


 , Silvana Mirella Aliberti 2,*,†, Antonio Barbieri 1, Paola Pentangelo 2, Ilaria Bisogno 3, Giovanni D’Arena 4, Emidio Cianciola 5, Michele Caraglia 6, Mario Capunzo 2
, Silvana Mirella Aliberti 2,*,†, Antonio Barbieri 1, Paola Pentangelo 2, Ilaria Bisogno 3, Giovanni D’Arena 4, Emidio Cianciola 5, Michele Caraglia 6, Mario Capunzo 21 Animal Facility, Istituto Nazionale Tumori - “Fondazione G. Pascale" - IRCCS, 80131 Naples, Italy
2 Department of Medicine, Surgery and Dentistry “Scuola Medica Salernitana”, University of Salerno, 84081 Salerno, Italy
3 Department of Radiological, Oncological and Anatomo-Pathological Science, University of Rome “Sapienza”, 00161 Rome, Italy
4 Hematology Service, San Luca Hospital, ASL Salerno, 84124 Salerno, Italy
5 Anesthesia and Intensive Care Unit, “Immacolata di Sapri" Hospital– ASL Salerno, 84073 Salerno, Italy
6 Department of Precision Medicine, University of Campania “Luigi Vanvitelli”, 80138 Naples, Italy
†These authors contributed equally.
Academic Editor: Graham Pawelec
Abstract
Breast cancer is the most common malignancy among women worldwide. Several studies indicate that, in addition to established risk factors for breast cancer, other factors such as cortisol release related to psychological stress and drug treatment with high levels of glucocorticoids may also contribute significantly to the initiation of breast cancer. There are several possible mechanisms by which glucocorticoids might promote neoplastic transformation of breast tissue. Among these, the least known and studied is the inhibition of the nuclear erythroid factor 2-related (Nrf2)-antioxidant/electrophile response element (ARE/EpRE) pathway by high levels of glucocorticoids. Specifically, Nrf2 is a potent transcriptional activator that plays a central role in the basal and inducible expression of many cytoprotective genes that effectively protect mammalian cells from various forms of stress and reduce the propensity of tissues and organisms to develop disease or malignancy including breast cancer. Consequently, a loss of Nrf2 in response to high levels of gluco-corticoids may lead to a decrease in cellular defense against oxidative stress, which plays an important role in the initiation of human mammary carcinogenesis. In the present review, we provide a comprehensive overview of the current state of knowledge of the cellular mechanisms by which both glucocorticoid pharmacotherapy and endogenous GCs (cortisol in humans and corticosterone in rodents) may contribute to breast cancer development through inhibition of the Nrf2-ARE/EpRE pathway and the protective role of melatonin against glucocorticoid-induced apoptosis in the immune system.
Keywords
- breast carcinogenesis
- glucocorticoids (GCs)
- glucocorticoid receptor (GR)
- glucocor-ticoid response element (GRE)
- nuclear factor erythroid 2-related factor 2 (Nrf2)
- antioxidant/electrophile response element (ARE/EpRE)
- melatonin (MLT)
Breast cancer is the most common malignancy among women and the second leading cause of cancer death among women worldwide [1, 2]. Unlike other malignancies that increase at the end of the fifth decade, breast cancer begins as early as the third decade of life. This may be due not only to the effects of estrogen but also of other ovarian hormones on breast tissue. Physiological and behavioral risk factors for breast cancer include: age, family history, reproductive history, BRCA1 and BRCA2 gene mutations, as well as ele-vated endogenous estrogen, hormone therapy, and certain types of benign breast disease [3, 4, 5]. In addition to these better known factors, other factors are suspected to confer an increased risk of breast cancer. These include alcohol consumption, tobacco abuse, physical activity, and certain aspects of diet such as meat and fat consumption [3, 4, 5]. Among the risk factors, prolonged exposure to high levels of estrogen may contribute significantly to the initiation and development of breast cancer. Indeed, recent experimental studies suggest that imbalanced oxidative metabolism of estrogen can generate genotoxic metabolites (e.g., estrogen-reactive quinones, oxygen free radicals) that can react with DNA to form unstable estrogen-DNA adducts in critical genes leading to cancer initiation [6]. In addition, the possible contribution of psychosocial stress on breast cancer initiation has been less studied. However, it is well known that neuroendocrine hormones (e.g., glucocorti-coids and noradrenaline) can significantly influence cancer biology [7, 8]. Animals and people are continuously exposed to a wide range of stresses (social or environmental) throughout their lifetime. Prolonged exposure to stressful conditions results in chronic activation of the hypothalamic-pituitary-adrenal (HPA) axis and subsequent release of glucocorticoids (GCs) from the adrenal cortex, which can lead to an increased risk of breast cancer [7, 8]. The HPA axis is linked to the circadian clock, resulting in modulation of GC levels in a diurnal pattern [9]. Evidence of alterations in circadian cortisol secretion is not a simple epiphenomenon alone but it is one of the most frequent signs of cancer-related desynchronization [10]. There is also evidence that an altered cortisol rhythm is prognostic for early breast cancer mortality because of its association with a more severe immunosuppressive state [11] or deficient pineal gland function that exerts antitumor activity [12, 13]. Other neuroendocrine alterations described in patients with breast cancer include progressive decline in nocturnal melatonin production (MLT) by the pineal gland, abnormally high levels of prolactin as well as altered daily patterns in estrogen signaling [14, 15, 16]. Several studies link GCs to increased incidence, progression, and recurrence of breast cancer, suggesting a likely link between circadian disruption and breast carcinogenesis [10, 17]. Regarding the treatment of human breast cancer, GCs may significantly contribute to decreased apoptosis of cancer cells, while increasing cancer cell survival and re-sistance to chemotherapy [18, 19]. An important role in these processes could be played by the transcription factor Nrf2 (NF-E2-related factor 2), which is tightly regulated by Keap1 (Kelch-like ECH-associated protein 1) mediated ubiquitination [6, 20, 21]. Specifically, Nrf2 is an important basic leucine zipper-containing transcription factor that controls gene expression of an elaborate network of cytoprotective proteins including antioxidant and detoxifying enzymes that defend cells from electrophiles and free radicals, playing a key role in preventing human carcinogenesis [6, 20, 21].
This review focused on the role of GC signaling in breast cancer biology. In particular, we discussed the potential mechanisms by which both glucocorticoid pharmacotherapy and endogenous GCs (cortisol in humans and corticosterone in rodents) may influence breast cancer initiation through inhibition of the Nrf2-ARE/EpRE pathway. We also examined the role of melatonin in protecting the immune system from apoptosis by reducing nuclear translocation of the glucocorticoid receptor (GR).
In addition to estrogen [6], glucocorticoids (GCs), known as stress hormones, may also play an important role in mammary carcinogenesis process [22]. GCs are produced and released from the adrenal cortex following activation of the hypothalamic-pituitary (HP) axis in both a circadian and highly impulsive manner [23]. These hormones exert their effects by binding to the glucocorticoid receptor (GR), which belongs to the nuclear receptor type 1 subfamily 3. Once bound to GR, the GC-GR complex translocates to the nucleus to modulate gene expression through both transactivation and transrepression of several genes implicated in hyperglycemia and immunosuppression. Alternatively, the GC-GR complex may also exert biological effects through direct protein-protein interactions in the cytosol [24]. There are several mechanisms by which glucocorticoids (GCs) may influence the development and progression of breast cancer. Among these, the least known and studied involves the Nrf2-ARE pathway.
As mentioned above, nuclear erythroid 2-related factor 2 (Nrf2) is a major leucine zipper-containing basic transcription factor that plays an important role in protecting cells from neoplastic transformation by increasing the expression of several cytoprotective enzymes including glutathione-S-transferase (GSTs), NAD(P)H: Quinone oxidoreductase 1,2 (NQO1,2), glutamate cysteine ligase, modifier subunit (GClm), superoxide dismutase (SOD1), catalase (CAT), glutathione peroxidase (GPX1) etc. capable of maintaining cellular redox homeostasis [6]. Specifically, when translocated into the nucleus, Nrf2 binds to the antioxidant/electrophile response element (ARE/EpRE) in the promoters of genes encoding endogenous antioxidants and proteins to enhance their transcription and affect particular cellular functions such as cell cycle and apoptosis, inflammation, inflammasome signaling, endoplasmic-reticulum stress (ER stress), and the unfolded protein response (UPR), autophagy and mitochondrial biogenesis [6, 21, 25, 26, 27, 28]. A loss of Nrf2 in response to high levels of glucocorticoids may lead to a significant decrease in the expression of these cyto-protective enzymes and proteins against oxidative stress that may be important for the initiation and development of breast carcinogenesis.
Below, we will summarize the main known molecular mechanisms by which glucocorti-coids inhibit Nrf2-ARE/EpRE signaling by promoting oxidative DNA damage and initiation of the breast carcinogenesis process.
In support of this notion, a previous study by Ki et al. [28] suggests
that dexamethasone (DEX), a synthetic glucocorticoid (GC) more potent than
cortisol, inhibits constitutive and oltipraz or tert-butylhydroquinone (t-BHQ)
inducible expression of glutathione-S-transferase A2 (GSTA2) in H4IIE cells,
binding to the glucocorticoid receptor (GR) and subsequent activation of the GR
without inhibiting nuclear translocation of Nrf2 and CCAAT/enhancer binding
protein
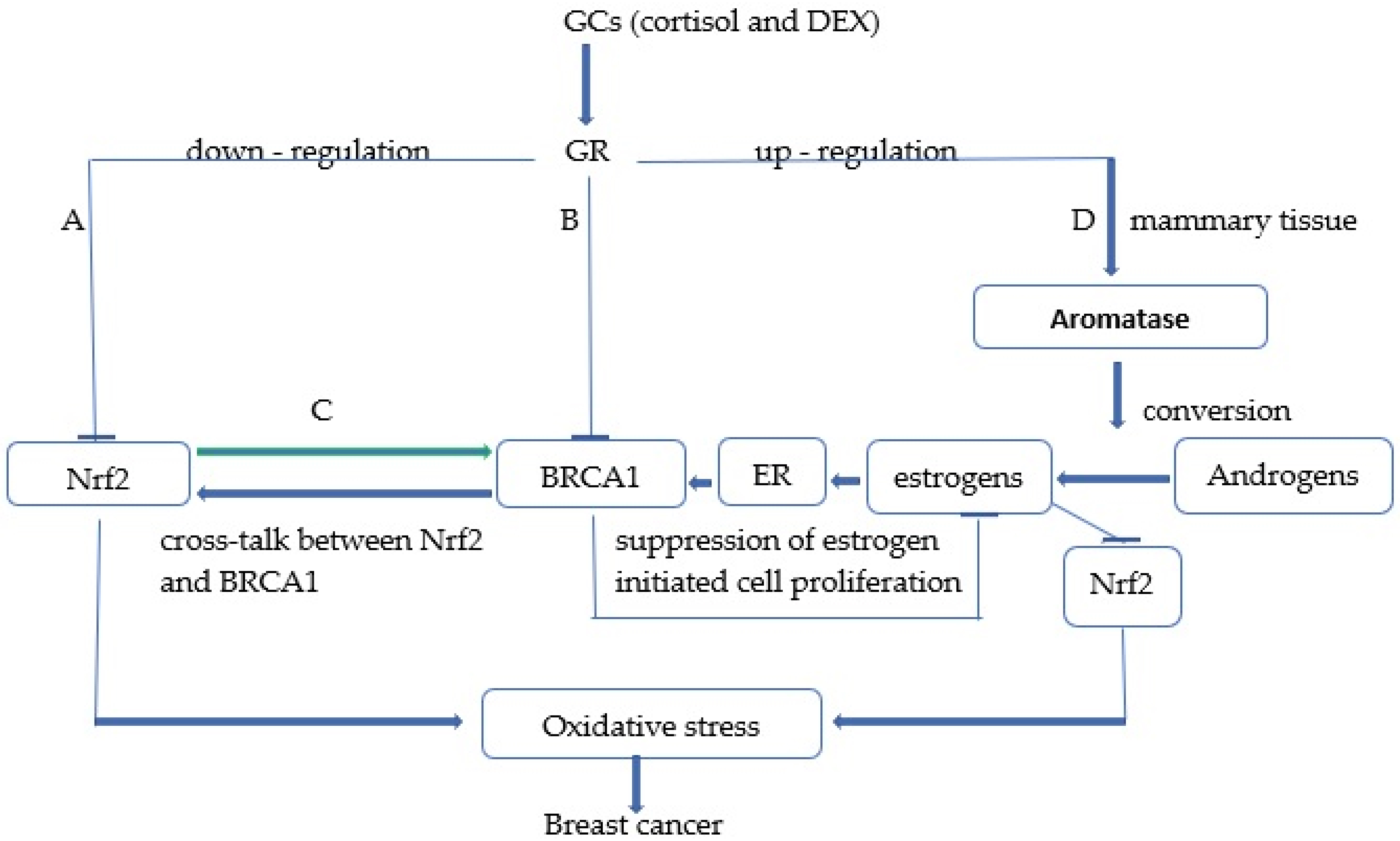 Fig. 1.
Fig. 1.Schematic representation of potential cellular mechanisms by which both endogenous glucocorticoids (GCs) and dexamethasone (DEX) pharmacotherapy promote breast cancer development through inhibition of Nrf2 signaling. Note: (A) High levels of glucocorticoids (cortisol, DEX) inhibit Nrf2 signaling through several mechanisms exhibited in the report. (B) GCs also inhibit expression of the tumor-suppressor gene breast cancer susceptibility gene 1 (BRCA1) that is involved in both activation of Nrf2 signaling by binding to the Nrf2 promoter and suppression of estrogen-initiated cell proliferation. (C) There is a reciprocal positive cross-talk between Nrf2 and BRCA1, so Nrf2-ARE/EpRE signaling can also stimulate BRCA1 expression, and both the Nrf2 and BRCA1-dependent pathways can be inhibited by high levels of GCs. (D) GCs indirectly inhibit Nrf2 signaling in breast tissue through activation of aromatase, which catalyzes the con-version of androgens to estrone that represses the Nrf2-ARE/EpRE pathway through estrogen receptor (ER)-dependent and -independent mechanisms.
The study by Kratschmar et al. [33] suggests that cortisol can suppress
Nrf2 signaling without requiring a GRE sequence in the promoter region. These
authors hypothesize that elevated 11
Recently, Alan et al. [41] also suggest that GR activated by DEX antagonizes the acti-vation of Nrf2 target genes through GR-Nrf2 interaction. This study indicates that im-pairment of the Nrf2-dependent defense system against oxidative stress accounts for glucocorticoid side effects, suggesting a new viewpoint for the pathogenesis of hypercortico-steroidism. Specifically, the authors demonstrated that DEX treatment during the antioxidant response increased GR recruitment to ARE/EpREs without affecting Nrf2 binding to ARE/EpREs, resulting in inhibition of histone acetyltransferase CBP (CREB-binding protein) recruitment and histone acetylation at ARE/EpREs that reduces Nrf2-mediated cytoprotection from oxidative stress. Because both CBP and GR share Neh4/Neh5 domains to bind Nrf2, it is expected that GR can not only put CBP away from Nrf2 but also recruit and interact with histone deacetylase 2 (HDAC2) to ARE/EpREs by associating with Nrf2 resulting in a decreased transcriptional activity of Nrf2 as a consequence of histone deacetylation. In support of this notion, the authors demonstrated that the inhibitory effect of DEX was significantly abrogated by treatment with HDAC inhibitors such as valproic acid (VA) and trichostatin A (TSA), suggesting that GR signaling inhibits the Nrf2-mediated antioxidant response through HDAC recruitment and subsequent histone deacetylation. These results collec-tively support the hypothesis that GR not only reduces Nrf2-dependent transcriptional activation by binding to Nrf2, which is referred to as transrepression, but also that it inhibits the expression of Nrf2 target genes regardless of the presence or absence of glucocorticoid response elements (GREs) (Fig. 1).
Glucocorticoids can also indirectly inhibit Nrf2 signaling in breast tissue by activating cytochrome 450 aromatase [42]. This microsomal enzyme catalyzes the conversion of androgen to estrone which is directly involved in the repression of the Nrf2-ARE/EpRE pathway through estrogen receptor (ER)-dependent and independent mechanisms [43, 44, 45]. Another important mechanism by which GCs may indirectly promote breast cancer development and progression is through inhibition of BRCA1 gene expression. Specifically, the breast cancer susceptibility gene (BRCA1) is responsible for half of all inherited cases [46]. Mutations in the BRCA1 and BRCA2 genes significantly increase the risk of developing breast and ovarian cancer in women and prostate cancer in men [47, 48, 49]. The function of the intact BRCA1 protein has been reported to prevent cancer development through several mechanisms including DNA repair, regulation of cell cycle progression, regulation of transcription, ubiquitination, and maintenance of genomic stability [50]. Importantly, studies by [51] have reported that the BRCA1 promoter contains an estrogen receptor response element (ERE) that can be stimulated by estrogenic activity. Because BRCA1 exerts an important role in suppressing estrogen-initiated cell proliferation and estrogen signaling pathways [52], increased expression of BRCA1 by estrogen is considered a key feedback mechanism by which rapidly proliferating cells modulate their growth [51]. Accordingly, factors that inhibit this control mechanism are considered essential in inducing dysregulated cell proliferation and breast cancer development. These authors argue that cortisol may act as one of these factors. Indeed, cortisol treatment of breast cells is expected to produce the loss of estrogen-induced stimulation of BRCA1 and the subsequent apoptotic effect by blocking the binding of the transcription factor GABP (GA-binding protein) to promoter regulatory sites (RIBS and UP regulatory elements) that are involved in the regulation of BRCA1 by cortisol [53]. Previous study by Kang et al. [54] also suggest that BRCA1 may increase the expression of Nrf2-modulated cytoprotective genes that are involved in the antioxidant and detoxifying cytoprotective response by binding to the Nrf2 promoter and regulating its transcription. In addition, Gorrini et al. [55] also reported that the tumor suppressor BRCA1 modulates Nrf2-dependent antioxidant signaling by physically interacting with Nrf2 and promoting its stability and activation. Accordingly, the authors demonstrated not only that mouse mammary cells lacking BRCA1 have increased ROS levels due to an altered Nrf2-dependent antioxidant response, but also that estrogen treatment can partially restore Nrf2 levels in the absence of BRCA1 to promote cell survival. These results collectively indicate that Nrf2 is downstream of BRCA1 and BRCA1 may promote Nrf2 transcriptional activity playing an important role in protecting cells against oxidative stress and carcinogens by Nrf2 [54, 56]. Further studies by Wang Q et al. [57] also demonstrated that Nrf2, plays an important role in the activation of BRCA1 transcription by binding directly to the BRCA1 promoter, suggesting the existence of a positive regulatory feedback loop between BRCA1 and Nrf2. Specifically, these authors suggest that increased Nrf2 expression significantly promoted not only the recruitment and binding of Nrf2, CBP and p300 active transcription complex to the ARE/EpRE (antioxidant/electrophile response element) site on the BRCA1 promoter but also the acetylation of histone H3 and H4 on the BRCA1 promoter. Both CBP and p300 are histone acetyltransferases (HATs) that act as key transcriptional coactivators [58] by catalyzing histone acetylation, chromatin remodeling, and promoting target gene transcription [59, 60]. These experimental results suggest that Nrf2-mediated epigenetic changes involving histone modifications and chromatin remodeling may be responsible for BRCA1 expression. Notably, reduced levels of wild-type BRCA1 expression have been identified in several sporadic breast cancers without BRCA1 gene mutations [61]. Therefore, somatic mutations in the BRCA1 gene may not play a decisive role in breast cancer [62, 63]. Decreased BRCA1 expression in sporadic breast cancer could be due to disturbances in gene regulatory mechanisms. For example, UHRF1 (Ubiquitin-like with PHD and ring finger domains 1) might be involved in epigenetic silencing of BRCA1 in sporadic breast cancer by inducing DNA methylation, histone modifications, and recruitment of the transcriptional complex on the BRCA1 promoter [64]. Therefore, elucidating the mechanism of regulation of BRCA1 expression by Nrf2 complexes could lead to a better understanding of breast cancer development. Taken together, these findings clearly indicate that high levels of glucocorticoids as a consequence of stressful life events or drug treatment may significantly contribute to the initi-ation of numerous diseases including breast cancer development and progression through direct and indirect mechanisms involving not only suppression of Nrf2 and BRCA1 expression but also activation of the aromatase enzyme involved in estrogen generation (Fig. 1).
Originally glucocorticoids were so named because of their ability to increase
blood glucose concentration. Indeed, adrenalectomized animals or individuals with
adrenocortical deficiency (Addison’s disease) suffer from persistent hypoglycemia
[65]. In humans, glucocorticoids increase circulating glucose levels and lipids
metabolites within minutes by acting simultaneously on the liver, skeletal
muscles and adipose tissue through the action of the GR [65, 66, 67, 68, 69, 70]. Human
individuals with higher circulating cortisol levels under resting (non-stressed)
conditions also have higher glucose and triglyceride levels and a higher score
reflecting insulin resistance and a pre-diabetic state [71]. As mentioned above,
chronic psychological stress activates the hypothalamic-pituitary-adrenal (HPA)
axis and increases circulating levels of cortisol [72]. The HPA axis also
stimulates the sympathetic nerves system (SNS) activity, which, in turn,
increases catecholamine levels [73]. Increased cortisol concentrations can be
considered as an important risk factor for the development and progression of
many disease conditions, including type 1 and 2 diabetes, cancer, and metastasis
[74, 75]. Indeed, elevated cortisol levels associated with stressful life events
[7] or diets based on high-glycemic index carbohydrates [76, 77] due to their
ability to create hyperglycemic conditions such as those found in diabetes, are
recognized to be responsible for high rates of protein glycation and the
subsequent formation and accumulation of advanced glycation end products (AGEs)
[78]. Increased protein glycation and gradual accumulation of AGEs are involved
in the pathogenesis and development of numerous chronic and age-related
inflammatory diseases, such as diabetes and diabetic complications,
cardiovascular diseases, Alzheimer’s disease, stroke and various types of
cancer, including breast cancer [77, 78, 79, 80, 81]. It is important to note that AGEs are a
heterogenous group of cross-linked irreversible components (e.g., pentosidine,
hydro-imidazolone, carboxymethyllysine) that are formed through nonenzymatic
post-translational modifications of macromolecules such as proteins, lipids and
nucleic acids by reducing sugars (glucose, fructose and pentose) through the
Maillard reaction. AGEs can be generated exogenously in food, particularly
through thermal processing, but also endogenously in the body [82]. More
commonly, the most potent reactive
In addition to the functions described above, high cortisol levels may also
contribute to the development and progression of breast cancer by altering immune
system function and elimination of transformed breast cells [102]. In particular,
glucocorticoids at pharmacological levels exert a potent antiinflammatory effect,
whereas prolonged exposure to glucocorticoids can lead to a variety of
undesirable side effects through several mechanisms, including immunosuppression
[102, 103, 104]. Indeed, both chronic stress conditions (when endogenous glucocorticoid
production is high) and glucocorticoid pharmacotherapy can significantly alter
immune cell differentiation, exerting immunosuppressive effects [105, 106, 107]. In
addition, glucocorticoids may also elicit a state of immunosuppression by
inhibiting the Nrf2-dependent antioxidant response and increasing apoptosis in
immune cells [28, 108, 109]. As mentioned above, Nrf2 signaling may protect
normal cells against DNA damage, ROS-induced mutagenesis, and carcinogenesis by
increasing the expression of several phase 2 detoxification enzymes and oxidative
stress-related proteins such as NQO1, heme oxygenase 1 (HO-1), GSTs,
UDP-glucuronosyl transferases (UGTs), superoxide dismutase’s (SODs) [21, 110, 111]. Activation of GRs by glucocorticoids has been reported to increase
apoptosis by affecting apoptotic proteins and through downregulation of the
Nrf2-dependent antioxidant response [33, 112]. Binding of GCs to GRs is known to
sensitize GRs for their nuclear translocation [113]. Indeed, many studies suggest
that GR transcriptional activity is regulated at every step of its activation,
including ligand binding, nuclear translocation, transcriptional cofactor
binding, and DNA binding [114, 115]. The level of Heat Shock Protein 90 (HSP90),
which is an important molecular chaperone of GR, is essential for translocation
and transactivation of activated GR [113, 116]. It appears that an increase in
the HSP90/GR ratio can significantly affect the interaction of GR with its ligand
(GC), and excess HSP90 blocks the binding of GR to its DNA response element
[117]. In particular, it has been shown that association of HSP90 with GRs is
essential to maintain the receptor not only in its steroid-binding conformation
but also in an inactive transcriptional state, even when GRs are located in the
nucleus [118]. Indeed, dissociation of HSP90 from the receptor allows binding of
the receptor to DNA and subsequent modulation of gene transcription [118]. Either
chronically stressful conditions or dexamethasone treatment may promote nuclear
GR translocation by facilitating dissociation of HSP90 from GR-HSP90 complex,
reduce Nrf2/HO-1 dependent antioxidant response and up-regulate apoptosis by
reducing Bcl-2 and increasing Bax expression respectively in immune cells [33, 112, 119, 120, 121]. It has also been reported that dexamethasone treatment reduced the
proliferative response by down-regulationg IL-2R expression and exerting an
inhibitory effect on IL-2 secretion from immune cells [122, 123, 124]. In contrast,
treatment with melatonin (MLT) alone or in combination with DEX, due to its
immunomodulatory property, significantly enhanced proliferative responses [125, 126]. In addition, MLT also blocked stress-dependent immunosuppression by
increasing IL-2 secretion from immune cells such as T-helper cells and peripheral
blood mononuclear cells (PBMCs) [127, 128]. Notably, melatonin is a chronobiotic
hormone produced primarily by the pineal gland only at night or more precisely in
the darkness from the amino acid tryptophan. There is also evidence that
melatonin is produced by the mitochondria of many other cells and organs,
including immune cells in a non-circadian manner [129]. Melatonin (MLT) exerts
considerable functional versatility with antioxidant, oncostatic, antiaging,
antistress, and immunomodulatory properties [130]. In mammals, rhythmic melatonin
production, in addition to being controlled by the central biological clock
located in the suprachiasmatic nuclei (SCN) of the hypothalamus via nocturnal
sympathetic input from noradrenaline, is also influenced by glucocorticoids
through modulation of the NF-kB transcriptional program [131]. Indeed, some
studies suggest that corticosterone increases nocturnal melatonin synthesis by
reducing the activity of NF-kB, a pivotal transcription factor that regulates the
expression of the key enzyme arylalkylamine N-acetyltransferase (AANAT) directly
involved in melatonin synthesis. Accordingly, the pineal gland is a target of GCs
as it expresses many glucocorticoid receptors [132]. In particular,
glucocorticoids exert a dual effect on melatonin production. Under stressful
conditions, characterized by concomitant
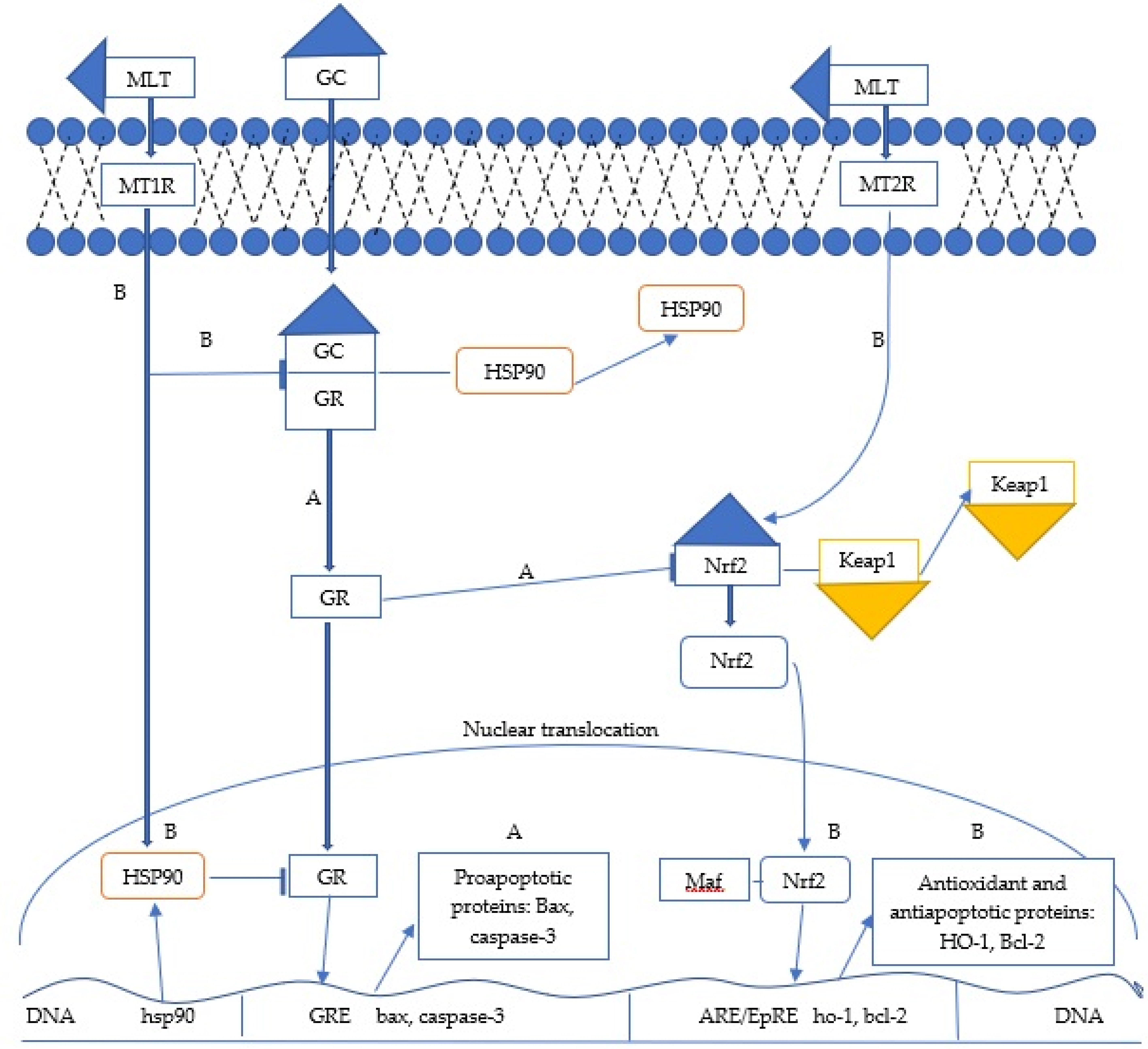 Fig. 2.
Fig. 2.Potential mechanisms by which melatonin inhibits immunosuppressive effects by either high levels of endogenous glucocorticoids (GCs) or dexamatasone (DEX) pharmacotherapy. Note: (A) Binding of GCs to the glucocorticoid receptor (GR) sensitizes the GR to its nuclear translocation by promoting dissociation of HSP90 from the GR-HSP90 complex, suppression of the Nrf2 signaling-dependent antioxidant response, and subsequent apoptosis in peripheral blood mononuclear cells (PBMCs) by increasing expression of the proapoptotic proteins Bax and cleaved caspase-3. (B) The binding of melatonin to its receptors (MT1,MT2) inhibits the immunosuppressive effects of GCs through several mechanisms such as : (1) blocking the dissociation of HSP90 from the GR-HSP90 complex; (2) increasing the expression of HSP90 protein in the nuclear fraction that attenuates the transcriptional activity of GR involved in the expression of apoptotic proteins; (3) stimulating the Nrf2 signaling-dependent antioxidant response and finally (4) increasing the expression of Bcl-2 and the Bcl-2/Bax ratio in PBMCs to maintain immune homeostasis under stressful conditions.
Although several genetic factors have been identified in breast cancer, most
cases are attributed to environmental factors. At least, it is widely accepted
that among risk factors, prolonged exposure to high levels of estrogen plays an
important role in the initiation and development of breast cancer [141]. Indeed,
studies in experimental animal models and cultured human cells strongly suggest
that estradiol (E2), its interconvertible metabolite estrone (E1), and their
estrogen quinones exert carcinogenic effects on breast tissue through several
mechanisms [142]. There are at least two main mechanisms involved in the
development and progression of estrogen-induced breast cancer: (i) estrogen
receptor mediated stimulation of abnormal cell proliferation that generates
random mutations; (ii) ER-independent mechanisms involving chemical (oxidative
pathway) inflammatory, epigenetic, and cancer stem cell pathways [143, 144, 145]. In
addition, although less well known and studied, either chronic stress conditions
(when endogenous glucocorticoid production is high) or dexamethasone treatment to
reduce inflammation may also significantly increase breast cancer risk. In our
review, we examined potential cellular mechanisms by which (both) glucocorticoid
drug treatment and psychological stress-related cortisol release may contribute
to breast cancer development through inhibition of the Nrf2-ARE/EpRE dependent
antioxidant response. For example, research has shown that cortisol treatment
significantly reduced the expression of the gene encoding the tumor suppressor
protein BRCA1 involved in the control of estrogen-induced breast cell
proliferation and DNA repair capacity. Because BRCA1 is able to influence
Nrf2-dependent transcriptional activation and the subsequent antioxidant
response, factors that inhibit this mechanism should also contribute to the
development of breast cancer in response to stress. There are also other
mechanisms by which glucocorticoids may influence breast cancer development and
progression by involving the Nrf2-ARE/EpRE pathway. For example, glucocorticoids
may antagonize the activation of Nrf2 target genes in a GR-dependent manner.
Specifically, dexamethasone (DEX) treatment can increase GR recruitment to
ARE/EpREs without altering chromatin binding of Nrf2, resulting in suppression
of CBP recruitment and histone acetylation to ARE/EpREs. Thus, GR signaling
significantly reduces Nrf2 transcriptional activation by decreasing
Nrf2-dependent histone acetylation, consequently impairing the Nrf2-dependent
cellular antioxidant response and predisposing to diseases such as immune
dysfunction and cancer [41, 146]. Studies by Ki et al. [28] also
indicate that after activation of GR by DEX, the GC-GR complex migrates to the
nucleus where it binds to GREs on the promoter region of GST. Subsequently,
recruitment of transcriptional repressors NCoR and SMRT to the DEX-GR complex on
GRE leads to suppression of the expression of phase II detoxifying enzymes such
as glutathione-S-transferase (GST) not only by regulating chromatin structures
through histone deacetylation but also by directly binding to the Neh 4/5 domain
of Nrf2, a protein motif essential forNrf2 transactivation. Accumulating
scientific evidence also indicates that circulating levels of GCs are regulated
by the hypothalamic-pituitary-adrenal axis, whereas their tissue levels are
controlled by en-zymes such as 11
AG, AB, GDA, EC, MCar and MCap designed the study. AG wrote the manuscript. SMA provided help and advice on grammar. PP and IB contributed to the literature search. SMA designed the figures and provided review and editing. AB, PP, IB, GDA, EC and MCar critically visualized the manuscript. MCap supervised the manuscript. All authors read and approved the final manuscript.
Ethical approval from the Human Research Ethics Committee was not required for this study. This is a retrospective study and is not directly associated with patients.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.