented a huge leap following ZFNs [31, 32, 33];
(3) The discovery of Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) paved the way to the development of the most widely applied genome editing method [34, 35, 36, 37]. Thanks to the simultaneous effort of several groups, the CRISPR/Cas system has been developed and is since employed by myriads of researchers to generate gene KO and KI models [38, 39, 40, 41, 42, 43, 44, 45]. Here, the bacterial endonuclease Cas9 is combined with a short guide RNA (sgRNA) to mediate its guidance to a specific genomic region [38, 39, 40, 41, 42, 43, 44, 45].
We envision the following aspects to be carefully considered when planning to use genome editing for therapeutic purposes (e.g., correction of a deleterious mutant):
(1) it needs to be efficient to reliably result in genome editing;
(2) it needs to be specific, because any undesired editing (i.e., off-target effect [46]) in the genome increases the risk of side effects;
(3) it needs to be feasible, i.e., target selection and manufacturing process need to be straight-forward and easy to apply;
(4) it needs to be affordable to enable accessibility to every patient, at least for the foreseeable future.
Taking all these aspects into account, a comparison of the three tools as potential therapeutic options can be made.
ZFNs resulted from basic research on zinc finger proteins [29, 47, 48, 49, 50]. Three zinc fingers, each recognizing 3 bp of DNA sequence on each strand, must bind to facilitate FokI dimerization to enable DNA cleavage [29, 47, 48, 49, 50]. The main caveats of ZFNs are low specificity, low efficiency, high time- and labor-intensity regarding target selection and manufacturing process, and low selectivity due to the high sequence prerequisites [51, 52]. It has to be mentioned, however, that ZFN target range, specificity and efficiency can be increased, e.g., by altering their structural properties [53].
TALENs are derived from TAL effectors, secreted virulence factors of the plant pathogen Xanthomonas [31]. As ZFNs, TALENs are fused to FokI, thus requiring a pair of TALENs facilitating FokI dimerization and DNA cleavage [31, 32, 33]. Compared to ZFNs, TALENs offer increased efficiency, easier design, higher selectivity because DNA recognition relies on single bases instead of triplets, and offer lower risk of off-target effects as well as reduced cytotoxicity [31, 54, 55]. Although some studies report CRISPR/Cas- or ZFN-mediated editing of mitochondrial DNA, TALENs are currently the only reliably demonstrated tool to target and manipulate mitochondrial genomes [56, 57, 58, 59]. A clear disadvantage of TALENs is their considerably larger size (3 kb) compared to ZFNs (1 kb), because larger plasmids can be challenging to assemble, clone and deliver into cells [60].
CRISPR/Cas-mediated genome editing is by far the most prominent and promising technique. It offers high editing efficiency and although risk of off-target effects in theory was initially higher compared to ZFNs and especially TALENs, sophisticated stringent guide selection algorithms have rendered it presumably the most specific of the three techniques to date [61, 62]. Combining these features with exceptional ease-of design and ease-of-use, CRISPR/Cas-mediated genome editing has made its way rapidly into everyday laboratory work [40]. Continuous improvements, especially regarding efficiency, target selection, off-target avoidance, and delivery (the latter is, e.g., reviewed here: [63]), have been implemented rapidly and are still ongoing.
In order to introduce a DSB, the Cas9 protein from Streptococcus pyogenes requires the binding of a sgRNA sequence termed protospacer to a matching genomic DNA sequence that is followed by a three nucleotide (NGG) protospacer-adjacent motif (PAM) [64]. CRISPR/Cas9 has been widely exploited as a versatile tool for genome editing and, with the above constraints, allows targeting of nearly any genomic region of interest. However, for those DNA regions that cannot be targeted with CRISPR/Cas9, other Cas variants (e.g., Cas12a) or engineered Cas that rely on different PAMs can be harnessed to overcome the described target restriction [65, 66, 67].
Regarding the economical aspect, it has to be noted that any therapeutic intervention based on genome editing is currently associated with tremendous cost. The conventional gene therapy product Zolgensma currently costs approximately 1.9 mio € per treatment course and can thus serve as an estimate for one-time administration of genome editing-based treatment [68]. In general, patent fees, the need of dedicated teams of scientists and physicians working together to treat single patients, and need for hospitalization and treatment of the patients strongly contribute to the high costs [69, 70]. In addition, manufacturing of genome-edited cells ex vivo, e.g., hematopoietic stem and progenitor cells (HSPCs), on a treatment scale requires large amounts of expensive reagents with good manufacturing practice grade as well as careful monitoring of cell quality prior reinfusion into patients [71]. Although this is certainly irrespective of the actual method applied, CRISPR/Cas has a huge edge here, not least because it is widely used, thus enabling tremendous progress in short time.
Choice of delivery is another important aspect with respect to genome editing-based medicine and a variety of methods including viral and non-viral vectors can be potentially used [72, 73]. Discussion of advantages and pitfalls of delivery for genome editing approaches is an extensively complex topic on its own and beyond the scope of this review. For a detailed overview, the interested reader is referred to a review addressing this aspect [74].
A summary of the properties of the three main established genome editing methods described here with respect to clinical applicability can be found in Table 1 (Ref. [61, 62, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84]).
| Genome editing method | ZFN | TALEN | CRISPR/Cas9 |
| Average editing efficiency |
Low | Mid–High | High |
| Editing specificity | High | High | High |
| Target design | Protein engineering for every target, requires optimization | Protein engineering for every target | Short guide sequence of around 20 nt length |
| Cost (including labor) | High | Medium | Low |
| Prerequisites of guide selection | High | Low–Very low | Low |
| Based on information from [61, 62, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84].
| |||
CRISPR/Cas-mediated genome editing and its potential application in human patients has made a lasting impact also in the public perception [85]. Remarkably, and in contrast to other prominent examples of “gene technology” such as human cloning [86], CRISPR/Cas has come to clinical application in record-breaking speed. And it has done so with great success. The individual stories of Victoria Gray and Jimi Olaghere, two patients suffering from sickle cell disease (SCD) who have recovered tremendously in response to treatment with CRISPR/Cas-mediated genome editing, are proof of that [87, 88]. Much hope thus lies in currently ongoing clinical trials. In addition, ZFN- and TALEN-based therapies are also under investigation and envisioned as potential therapeutic regimen in clinical trials. Although they are less popular compared to CRISPR/Cas because of the limitations listed above, the theoretically higher specificity due to more restricted target recognition could render them safer for clinical application. The current status of clinical trials employing either of the aforementioned genome editing methods is described in the next section.
To analyze the current status of clinical trials, we accessed the National Institute of Health (NIH) ClinicalTrials.gov study database. Although we cannot rule out to miss single studies by using this approach (e.g., not all studies need to be registered), the database includes worldwide studies, and thus provides a comprehensive, easily accessible, and publicly available overview. As of 2022-06-01, the NIH study database returns a list of 88 hits when queried for the search terms “CRISPR OR TALEN OR ZFN OR ‘zinc finger nuclease’ OR ‘genome editing”’. Manual correction for long-term follow-up studies of already completed studies (8), studies withdrawn before initiation (5), or purposes unrelated to treatment (21), such as the use of genome editing for diagnostic test development or disease modelling, yields 54 hits.
The majority of these studies is associated to CRISPR (37), followed by ZFN (12), and TALEN (5) (Fig. 1A). For the sake of simplicity, we assigned the one study planning to evaluate both CRISPR and TALEN to both CRISPR and TALEN, whereas we assigned the single study planning to employ CRISPR base editing to CRISPR.
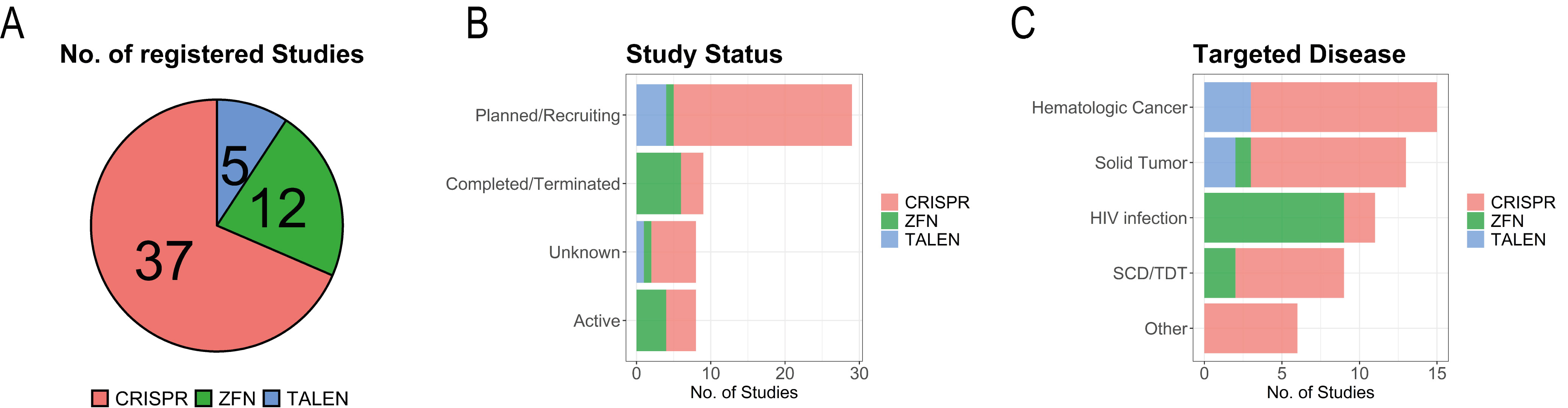 Fig. 1.
Fig. 1.Studies enlisted in the clinical trials database. Details are shown for distribution of genome editing methods by total number of studies (A), by study status (B), and by targeted disease (C).
Most of the studies (Fig. 1B), however, have not yet started and are in the phase of planning or recruiting patients (34). Eight studies are listed as completed, the majority investigating ZFNs for a potential cure of human immunodeficiency virus (HIV) infection (6), and two studies investigating CRISPR/Cas-mediated genome editing for treatment of solid cancer. Eight studies are listed as active, half of them employing ZFNs and the other half employing CRISPR-based treatment. The ZFN-based studies aim at treatment of HIV infection (3), and Transfusion-Dependent Beta-thalassemia (TDT, 1). The CRISPR/Cas-based studies aim at treatment of TDT (2), SCD (1), and refractory viral keratitis (1). Five studies planning to treat cancer were withdrawn before initiation, the majority of them CRISPR/Cas-based (4) and one based on TALENs.
Three studies employing ZFNs to treat Mucopolysaccharidosis I, Mucopolysaccharidosis II, and Hemophilia B were listed as terminated with the subjects being treated moved to long-term follow-up studies. Additionally, one study employing CRISPR/Cas-mediated genome editing for the treatment of advanced cancer was listed as terminated, although results of this study are actually published [89]. The eight studies with unknown status are comprised of CRISPR/Cas- (6), TALEN- (1), and ZFN-based (1) genome editing. The 29 studies listed as planned/recruiting are mainly comprised of CRISPR/Cas-mediated genome editing approaches (24), with the vast majority (14) of them employed to treat hematologic malignancies (9), solid tumors (5), and SCD/TDT (4). The remaining studies listed as planned/recruiting include one application of ZFNs to treat SCD and four studies employing TALENs to treat hematologic malignancies (3) and solid tumors (1).
In terms of disease (Fig. 1C), the majority of registered CRISPR/Cas-based studies targets cancer (total: 23, hematologic: 13, solid: 8, both hematologic and solid: 2), followed by SCD (4), TDT (3), and HIV infection (2). Nine out of 17 studies listed for ZFN target HIV infection, whereas the five studies employing TALENs aim at curing cancer (3) or treat precancerous lesions (2).
In contrast to the many studies listed in the clinical trials database of the NIH, only few (7) are associated with peer-reviewed publications describing their results or at least partial results thereof. To provide the most reliable overview of the current state of genome editing-based trials, we focused on these studies. There are certainly many interesting approaches in the pipeline or already applied in clinical studies, but the outcome cannot be certain and available data are limited. Results of four further studies are either submitted (2) or already available (2) in the clinical trials database. Whether this implies failure of the majority of studies supposed to be already completed or whether their outcomes simply have not been reported yet, remains to be seen. Nonetheless, the results that are published are promising, especially regarding the safety of the approaches. A summary of the study results described in the following sections can be found in Table 2 (Ref. [89, 90, 91, 92, 93, 94, 95]).
| Study-associated publication | n | Method | Disease | Cell Type | Target gene (gene symbol) | Outcome | Delivery of genome editing components | Editing efficiency | NCT Number (Status) |
| [89] | 3 | CRISPR | refractory advanced myeloma (2), refractory metastatic sarcoma (1) | T cells | TRAC/TRBC/PDCD1 | safe & feasible | RNP by electroporation | 45/15/20% | NCT03399448 (T) |
| [90] | 12 | ZFN | HIV | CD4 |
CCR5 | safe & feasible | adenovirus | 11–28% | NCT00842634 (C) |
| [91] | 14 | ZFN | HIV | CD4 |
CCR5 | Delay in viral rebound after ART withdrawal; safe & feasible | mRNA by electroporation | 10–34% | NCT02388594 (C) |
| [92] | 12 | CRISPR | NSCLC | T cells | PDCD1 | safe & feasible; low response rate | plasmid DNA by electroporation | 0.4–24.9% | NCT02793856 (C) |
| [93] | 45 (2*) | CRISPR | TDT, SCD | HSPCs | BCL11A | Strong improvement of health status | RNP by electroporation | 69/78–83% | NCT03655678/NCT03745287 (O/O) |
| [94] | 74 (6*) | CRISPR | TTRA | Hepatocytes | TTR | Strong reduction of protein serum concentration, safe | sgRNA and Cas9 mRNA by ApoE-coated lipoparticles | 73% (whole liver of cynomolgus monkeys) | NCT04601051 (O) |
| [95] | 5 (1*) | CRISPR | HIV + ALL | HSPCs | CCR5 | Remission of ALL, no cure of HIV infection | Plasmid DNA by electroporation | 18% | NCT03164135 (U) |
| *: Number of patients described in the associated study. Table is ordered by study status. Study status abbreviations: T, Terminated; C, Completed; O, Ongoing; U, Unknown. | |||||||||
The first published results involving genome editing for the treatment of
HIV infection aimed at disrupting the C-C motif chemokine
receptor 5 (CCR5) gene encoding for an important viral co-entry
receptor by adeno-associated virus (AAV)-mediated transfer of a
CCR5-targeting ZFN [90]. The reinfusion of edited autologous CD4
In another published case report, the authors described CRISPR/Cas-based treatment of a patient with acute lympathic leukemia (ALL) and a coexisting HIV infection [95]. Transplantation of healthy donor cells with a genome-edited CCR5 locus into the patient led to full leukemia remission [95]. Unfortunately, however, cure of HIV infection was not achieved, presumably because of low gene editing efficiency (18%), requiring continuation of ART [95].
Most of the currently active and recruiting studies aim at treating a variety of hematologic cancers and solid tumors. Chimeric antigen receptor (CAR) T (CAR-T) cell therapy already provides a means for treatment of certain large B cell lymphoma after failed systemic treatment regimen [97], and several promising reports show how genome editing may aid in improving efficiency of these therapies. Particularly, targeting the checkpoint gene programmed cell death 1 (PDCD1) is the focus of current research in this context. One study reported electroporation of CAR-T cells with plasmids encoding for Cas9 and sgRNAs targeting PDCD1 resulting in editing efficiencies of 0.4–24.9% [92]. The infusion of autologous genome-edited T cells was well tolerated, but in terms of therapeutic potential only a small impact could be observed in the respective study group [92].
Improving T cell receptor (TCR)-based therapy [98] by CRISPR/Cas-mediated genome editing is another major research target. A report from a terminated study including three patients with refractory cancer, described the electroporation of ribonucleoprotein (RNP) complexes consisting of Cas9 and sgRNAs targeting T cell receptor alpha constant, T cell receptor beta constant and PDCD1, achieving editing efficiencies of 45, 15 and 20%, respectively [89]. This is inasmuch remarkable as three genes were targeted simultaneously and still moderate editing efficiencies were achieved. The infusion of autologous T cells was reported to be safe and one patient showed signs of tumor remission [89].
Another group of diseases targeted by genome editing enlisted in the NIH study
database are monogenic diseases which offer the potential for cure if the
respective responsible gene can be repaired or knocked out. One of the most
prominent examples includes two patients, one suffering from TDT and the other
from SCD. Both diseases are caused by mutations in the
Taken together, the listed studies show promising initial results and will further accelerate application of CRISPR-based therapeutic cures for a variety of diseases. Additional clinical trials conducted by pharmaceutical companies are also in different stages of development, but the data thereof are most often based on company press releases and are thus either limited or inaccessible.
Although the potential of CRISPR/Cas-based therapeutics appear to be limitless, failures of previous gene therapy-based clinical trials [108] demand caution for a treatment option. Two major issues are important in this regard: (1) off-target cleavage caused by unspecific sgRNA binding [109, 110, 111, 112, 113], and (2) predominant activation of error-prone NHEJ repair following the induction of genomic DSBs [114, 115]. Although there is currently no consensus about an off-target edit threshold and this is likely cell type-dependent, testing the sgRNA in patient-derived cells followed by targeted deep sequencing [116] is most likely necessary since human lives are at stake.
Stringent bioinformatics-based sgRNA selection, usage of FokI fused to catalytically dead Cas9 (dCas9), usage of naturally occurring or designed Cas variants, as well as structural optimization of sgRNAs, e.g., by varying length or chemical modification, have shown to result in greatly enhanced on- vs off-target binding and cleavage [117, 118, 119, 120, 121, 122]. In addition, the presence of SNVs for a given target complicates allele-specific target design and can decrease or even abolish CRISPR efficiency if not considered [123, 124, 125]. Patient-specific sequencing of the genomic region surrounding the sgRNA target sequence should thus be standard practice to optimize sgRNA target selection. Another way of reducing off-target edits is the Cas9 nickase approach, which makes use of engineered Cas9 producing genomic single strand breaks (nicks) by mutating one of the two catalytically active endonuclease domains of Cas9, each of which targets only one DNA strand [126]. Therefore, DSBs and thus NHEJ/HDR are only activated if both strands are nicked in close proximity, otherwise non-mutagenic high-fidelity base excision repair (BER) prevents genomic mutations [126, 127]. This approach, however, requires target-restricting selection of two sgRNAs and transfection of two considerably-sized plasmids or RNPs [128, 129].
Although HDR does not strictly rely on DSBs [130], they strongly increase its efficiency [131]. A certain degree of risk due to random integration of donor templates should be considered in addition [132]. The delivery of single-stranded DNA oligonucleotides can solve this risk, but is only feasible for the correction of few nucleotides [133].
The more pressing problem especially when precise editing is required, however, is related to the induction of DSBs and consequent activation of the NHEJ pathway in general [114, 134, 135]. Although repair is not literally random [136], indels are always distributed along patterns [137]. One individual desired specific indel cannot be reliably predicted and thus the repair is, in the practical sense of the word, random after all. This is inherent to any DSB-based genome editing method. While certainly tolerable in a research laboratory setting or even in case of gene KO-based treatment, NHEJ precludes precise and predictable repair, e.g., tailored repair of disease-causing SNVs [138, 139].
CRISPR/Cas-mediated genome editing options independent of DSBs can circumvent
this issue. The concept of base editing (BE) relies on deamination of either
deoxycytidine (cytosine base editors: CBE) or deoxyadenosine (adenosine base
editors: ABE) in the sgRNA-defined target region [140, 141, 142]. The resulting
“mutagenic intermediates” deoxyuridine (in case of CBE) or deoxyinosine (in
case of ABE) are repaired mainly by cellular BER, resulting in C/G -
Because BE-mediated genome editing does not induce DSBs, the prevalence of indels is negligible [140]. The main disadvantages of BE-based genome editing are in consequence unintentional deamination due to unspecific sgRNA targeting or unspecific deaminase activity, and introduction of bystander mutations by potential conversion of any mutable nucleotide in the editing window [141, 151, 152, 153, 154]. Efficiency of CBE has been improved by fusing an uracil glycosylase inhibitor to dCas9, while general BE efficiency is elevated by employing Cas9 nickase thereby favoring mutagenic intermediates as repair templates [142, 155]. Usage of deaminase variants, e.g., with narrower editing windows, or employment of Cas variants to extend the target potential limited by genomic PAMs, also aim at improving editing efficiency [150, 152, 156, 157, 158, 159, 160, 161]. However, they are not able to overcome the reliance on C and/or A to initiate editing events which drastically limits target selection.
The development of prime editing (PE) by David Liu and colleagues thus represents a major step towards seamless genome editing: here, the Cas9 nickase H840A is fused to a reverse transcriptase (RT) [162]. The equivalent to the sgRNA termed PE guide RNA (pegRNA) additionally contains a 3’ extension consisting of a primer binding site and a RT template containing the desired edit [162, 163]. Upon binding, the non-complementary strand is nicked, the primer binds the exposed strand and the RT introduces the template in 3’ direction [162]. The newly synthesized 3’ flap does not allow complementary binding to the unedited 5’ flap which is removed by endonuclease activity resulting in edit of both strands [162]. Third generation PE introduces an additional nick in the unedited strand ensuring that the edited strand is used as a repair template thereby increasing efficiency and specificity [162, 164, 165, 166]. PE was already applied to correct mutations in patient-derived organoids, induced pluripotent stem cells, and in mice [167, 168, 169].
Taken together, PE is not only an alternative to BE when there is a risk of bystander mutations, but greatly enhances the target spectrum as it can make use of the full selectivity of CRISPR/Cas-based genome editing. For a more detailed overview on CRISPR/Cas genome editing tools including development of PE leading to the current generation of PE3 the reader is referred to the comprehensive review of Anzalone, Koblan, and Liu [170].
Since the efficiency of PE is still rather low compared to BE [171], research aimed at improving efficiency is paramount. Usage of engineered, more stable pegRNAS or interference with the endogenous mismatch repair system have been reported to increase editing efficiency [172, 173]. Increasing the efficiency of such targeted manipulation coupled with low off-target effects will further enable clinical research and application. Once all these limitations are overcome, PE will likely be the game-changer for precise correction of disease-causing genomic mutations.
One of the major issues regarding the use of genome editing in clinical applications is of regulatory nature. Currently, there seem to be no clearly set standards regarding the evaluation of safety in preclinical animal models [174, 175]. On the other hand, providing data about safety in a preclinical animal model can take years [176], precluding fast verification on an organismal level. Genetic variability within a population can also affect off-target cleavage and needs to be considered for a given disease [177]. Since the technology is rather novel, defining such standards is an equally laborious and essential task, because guidelines for development of conventional clinical therapies were developed far before the technology even existed. The FDA, which is the responsible agency in the US, has recently published a draft, containing recommendations for industry sponsoring clinical trials [178]. In addition, the World Health Organization (WHO) published recommendations for human genome editing from an expert advisory committee [179]. It has to be noted, however, that the WHO as such cannot set legally binding rules. The nature of these recommendations thus shows the difficulties of advisory committees and regulatory agencies to set standards. Conceivably, this has to be an international effort which will be complex enough a task given already the differences in definition of gene therapy and genome engineering/editing on different continents [180].
A technology as powerful as genome editing, especially with the advent of CRISPR/Cas-mediated genome editing, will undoubtedly revolutionize medical therapy. On the other hand, there is also great potential to abuse it, e.g., to eradicate unwanted genetic traits or enhance others without any medical implication. Therefore, it is absolutely essential to agree on a set of ground rules when applying genome editing in human beings. Obviously, this is not the subject of scientific discussion alone, but should involve the public, lawmakers, politicians and philosophers. The difference in treating manifested diseases in adults versus the treatment of hereditary diseases prenatally comes to mind, which will definitely raise very different arguments. As the infamous “CRISPR-babies” have shown, the discussion needs to happen [181] and it needs to happen now. The discussion of whether or not to apply genome editing in unborns to prevent rather than treat a disease will likely not be harmonic resulting in a worldwide consensus, but neglecting the necessity of it will certainly be worse.
Genome editing is already “business as usual” in the laboratory setting. Particularly CRISPR/Cas-mediated gene editing has rapidly and nearly completely replaced other genome editing techniques. Application in human beings, however, is obviously a different matter with editing efficiencies being substantially lower and side effects apart from off-target editing not easy to predict. Therefore, clinical trials are the most important tool to gain further insights into the behavior of genome-edited patient’s cells. Unfortunately, trials take time, which explains the rather limited available reliable amount of information with many studies still ongoing. This also explains why many of the studies described herein rely on ZFNs which are the somehow “older” technology compared to CRISPR. As mentioned in the introduction, high cost is another factor limiting broad use right now. However, these issues are not unusual for new treatment options and will benefit from both technological improvements as well as rise of new competitors providing materials and methods for genome editing. This clearly highlights the need for ongoing research dealing with optimization of editing efficiency especially aiming at editing in patient-derived primary cells.
As of now, there are no clearly set regulatory guidelines specific for these recently developed therapies. Since most of the trials are in Preclinical or Phase I stage, such guidelines will likely be introduced based on accumulating adverse events. This will further delay the entry of these products into regular use.
As long as a gene target is known which is either causative or at least the main disease driver, genome editing is a valid treatment option. Considering that there is, e.g., no curative treatment for neurodegenerative disorders such as Huntington’s or Alzheimer’s disease [182], these patients might benefit from any improvement.
Especially the results from TDT and SCD ongoing studies are really intriguing considering the therapeutic potential of genome editing [93]. Taken together, genome editing will continue its triumphant path from “bench to bedside”.
Conceptualization: JD, HR AR, Supervision: AR, Visualization: JD, Writing — original draft: JD, HR, Writing — reviewing and editing: JD, HR, AR.
Not applicable.
We thank S. Hoffmann for discussion and comments on the manuscript.
Funding for open access charge: IUF – Leibniz Institute for Environmental Medicine.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.