
Academic Editor: Roberto Bei
Ferroptosis is an emerging form of non-apoptotic, regulated cell death that is mechanistically dependent on aberrant iron accumulation and excessive lipid peroxidation. Further evidence indicates that ferroptosis plays a crucial role in the efficacy of tumor immunotherapy. Ferroptosis is often constrained by tumor-associated macrophages (TAMs), and this poses a challenge to clinicians aiming to exploit the potency of immunotherapy to treat various forms of cancer. Current advances revealed a dual character to TAMs in regulating tumor ferroptosis. Specifically, some signaling molecules released from cells undergoing ferroptosis can exert effects on TAM polarization. In this review, we summarize the currently characterized mechanisms of macrophage-ferroptosis crosstalk, discuss how macrophage-ferroptosis crosstalk affects the outcome of tumor immunotherapy, and provide an overview of current advances that seek to leverage this crosstalk to improve cancer immunotherapy efficacy. Despite the fact that further efforts are still required to achieve a more comprehensive understanding of the mechanisms that control this signaling, targeting macrophage-ferroptosis crosstalk has clear potential for reversing immunotherapeutic resistance and may shed light on new therapeutic strategies to overcome some advanced and metastatic malignancies.
Tumors constantly evolve to meet the demands of their surrounding stromal and immune cells, forming a basis for an intricate, and complex network of cellular and molecular interactions termed the tumor immune microenvironment (TIME). In the early stages of tumor progression, the TIME exerts an anti-neoplastic effect on the developing neoplasm, but later serves as an accomplice in promoting tumor growth, metastasis, and therapeutic resistance. Thus, the TIME presents a challenge to effective tumor management [1, 2, 3, 4]. To reshape TIME to favor its tumor-suppressive capacities, immunotherapy has emerged as a promising strategy for some refractory malignancies, most notably evidenced by its approval to treat advanced melanoma in 2011 [5]. Despite manifold targets and mechanisms, most existing immunotherapies share a common tumor-killing ability by triggering regulated cell death (RCD) [6]. Ferroptosis, an iron-dependent and lipid-peroxide-driven form of RCD [7], exerts a prominent effect during immunotherapy [8].
First discovered when studying erastin-induced cell death in 2012, ferroptosis has aroused ongoing interest owing, in part, to its property of selective tumor scavenging [9, 10]. Unlike other forms of RCD, ferroptosis does not rely on the caspase system, and demonstrates unique morphological characteristics including a condensed mitochondrial membrane, vanished mitochondria cristae, and a ruptured outer mitochondrial membrane. However, retention of a relatively intact nucleus occurs during ferroptosis [11]. Aberrant iron accumulation plays an essential role in initiating ferroptosis through a Fenton reaction and iron-activated enzymes, whereas excessive lipid peroxidation products eventually lead to the oxidative destruction of intracellular biomembranes [12]. Although many signaling molecules are characterized as participating in regulation of ferroptosis, the endogenous antioxidant glutathione peroxidase 4 (GPX4), which is widely expressed and requires glutathione (GSH) to reach its optimal activity, is viewed as a key controlling molecule in preventing the activation of ferroptosis during both physiological and pathophysiological conditions [10]. Intriguingly, CD8+ T cell-mediated ferroptosis is widely observed in most tumor cells during immunotherapy treatment, revealing potential therapeutic values for ferroptosis in promoting cell killing during tumor immunotherapy [8, 13, 14].
Another key factor influencing the efficacy of tumor immunotherapy is tumor-associated macrophages (TAMs). It is well accepted that TAMs function as a double-edged sword within the TIME [15]. This range in TAM function owes to two distinct cell polarizations, defined as the classic phenotype (termed M1) and the alternative phenotype (termed M2), which have opposing functions in tumor progression. M1 macrophages function in antigen presentation, induce inflammatory responses, scavenge pathogenic microorganisms, and exert anti-neoplastic effects within the TIME [16, 17]. In contrast, M2 macrophages are able to limit the inflammatory response and contribute to tumor progression by stimulating proliferation, angiogenesis, and metastasis [18]. Notably, M2-like TAMs are highly associated with therapeutic resistance and are widely viewed as providing a barrier to effective tumor immunotherapy [19, 20].
Intriguingly, TAMs and ferroptosis share an intricate crosstalk signaling network as compelling evidence from integrative bioinformatic analyses highlights their intimate association at many levels. Hence, it is of great necessity to profile and interpret their interplay in a more comprehensive view. Herein, we endeavor to highlight and integrate the sophisticated crosstalk pathways between TAMs and tumor ferroptosis, and subsequently discuss how their crosstalk affects the outcomes of tumor immunotherapy. Finally we seek to summarize current advances associated with macrophage-ferroptosis crosstalk in tumor elimination in anticipation that this will shed light on new therapeutic strategies for as-yet undruggable malignancies.
Ferroptosis starts with aberrant iron metabolism and excessive lipid
peroxidation, and ends with disrupted lipid bilayers [6]. Physiologically,
extracellular Fe
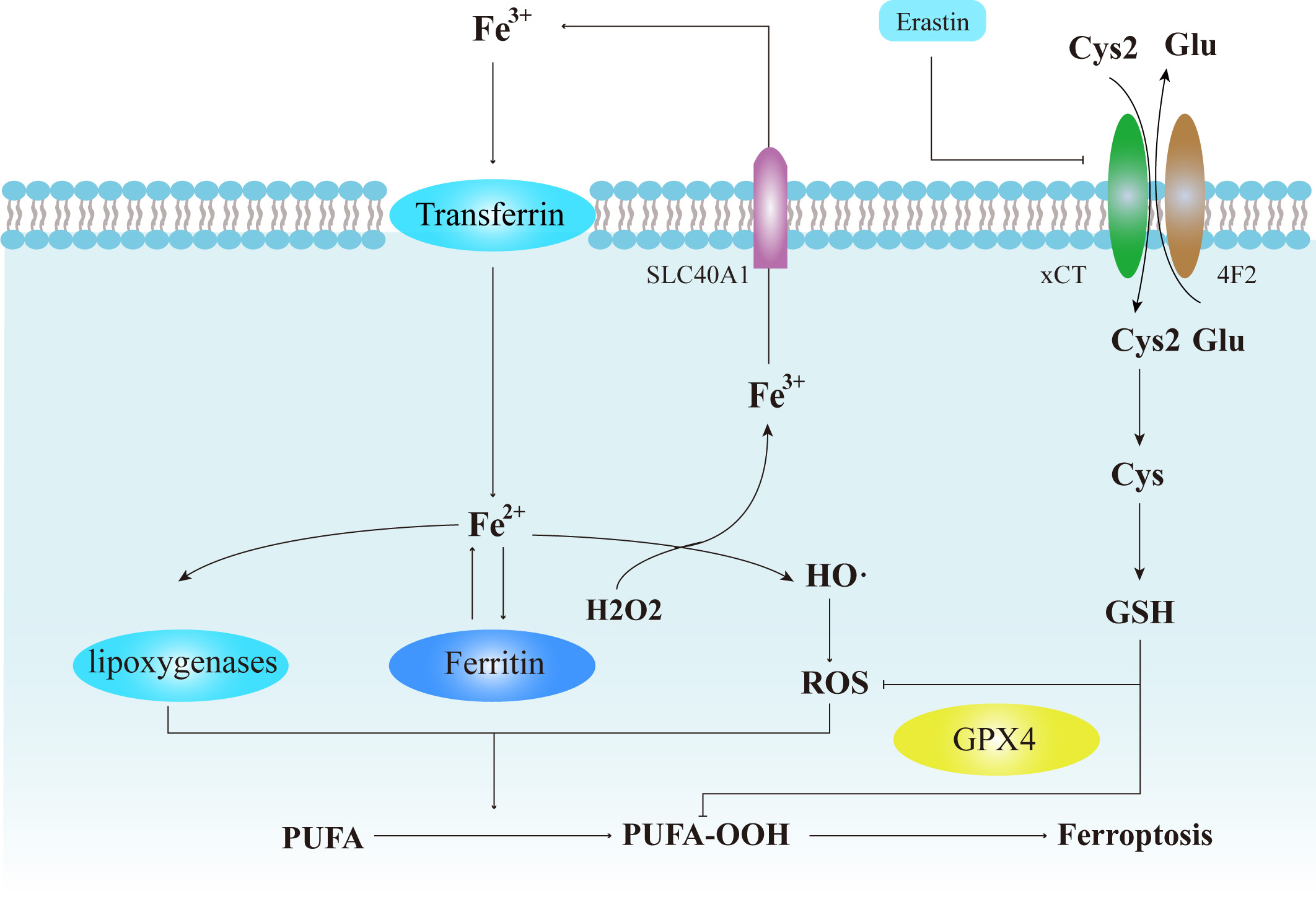 Fig. 1.
Fig. 1.Cellular events in the regulation and process of ferroptosis.
Dysfunction within iron metabolism leads to excessive release of free
intracellular Fe
Within the TIME, ferroptosis is co-regulated by intracellular and extracellular
pathways. The pivotal intracellular system in regulating ferroptosis is the
system Xc
Beyond intracellular mediators, ferroptosis within the TIME is principally
induced by CD8+ T lymphocytes. In 2019, Wang et al. [14] first reported
that CD8+ T cells can induce tumor ferroptosis as a result of immunotherapy by
producing IFN-
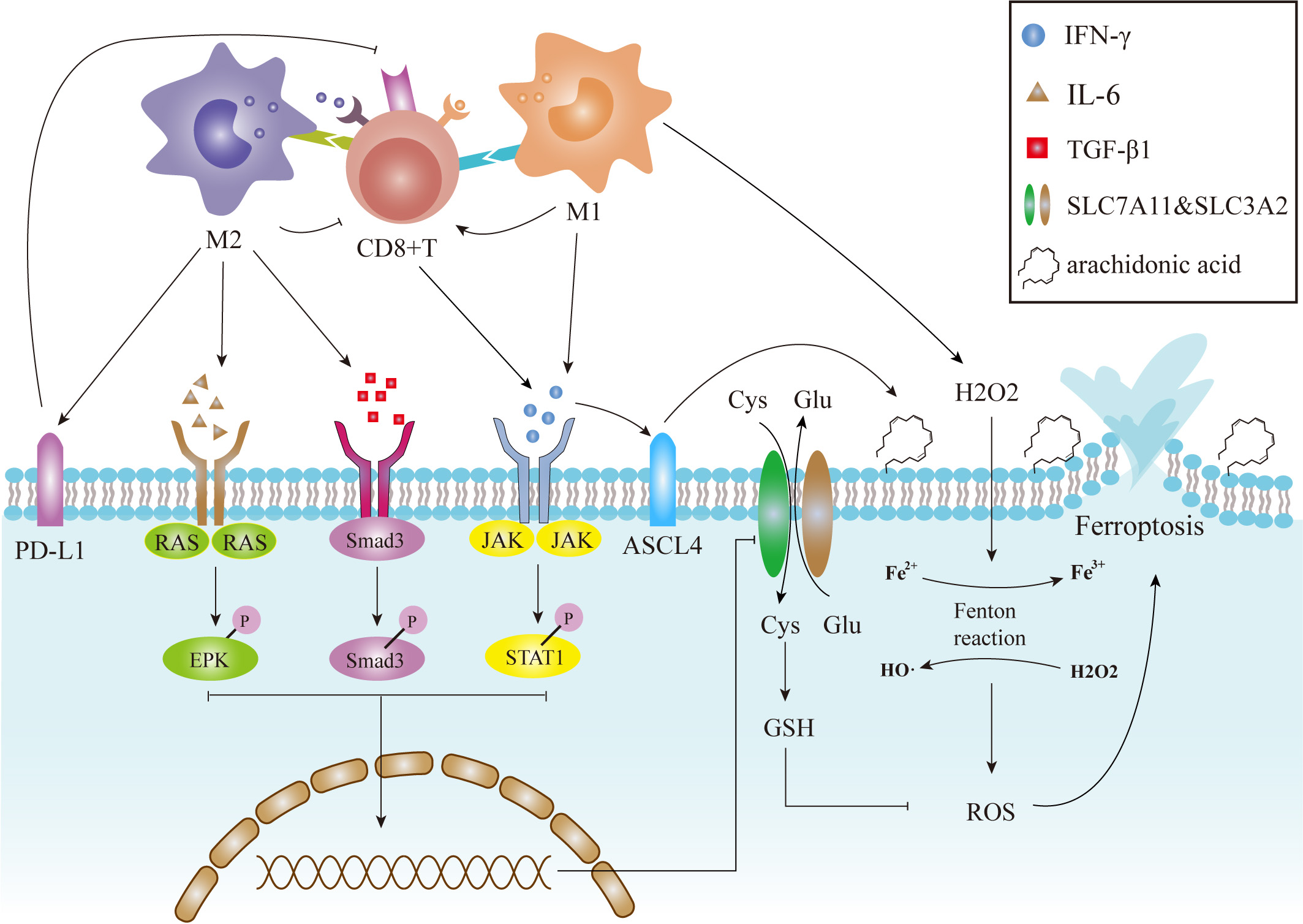 Fig. 2.
Fig. 2.Dual effects of TAMs on ferroptosis. M1-like TAMs can trigger tumor ferroptosis through at least three distinct mechanisms: activation of CD8+ CTLs, release of proinflammatory cytokines, and providing peroxides to accelerate Fenton reactions. The former two pathways downregulate the expression of SLC3A2 and SLC7A11 at the transcriptional level, resulting in downregulation of an endogenous anti-ferroptosis system. The latter mechanism contributes to excessive ROS production which, in turn, promotes tumor ferroptosis. M2-like TAMs can indirectly suppress ferroptosis either by inactivating CTLs or by upregulating the expression of oncogenic PD-L1. JAK, Janus kinase; STAT1, signal transducer and activator of transcription 1; Smad2, small mothers against decapentaplegic homolog 2; Smad3, small mothers against decapentaplegic homolog 3; Smad4, small mothers against decapentaplegic homolog 4; RAS, rat sarcoma viral oncogene homolog; SLC3A2, Solute Carrier Family 3 Member 2; SLC7A11, Solute Carrier Family 7 Member 11; PD-L1, programmed cell death 1 ligand 1.
To date, TAMs are well recognized as a predominant regulator within the TIME and whose functional status is prone to remodeling by exogenous signals due to the wide distribution of diverse signal receptors on their cell surface [37]. It has been well-established that macrophages are able to trigger ferroptosis via multiple signaling pathways, and, in turn, ferroptotic products can regulate the polarization of TAMs [6, 38, 39]. These findings imply an underlying functional existence of macrophage-ferroptosis crosstalk which may influence the efficacy of tumor immunotherapy. Therefore, it is necessary to fully delineate macrophage-ferroptosis crosstalk to provide critical insight into factors that can influence the therapeutic effects of immunotherapy.
The two distinct TAM differentiation states govern TAM effects on tumor ferroptosis. As a major anti-tumor effector within the TIME, M1 macrophages can promote tumor ferroptosis by enhancing tumor vulnerability to oxidative damage and activating a tumor-eradicating immune response within the TIME [40]. Recent studies have suggested the existence of at least three different mediators in this process, including CD8+ cytotoxic T lymphocytes (CTLs), various cytokines, and peroxides resulting from the respiratory burst (Fig. 2) [41, 42].
The CTL-mediated pathway is considered to have a major contribution to
initiating tumor ferroptosis. It is well established that M1 macrophages utilize
cell contact-dependent signaling to activate CTLs [43], which plays a crucial
role in triggering ferroptosis during tumor immunotherapy. As discussed above,
interferon
Although bioinformatic analyses have identified a significant negative
correlation between tumor ferroptosis and M2 infiltration [39, 47, 48, 49, 50, 51, 52],
there are few studies exploring the direct effects of M2 macrophages on tumor
ferroptosis. Instead, M2 macrophages are more likely to play an indirect role in
regulating ferroptosis by disrupting the ferroptosis-promoting function of CTLs
[53]. It is well documented that M2 macrophages can prevent the recruitment and
activation of CTLs via the inhibition of numerous chemokines such as C-X-C motif
chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10) and C-X-C
motif chemokine ligand 12 (CXCL12) [54, 55]. Together, these molecules disable
major mechanisms that promote tumor ferroptosis during immunotherapy. In
addition, M2 macrophages can enhance tumor-derived resistance to ferroptosis by
upregulating the expression of PD-L1 on neoplastic cells [56, 57], which can
engage with PD-1 to initiate programmed cell death in CTLs [53, 58]. Furthermore,
Xu and colleagues, using a combination of in silico and
in vitro experimental approaches, recently identified a novel
molecular matrix remodeling-associated protein termed MXRA8, which can decrease
the level of intracellular ferrous iron (Fe
It is noteworthy that some M2-associated cytokines may serve to promote
ferroptosis. For example, TGF-
In summary, M1 macrophages function in the activation of tumor ferroptosis through both direct and indirect mechanisms, whereas M2 macrophages principally act by opposing inactivation of CTLs in TIME. Despite the need for more information to fully appreciate the intricacies of these signaling pathways, there is little doubt that TAMs play a vital role in regulating tumor ferroptosis. This supports the emerging view of TAMs are valuable targets to increase neoplastic sensitivity to ferroptosis-inducing immunotherapy.
While macrophages are involved in the regulation of ferroptosis, ferroptosis is also capable of affecting macrophage polarization through several mechanisms. Current advances in bioinformatics have highlighted significant correlations between ferroptosis-related genes (FRGs) and TAM infiltration in several malignancies. Such advances were possible with the assistance of newly-emerging algorithms such as Cibersort, TIMER, quanTIseq and xCell (Fig. 3). Generally speaking, ferroptosis-promoting FRGs are more likely to serve in an anti-oncogenic capacity, while ferroptosis-blocking FRGs are predisposed to contribute to tumor progression, and thereby may be linked to TAM infiltration. For example, ferroptosis-promoting FRGs such as HIF1A, SLC7A11, GPX4, FTH1 are highly associated with poor prognosis as well as TAM infiltration in ovarian cancer. Other ferroptosis-promoting FRGs, including FGFR3, CDGSH and CISD1, which are directly involved in either iron storage or inhibiting iron uptake, are also associated with an increased proportion of tumor-infiltrating macrophages in bladder cancer [39, 67, 68, 69, 70, 71]. The ferroptosis driver SOCS1 and suppressor FTH1 correlate with M1 and M2 infiltration, respectively, in head and neck squamous cell carcinoma (HNSCC) [39] which implies the probability of ferroptosis-mediated macrophage polarization within the TIME. Moreover, some ferroptosis-associated lncRNAs are also implicated in TAM infiltration in hepatocellular carcinoma [68]. In conclusion, many differentially expressed FRGs and lncRNAs are highly associated with tumor prognosis, clearly indicating the potential involvement of ferroptosis-related signals in tumor progression [39, 69, 70, 71]. However, further studies are required to identify the specific effect of FRGs on TAM polarization.
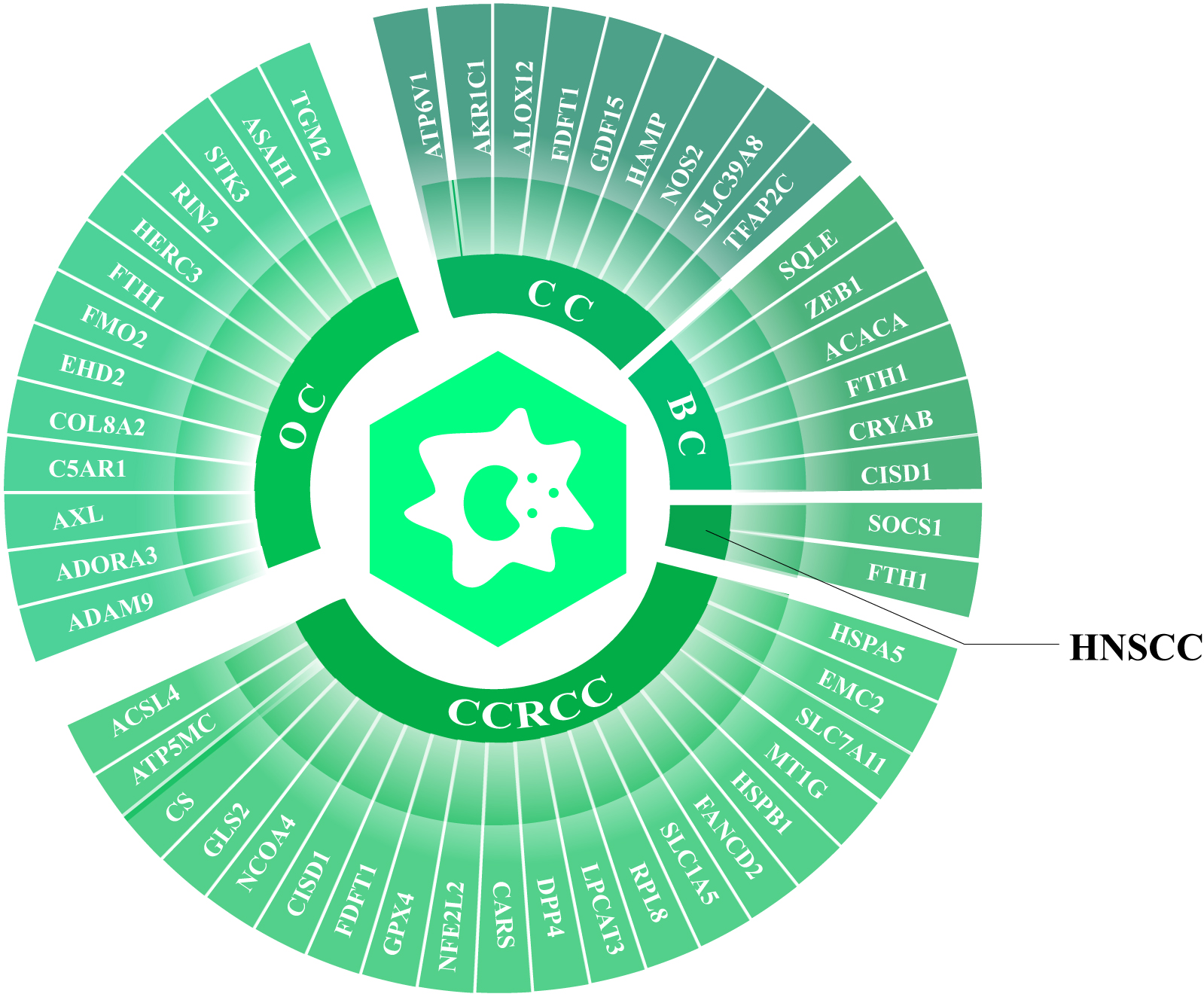 Fig. 3.
Fig. 3.Ferroptosis-related genes (FRGs) in different tumors are associated with TAM infiltration. Integrative analysis with bioinformatic tools highlight comprehensive differentially expressed FRGs that are associated with TAM infiltration in different cancers. OC, ovarian cancer; CCRCC, clear cell renal cell carcinoma; HNSCC, head and neck squamous cell carcinoma; BC, bladder cancer; CC, colorectal cancer.
Recent studies of damage-associated molecular patterns (DAMPs) have revealed one of the feasible means to mediate ferroptosis-related TAM polarization at the molecular level. By definition, DAMPs are an array of molecular products from damaged tissues, whose counterparts, the pattern recognition receptors (PRRs), are located on the surface of macrophages. PRRs function to interpret the messages delivered by DAMPs and produce responses to tissue damage [72]. Interestingly, different DAMPs released by tumor cells undergoing ferroptosis may convey completely distinct messages, either as an “eat-me” signal or as a “save-me” signal, depending on the specific type of DAMPs and neoplastic subtypes (Fig. 4).
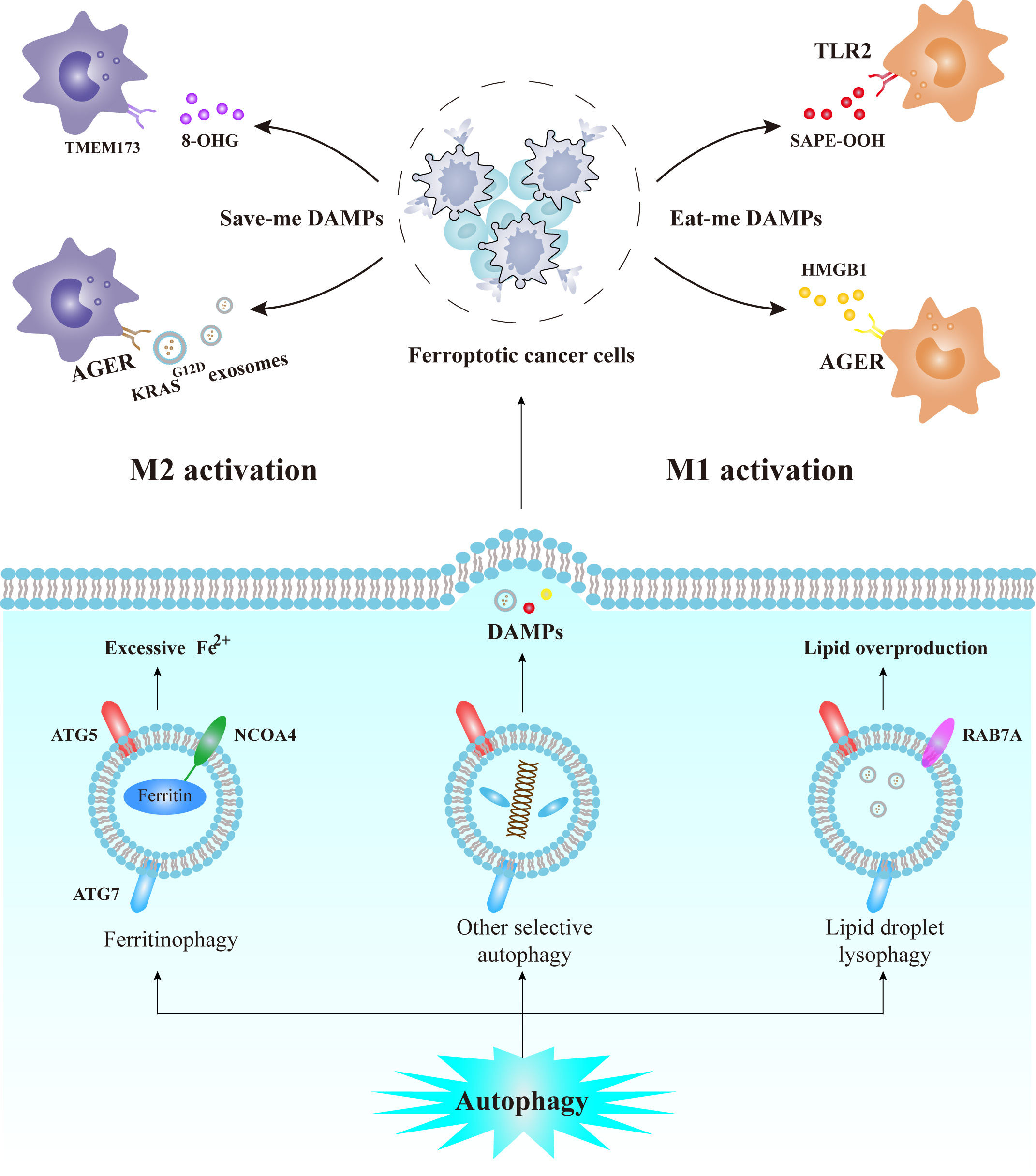 Fig. 4.
Fig. 4.Relationship between selective autophagy, tumor ferroptosis,
ferroptotic DAMPs, and the TAM polarization. The NCOA4-mediated ferritinophagy
and the RAB7A-mediated lysophagy of lipid droplets could provide labile Fe
The “eat-me” DAMPs allow navigated immune clearance of ferroptotic cells and
promote anti-tumor immunity. For example,
1-steaoryl-2-15-HpETE-sn-glycero-3-phosphatidylethanolamine (SAPE-OOH), one of
the oxygenated lipid products released from cells undergoing ferroptosis, serves
as an “eat-me” DAMP by interacting with toll-like receptor 2 (TLR2) on
macrophages. Within the context of ovarian cancer, this event has been shown to
promote M1 activation [73]. Another important “eat-me” DAMP is high mobility
group box 1 (HMGB1), whose release relies on ferroptosis-related autophagy [72].
HMGB1 can not only promote M1 polarization but also can upregulate advanced
glycosylation end product-specific receptor (AGER)-mediated TNF-
Alternatively, the “save-me” DAMPs initiate a repair process in neoplastic
tissues, resulting in immunosuppression within the TIME and enhanced resistance
to ferroptosis [8]. It was reported that ferroptotic cells in pancreatic ductal
adenocarcinoma (PDAC) can package an oncogenic form of the protein KRAS
Notably, ferroptosis is often accompanied by autophagy, which plays an essential
role in regulating ferroptosis (Fig. 4). Some ferroptosis-associated DAMPs,
including HGMB1 and 8-OHG, originate from the autophagy of specific cellular
components, whereas knockout of autophagy-dependent genes, such as ATG5 and ATG7,
will inhibit the release of DAMPs [38, 79]. Further, the selective autophagy of
ferritin and lipid droplets will release dramatic amounts of labile iron and
lipoperoxides, thus providing a significant pool of molecules to trigger
ferroptosis. For example, ferritin can specifically bind to the C-terminus of
nuclear receptor coactivator 4 (NCOA4) which will subsequently be trafficked into
autophagosomes [80]. Similarly, ras-related protein Rab-7a (RAB7A) can contribute
to lipid phagocytosis by recruiting lipid droplets for delivery into lysosomes
[81]. Of note, some classical autophagy-associated molecules can inactivate the
core components of the intracellular antioxidant system in an
autophagy-independent manner. Specifically, AMPK-mediated Beclin 1 (BECN1)
phosphorylation can trigger the formation of the BECN1/SLC7A11 complex and
inhibit the transport activity of system Xc
In sum, the core mechanism of ferroptosis-mediated macrophage polarization can be summarized into three basic steps: (1) DAMPs are produced by cells undergoing ferroptosis; (2) DAMPs are released from cells in a specific form; (3) DAMPs are ultimately uptaken by macrophages to induce a series of phenotypic alterations (Fig. 5). On the basis of these three steps, we can develop a series of therapeutic strategies to favor the “eat-me” DAMPs and suppress the “save-me” DAMPs. It is noteworthy that autophagy is vital in the production of some DAMPs, and that exosomes can serve as a crucial DAMP carrier for tumor/macrophage communication. Thus, targeting autophagy-related signaling molecules, as well as the formation of exosomes, may also contribute to resetting DAMP release in immunotherapy.
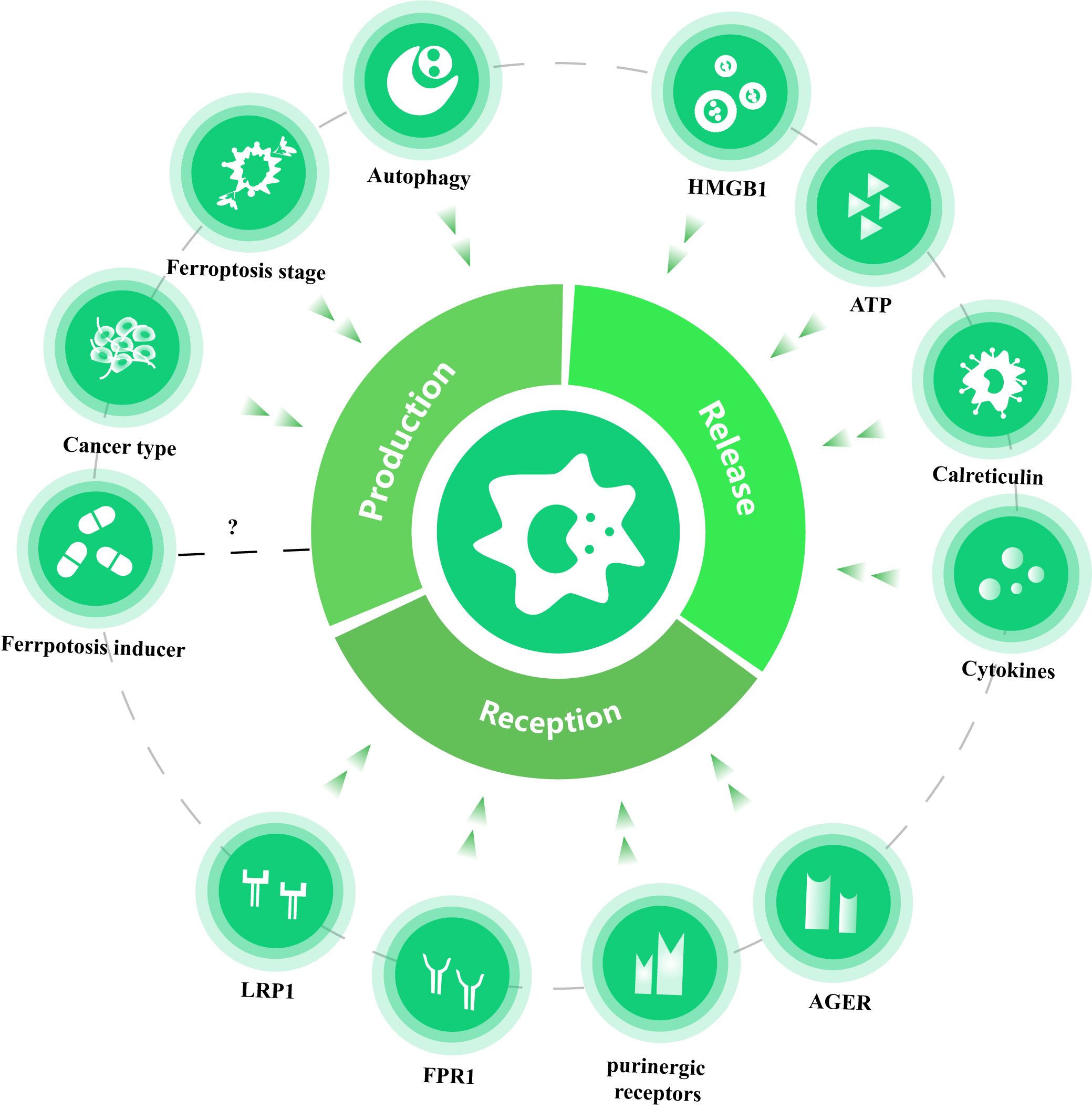 Fig. 5.
Fig. 5.Three basic steps of DAMP-mediated TAM activation and the factors affecting the immunogenic property of ferroptotic tumor cells. Ferroptotic cancer cells exert the immunostimulatory effects to activate TAMs by the production, release and TAM recognization of DAMPs (taking cytokines as a special kind of inducible DAMPs). Factors affecting DAMP production could largely impact the type of ferroptotic DAMPs, and different types of DAMPs and PRRs will activate distinct signaling pathways, thereby determining the immunogenic property of ferroptosis. Of note, it is still unclear whether the type of ferroptosis inducers could impact DAMP production. PD-L1, programmed cell death 1 ligand 1; CTL, Cytotoxic T Lymphocyte; LRP1, low density lipoprotein receptor-related protein 1; FPR1, formyl peptide receptor 1; HMGB1, high mobility group box 1; AGER, advanced glycosylation end-product specific receptor.
Ferroptosis is a necrotic process accompanied by oxidative damage and autophagic degradation, and which generates a multitude of compounds with strong immunogenicity and adjuvanticity [83]. Accumulating evidence has established that ferroptotic tumor cells are abundant sources of DAMPs such as HMGB1, ATP and HSP90, which could induce activation and maturation of the antigen-presenting cells (APCs) and lead to secondary secretion of proinflammatory cytokines [84]. Conversely, inhibiting either the release of DAMPs or blocking binding to PRRs will result in dysregulation of anti-tumor CTLs within the TIME [85]. Thus, these findings suggest that ferroptosis exhibits hallmarks of immunogenic cell death (ICD), and plays a crucial role in activating cancer-killing immune responses within the TIME.
Ferroptosis exerts immunostimulatory effects mainly through DAMP production and
major forms of ferroptotic DAMPs can vary in a time-dependent way. For example,
early in the process of ferroptosis, cells produce higher levels of HMGB1 but at
later stages, feroptotic cells tend to generate more oxidized lipids. These
oxidation products decrease phagocytosis and antigen cross-presentation by
dendritic cells [86]. Such a distinct immunogenic propensity between early and
late ferroptotic cells is presumably because ferroptosis-associated autophagy may
be the dominant biological event during the early stages of ferroptosis, but at
later stages this may be replaced by ROS generation and lipid peroxidation. Other
than different temporal stages of ferroptosis, different cancer cell types appear
to have preferences for a specific type(s) of ferroptotic DAMPs, such as the
PDAC-specific KRAS
Immunostimulatory efficiency relies on the specific type of byproduct produced by cells undergoing ferroptosis. Generally speaking, cytokines are more effective than DAMPs as they function at lower concentrations, generally at the picogram level [87]. Owing to the similarities between cytokines and DAMPs, Yatim et al. [88] proposed that cytokines can be seen as inducible DAMPs (iDAMPs), and both DAMPs and iDAMPs are required for efficient immune activation. However, most iDAMPs are stimulated by DAMPs in the later stages of activation; therefore, DAMPs play a vital role in early immune activation. Of the various ferroptotic DAMPs, HMGB1 demonstrates higher immunostimulation efficiency and enhanced stability in the extracellular environment compared with ATP, which is rapidly depleted after 24 h of ferroptosis induction [86]. However, Elliott et al. [89] discovered abolishment of APC recruitment when ATP fails to bind to the purinergic receptors, suggesting that ATP plays an indispensable role in activation anti-tumor immunity. Additionally, calreticulin exposure is also required for immunostimulation during the triggering of ferroptosis-like cell death exhibited by TRAMP-C1 cells [90]. Taken together, these findings revealed at least three ferroptotic DAMPs, namely HMGB1, ATP and calreticulin, are essential in mediating the activation of anti-tumor immunity.
Finally, different PRR pathways have been characterized as having distinct effects. In general, DAMPs have higher affinity to TLRs [91], which can accept a wide range of ligands and activate serval classical signaling pathways to promote innate immunity. Some PRRs can activate specific pathways to exert distinct effects. For example, CRT and HSPs can bind to LRP1 and drive immunogenic phagocytosis [92, 93], while ATP can stimulate inflammasomes through purinergic receptors [85]. Notably, certain immunostimulatory DAMPs do not signal through PRRs but rather bind to other immunostimulatory receptors like purinergic P2 receptors, low density lipoprotein receptor-related protein 1 (LRP1), formyl peptide receptor 1 (FPR1), or AGRE to propagate danger signals [94]. Conversely, some DAMPs may not always act as danger signals but as “bystanders”. For example, HSP90 does not exert any immunogenic effects despite being commonly exposed on the surface of cancer cells [95]. Taken together, ferroptosis is an indispensable part of ICD, and presents a promising therapeutic strategy by targeting ferroptotic DAMPs and relevant immune signaling pathways to enhance immunogenic effects and improve cancer immunotherapy (Fig. 5).
Since the initial approval of anti-cytotoxic T lymphocyte-associated protein 4 (anti-CTLA-4) for clinical use in melanoma patients in 2011, immune checkpoint inhibitors (ICIs) have been a predominant strategy for treating some advanced malignancies [96, 97]. One of the most widely-used ICIs, anti-PD-L1 antibody, blocks PD-L1, a CTL inhibitor expressed on neoplastic cells. This approach exploits the anti-tumor potential inherent in CTLs and included in this response is promotion of tumor ferroptosis [98]. ICIs were once considered to be the ultimate solution to metastatic malignancies until drug tolerance was reported in clinical cohorts [99, 100]. It is well recognized that many of the intrinsic properties of tumors including tumor heterogeneity, tumor mutation burden, and metabolic reprogramming are associated with the therapeutic effects of ICIs [101]. Just as clearly, TIME components, especially TAMs, play a crucial role in tumor resistance to ICIs. Various cytokines from TAMs can both disable and deplete CD8+ T cells as well as interact with tumor cells resulting in metabolic reprogramming and even promote irreversible alterations in the cell’s genetics [102]. Such typical changes include overexpression of oncogenic ICIs and insensitivity to immunogenic cell death [103, 104]. Notably, a predominant population of M2-like TAMs is observed in tissue samples taken from non-responders to immunotherapy, indicating that M2-like TAMs may play a central role in contributing to cancer-acquired resistance to immunotherapy [105, 106]. However, some drug-resistant tumor cells undergoing epithelial-mesenchymal transition (EMT) may display heightened sensitivity to ferroptosis due to GPX4 system downregulation and the activation of inflammatory pathways that occur during EMT [107].
On basis of these findings, we can propose a hypothesis to account for the cancer-acquired immunotherapeutic tolerance stemming from macrophage-ferroptosis crosstalk. Specifically, in the early therapeutic stages, anti-PD-L1 therapies can alert a significant number of CTLs in a very short time, and this can activate a potent tumor ferroptotic response [14]. This response may lead to a predominant release of “save-me” DAMPs and thereby promote an overwhelming accumulation of M2 macrophages within the TIME. These dominant M2 macrophages can, in the long term, elevate the expression of tumorigenic PD-L1 and activate feedback signaling to suppress CTL-mediated ferroptosis during immunotherapy (Fig. 6) [20, 108, 109]. Although such negative feedback may slow down as tumor resistance to ferroptosis gradually develops, it will be irreversible once therapeutic resistance is rooted within the genome of the cancer cells [104]. This may be an important cause for iron-induced immunotherapeutic resistance in PDAC [38, 110]. However, given the cancer type-dependent property of DAMP production, whether such a mechanism of immunologic tolerance exists in other types of malignancies is still unclear. While further efforts are clearly required to veraciously test this hypothesis, it is very likely that the macrophage-ferroptosis axis can provide key insight into mechanisms that govern the development of immunotherapeutic resistance. Rebalancing macrophage-ferroptosis crosstalk to favor M1 macrophage polarization may provide a necessary conceptual framework to understand, and ultimately overcome, the current therapeutic resistance dilemma.
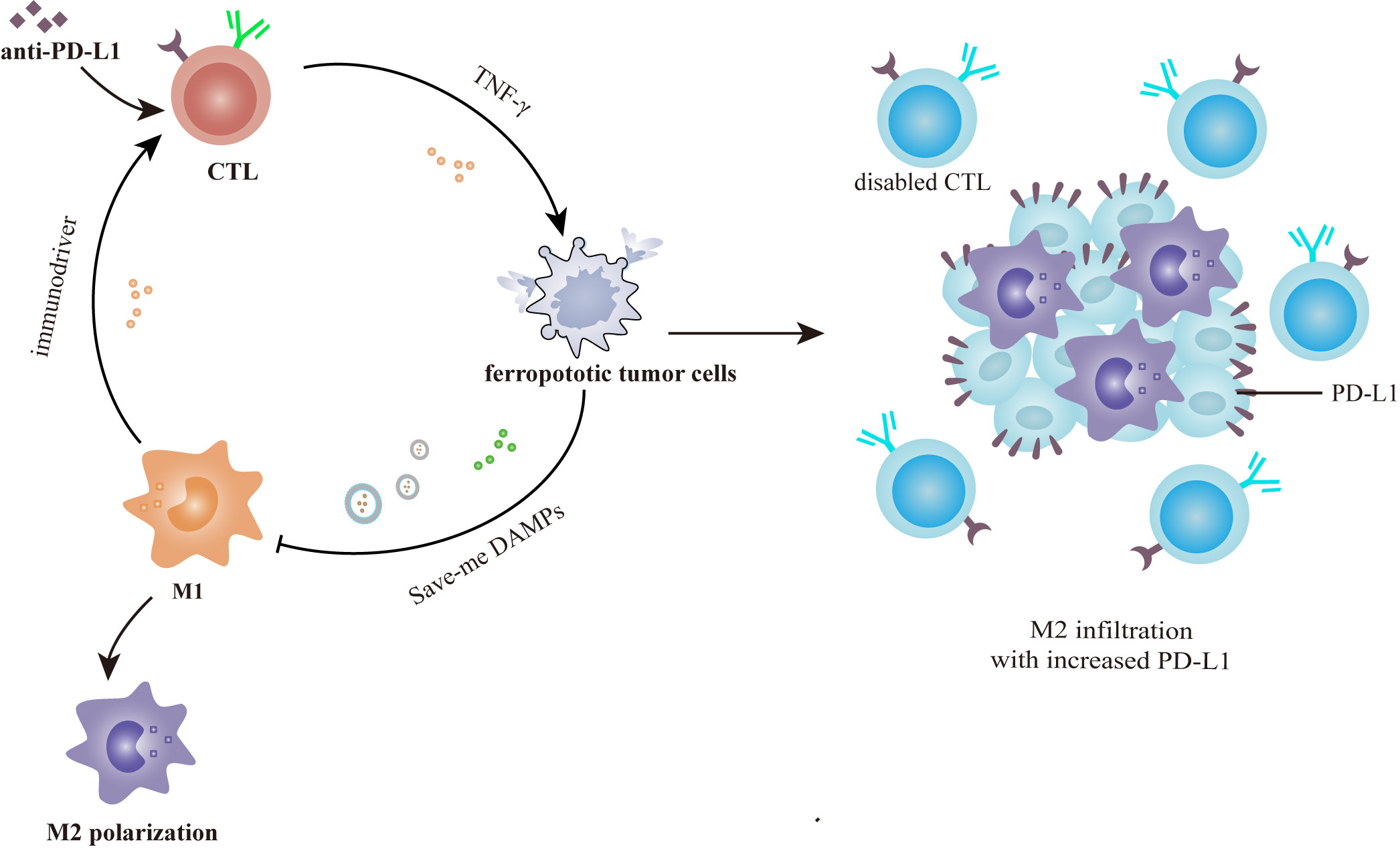 Fig. 6.
Fig. 6.Immunotherapeutic tolerance caused by the macrophage-ferroptosis crosstalk. Tumor resistance to anti-PD-L1 antibody derives from negative feedback signaling between ferroptotic cells and macrophages. Anti-PD-L1 immunotherapy causes intensive tumor ferroptosis in the early stage, which in return promotes M2 infiltration and tumorigenic PD-L1 expression via the “save-me” DAMPs. This may account for the iron-induced resistance of advanced PDAC to immune checkpoint inhibitors.
Recently, drugs targeting ferroptosis have shown promise as effective
anti-cancer therapeutics. Currently, ferroptosis inducers can be divided into 4
types based on targets and mechanisms (Table 1, Ref.
[9, 111, 112, 113, 114, 115, 116, 117, 118, 119]). Type I ferroptosis inducers directly
bind to subunits of system Xc
| Type | Agent | Mechanism | Application | References |
| Type I | erastin | inhibits system Xc |
preclinical experiments | Dixon et al. [9] |
| sorafenib | inhibits system Xc |
clinical treatement for several advanced cancers | Sun et al. [115] | |
| sulfasalazine | inhibits system Xc |
clinical treatement for lymphoma, SCLC and PC | Gout et al. [116] | |
| Type II | RSL3 | inactivates GPX4 | preclinical experiments | Sui et al. [111] |
| Yang et al. [117] | ||||
| altretamine | inactivates GPX4 | clinical treatement for OC | Woo et al. [112] | |
| Type III | FIN56 | depletes GPX4 and CoQ10 | preclinical experiments | Sun et al. [118] |
| statins | depletes GPX4 and CoQ10 | undergoing clinical trials for HCC and AML | Viswanathan et al. [119] | |
| Type IV | FINO2 | increases the level of labile iron; inactivates GPX4 | preclinical experiments | Gaschler et al. [113] |
| Others | ferroptocide | depletes thioredoxin | preclinical experiments | Llabani et al. [114] |
| CC, Hepatocellular carcinoma; AML, Acute myeloid leukemia; SCLC, small-cell lung cancer; OC, ovarian cancer; PC, prostate cancer. | ||||
Sorafenib is currently the FDA-approved ferroptosis inducer for treatment of
certain advanced cancers [120]. Unlike other type I ferroptosis inducers,
sorafenib acts as a multi-kinase inhibitor which inactivates kinases essential
for system Xc
Type II ferroptosis inducers inhibit the cellular antioxidant system by directly inactivating GPX4, and drugs of this type include RSL3 and altretamine. Of these, RSL3 is widely used as a complement to erastin in parallel experimental designs [111], while altretamine is clinically approved for treating ovarian cancer [112]. Slightly different from type II ferroptosis inducers, the type III ferroptosis inducers deplete GPX4 by mediating the degradation of GPX4 and CoQ10, and drugs of this type are FIN56 and the statins. It remains unclear whether GPX4 depletion is caused by statins or via statin-mediated decreases in cholesterol [6]. Further, type IV ferroptosis inducers can accelerate lipid peroxidation by increasing cellular levels of labile iron. Of note, FINO2, a type IV ferroptosis inducer, can synergistically trigger the oxidation of labile iron and inactivation of GPX4 [113]. Finally, some atypical ferroptosis inducers can target the non-classical pathways that trigger ferroptosis. One such drug is ferroptocide which targets thioredoxin to produce excessive ROS [114]. Taken together, this evidence indicates that targeting ferroptosis can provide an innovative and promising therapeutic strategy for malignant tumors as a complement to traditional chemotherapy.
TAMs are enriched with diverse types of signaling receptors that are sensitive to glycolipids and cytokines present within the TIME, and these provide TAMs with great metabolic and functional plasticity [123, 124]. It is well undeerstood that macrophage polarization is a reversible process with modulators from the local microenvironment intervening in the reprogramming process at multiple levels [125, 126]. To reprogram M2-like TAMs to favor activation of tumor ferroptosis, a series of bioengineered nanoparticles was developed to target various pathways. In brief, currently-developed nanoparticles targeting TAM repolarization can be divided into three main subtypes: nanocarriers, magnetic nanoparticles, and cell membrane-derived bionic nanoparticles (Fig. 7).
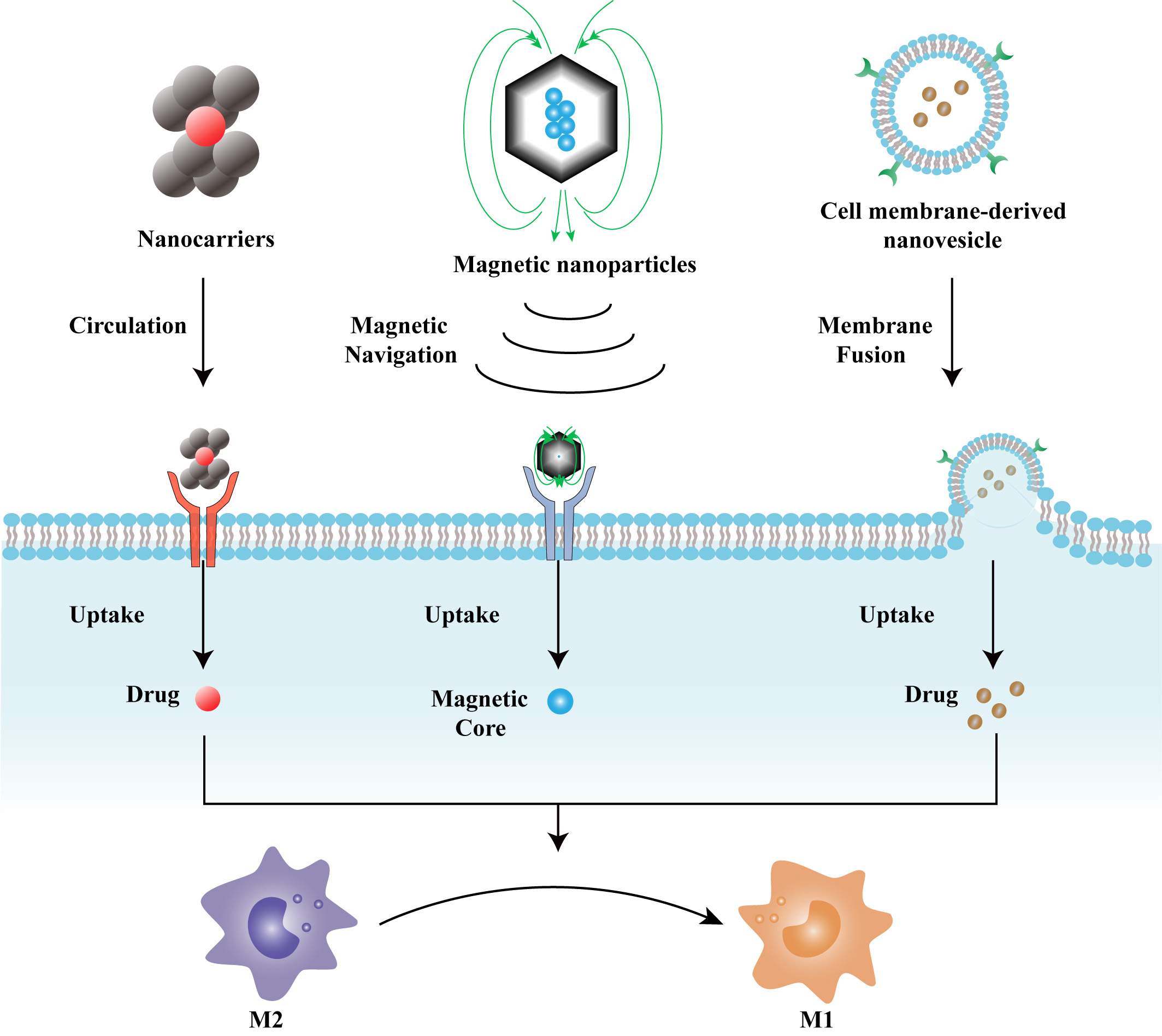 Fig. 7.
Fig. 7.Structural and functional characteristics of TAM repolarization-targeting nanoparticles. Nanocarriers can be specifically recognized and uptaken by TAMs, where the drug molecules are released to exert their effects. Magnetic nanoparticles can be efficiently delivered via magnetic navigation and can be captured through coat/receptor interaction. Cell membrane-derived nanovesicles are naturally compatible with the structure of lipid bilayers, and, as such, can be easily uptaken by TAMs.
Nanocarriers work by targeted drug delivery or tumor-killing nano-scale effects such as cytotoxic photodynamic and photothermal effects [127, 128]. These drugs are usually designed with unique geometrical structures which can be specifically recognized and uptaken by TAMs rather than by other normal tissues, thereby exhibiting low general cytotoxicity and high biocompatibility [129, 130]. For example, Fu and colleagues [131] developed a novel nanoparticle using a polymer of poly (styrene-co-maleic anhydride) (PSMA), and a polymer of poly [2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (PPV). This nanoparticle exhibited high affinity for TAMs and can repolarize TAMs into the tumoricidal M1 phenotype, likely owing to its lipopolysaccharide-like structure. In addition, by taking advantage of the acidic metabolic feature of the TIME, Xiao’s team [132] ingeniously designed a micellar nanodrug with M2-targeting peptides hidden in the pH-sheddable polyethylene glycol (PEG) corona so that the repolarizing drugs inside in nanoparticle can only target macrophages within the TIME. Furthermore, Qian et al. [133] developed an efficient siRNA and CpG oligonucleotide (ODN) delivery system that facilitates TAM remodeling by directly silencing M2 macrophage-related genes by using a nucleic acid step-wise, self-assembly technique. Inspiringly, CpG-siRNA-tFNA could effectively trigger proinflammatory cytokine secretion and activate NF-kB signal pathway, thus inducing dramatic antitumor immune responses in 4T1-bearing mice.
Magnetic nanoparticles with functional coatings are the next-generation version
of traditional nanocarriers. These nanoplatforms are endowed with integrative
functions such as magnetic navigation-based targeted delivery, nano-scale
therapeutic effects, and use in magnetic resonance imaging [134, 135, 136]. Iron
oxide-based magnetic nanoparticles can be released as free iron ions which can
subsequently directly activate tumor ferroptosis [136, 137]. In 2020, Rao’s group
[138] developed novel magnetic nanoparticles coated with genetically engineered
cell-membranes (gCM-MNs) which can precisely target the TIME through the guidance
of magnetic navigation. Not only can gCM-MNs block the CD47-SIRP
Composed of phospholipid bilayers, saccharides, and proteins, the cell membrane
is naturally compatible with the in vivo environment and
possesses a remarkable affinity to homogeneous cells [139]. Therefore, cell
membrane-derived bionic nanoparticles are the current optimal nanoplatform for
targeted therapy. Of note, some M1-derived exosome mimics can simultaneously act
as both an M2 repolarizer and ferroptosis inducer, providing an ideal biomaterial
to achieve the perfect combination of these two anti-cancer properties. The case
in point is CCR2(+)-Fe-M1-nanovesicles (Nvs), which are bioengineered with
up-regulated C-C motif chemokine receptor 2 (CCR2) expression and are loaded with
Fe
Despite their great promise, there are great challenges to the clinical translation of these nanoparticles. For one, it is hard to guarantee the specificity of observed therapeutic effects in large-scale clinical cohorts due to individual variation and tumor heterogeneity. For another, techniques used to prepare nanomedicines in the lab must be improved to meet the demands and requirements for large-scale production [143]. In this context, more effort is needed in the field of translational medicine to equip these nanodrugs with critical properties such as stable therapeutic effects and higher biosafety.
Despite great progress made in exploring independent roles of TAMs and ferroptosis in tumor immunotherapy, there are few studies focused on the integrative effects of macrophage-ferroptosis crosstalk in tumor immunotherapy. In fact, TAMs and ferroptosis are coordinately regulated, and thus co-regulate response to tumor immunotherapy. In this review, we profiled the molecular landscape of macrophage-ferroptosis crosstalk in the TIME with a focus on the different roles of M1 and M2-like TAMs. We also discussed the significant roles for macrophage-ferroptosis crosstalk in immunotherapeutic tolerance, and summarized the latest advances in treatment strategies that seek to leverage ferroptosis and TAM repolarization. Although these combined strategies have demonstrated dramatic anti-tumor effect, related clinical studies are still in their infancy and the detailed mechanisms governing macrophage-ferroptosis crosstalk still remain largely unexplained. Thus, further efforts are needed to better understand the signaling network that links TAMs and ferroptosis, and to promote the clinical translation of treatment strategies that seek to leverage macrophage-ferroptosis signaling for clinical benefit. We believe that synergistic strategies that target macrophage-ferroptosis crosstalk will dramatically improve the clinical application of tumor immunotherapy in the future.
ZZ and WY together conceptualized the idea. BX and NH collected reliant literatures. ZZ integrated useful information and wrote the manuscript. ZG and WB created the figures. BS and WY offered advice on polishing the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We would like to apologize to colleagues whose work is not cited due to space constraints. And we also thank anonymous reviewers for excellent criticism of the article and all the authors in the reference list.
This work was funded by the Sichuan University-Panzhihua City 2021 Campus Cooperation Special Fund Project (2021CDPZH-7) to WY, and the College Students’ Innovative Entrepreneurial Training Project (S202210610544) to NH.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.