


1 Department of Endocrinology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, 310003 Hangzhou, Zhejiang, China
†These authors contributed equally.
Academic Editor: Graham Pawelec
Abstract
Obesity has become an urgent and serious public health challenge with an overwhelming increase over the decades worldwide. The rate of obese children and adolescents has recently accelerated, especially in China. Obesity is closely related to unbalanced cellular energy metabolism. Mitochondria, as the main organelles of energy metabolism, play an important role in the pathophysiology of obesity. Recent researches have revealed that mitochondrial dynamics with constant fission and fusion, can alter mitochondrial structure, organelle connections, ROS production, neuronal activity, and OXPHOS system as well as adipose tissue thermogenesis, which ultimately lead to obesity. In this review, we will update the latest findings about mitochondrial fission/fusion related GTPase proteins and discuss the effects of mitochondrial dynamics in the pathophysiology of obesity.
Keywords
- mitochondrial dynamics
- fission
- fusion
- obesity
- Opa1
- Drp1
- Mfn1
- Mfn2
Obesity has become an urgent and serious public health challenge with an overwhelming increase worldwide over the past 50 years. A total of 1.9 billion and 609 million adults, approximately 39% of the world’s population, were estimated to be overweight and obese in 2015 [1, 2]. The total number of worldwide obese population has nearly tripled while the number of obese children and adolescents (aged 5 to 19 years) worldwide has risen tenfold since 1975. Over 41 million children under the age of 5 and over 340 million children and adolescents aged 5–19 years old were reported overweight or obese in 2016 [3, 4].
Obesity has physical and psychological health impact throughout all stages of life [5]. Obesity itself can cause high blood pressure, high cholesterol, fatty liver disease, increasing the risk of impaired glucose tolerance, insulin resistance, and type 2 diabetes, as well as the metabolic syndrome in childhood [6, 7, 8, 9]. Alarmingly, childhood obesity increases the likelihood of adult obesity with an additional increased risk of cardiovascular morbidity and mortality [10, 11, 12].
Excessive energy intake and reduced energy consumption contribute to the development of obesity. Obesity is closely related to unbalanced cellular energy metabolism, especially mitochondrial metabolism [13]. Mitochondrial dysfunction is highlighted in the pathophysiology of obesity, including mutation of mitochondrial DNA, reduction in mitochondrial content and/or biogenesis, impaired dynamics (fission/fusion), impaired mitophagy, failure in bioenergetics, reduced enzyme activity, and augmented oxidative stress [14]. It has becoming increasingly well-acknowledged that mitochondria are dynamic organelles. Mitochondrial shapes can be club-shaped, spherical, filamentous and so on. One shape of mitochondria may transform into any other type, by either fusion or division (fission) [15, 16]. Recent researches have revealed that mitochondrial dynamics as a conserved mechanism can regulate mitochondrial remodeling with the fission and fusion processes, thus affecting the pathophysiology of metabolic diseases, including obesity and type 2 diabetes [17, 18, 19].
This review focuses on the new aspects of mitochondrial dynamics and pathophysiology of obesity. We will summarize the recent findings of mitochondrial dynamic regulatory proteins, and discuss the effects of the unbalanced mitochondrial fission and fusion by reviewing the updated literature on their dysregulations in obesity so as to further understand the pathophysiology of obesity and provide new insights for future treatments.
Mitochondrial dynamics are regulated by a family of GTPase proteins. Dynamin-related protein 1 (Drp1) is the main fission protein that clefts mitochondrial membrane [20]. Besides Drp1, mitochondrial fission 1 protein (Fis1) also regulate the mitochondrial division [21, 22]. On the other hand, mitochondrial dynamin like GTPase optic atrophy 1 (Opa1) leads to the mitochondrial fusion of mitochondrial inner membrane [23], while mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) are responsible for the fusion of mitochondrial outer membrane [24]. Membrane-associated RING-CH-type finger 5 (March5) is a newly discovered mitochondrial fusion regulatory protein [25]. Mitochondrial dynamic related GTPase proteins and the main functions are listed in Table 1. Fig. 1 presents the interactions between mitochondrial fission/fusion and regulations between organelle/cell in the pathophysiology of obesity (Fig. 1).
| Fission/fusion protein | Change | Model | Organ/tissue | The main mechanisms | Influence on obesity | |
| fission | Drp1 | Ser-600 phosphorylation | mice | BAT | increased thermogenesis | protective |
| Ser-637 hyperphosphorylation | mice | skeletal muscle | increased oxidative metabolism | protective | ||
| transcription inhibition | mice | WAT | damaged adipocyte |
accelerative | ||
| tissue specific ablation | mice | adipose tissue | failure of LDs release | accelerative | ||
| tissue specific ablation | mice | liver | increased expression of FGF21 | protective | ||
| tissue specific ablation | mice | muscle tissue | reduced the phosphorylation of P38 | protective | ||
| Fis1 | reduced level due to maternal obesity during pregnancy | mice | whole body | reduced energy metabolism | accelerative | |
| reduced expression | mice | adipocytes | reduced TG content | protective | ||
| fusion | Opa1 | abnormal Oma1-Opa1 system | mice | BAT | abnormal thermogenesis | accelerative |
| IL-1R-MyD88-IRAK2-PHB/OPA1 pathway | mice | adipose tissue | reduced |
accelerative | ||
| tissue specific downregulated expression | mice | BAT | abnormal mitochondrial cristae structure | protective | ||
| tissue specific gene inactivation | mice | POMC neurons | decreased lipolysis of WAT | accelerative | ||
| tissue specific gene deletion | mice | muscle tissue | increased secretion of FGF21 | protective | ||
| Mfn1/2 | MFN2 p.R707W mutant | human | whole body | inhibition of leptin expression | accelerative | |
| MFN1/2 tissue specific gene deletion | mice | Agrp neurons | impaired electrical activity of neurons | protective | ||
| MFN2 tissue specific gene deletion | mice | POMC neurons | loss of mitochondria-ER contact | accelerative | ||
| increased Mfn1/2 expression induced by PGC-1 |
mice | adipose tissue | increased thermogenesis | protective | ||
| Mfn2 cell specific dysfunction | Human/mice | Hela/muscle cell | repressed nuclear-encoded subunits of OXPHOS complexes | accelerative | ||
| Mfn2 tissue specific reduced expression | mice | adipose tissue | reduced fatty acid transfer | accelerative | ||
| cell MFN2 knockout | human | Hela/muscle cell | inhibit autophagic-lysosomal fusion | accelerative | ||
| March5 | cell specific knockout | mice | adipocytes | increased lipid uptake and synthesis | protective | |
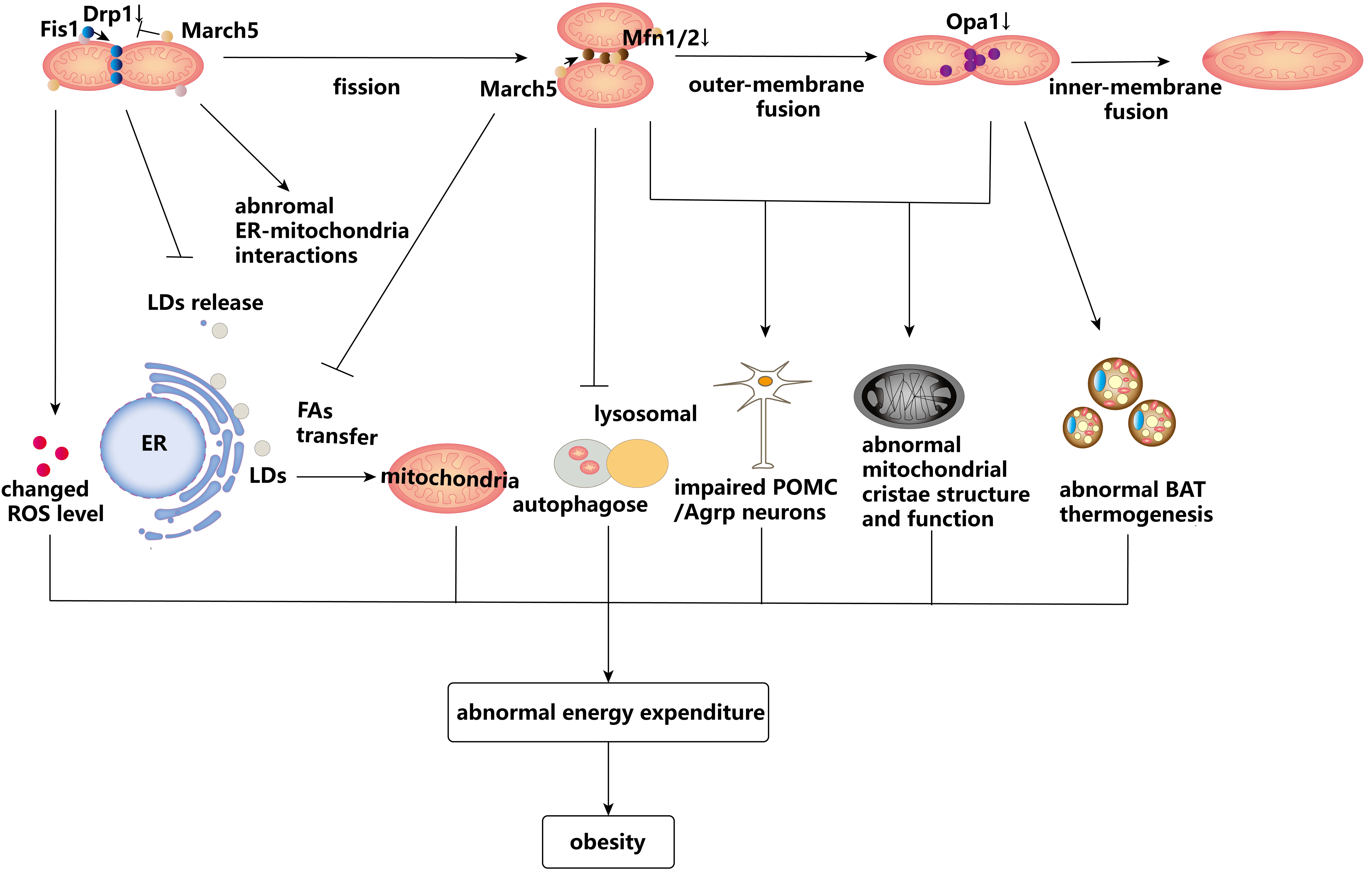 Fig. 1.
Fig. 1.Mitochondrial dynamics and organelle/cell interactions on obesity.
Drp1 is the main mitochondrial fission protein that is responsible for the
cleavage of mitochondrial inner and outer membranes in mammals. Mutations in the
GTPase domain can specifically cause alterations in the mitochondrial morphology
and mitochondrial collapse [26]. Drp1 is proved to participate in the
mitochondrial fission in mammalian cells as well as in C. elegans [27, 28]. Drp1
alone has the ability to constrict and sever cell membranes. Cutting requires the
membrane binding, self-assembly and GTPase activity of Drp1, which plays the
dominant role in the division of mitochondria and peroxisome [20]. Drp1 also
plays a role in mitochondrial cristae remodeling mediated by the endoplasmic
reticulum (ER) Ca
The mitochondrial fission function of Drp1 is related to protein molecular
modification and interactions. Norepinephrine (NE) mediated phosphorylation of
Drp1 Ser-600 mediates the mitochondrial division and induces the mitochondrial
uncoupling in brown adipose tissue (BAT), which results in increased heat
production and energy expenditure. The mitochondrial division in BAT may be a
novel potential therapeutic target to treat obesity [30]. In contrast, the
hyperphosphorylation of Drp1 Ser-637 enhances the mitochondrial respiration by
increasing mitochondrial fusion and lengthening mitochondria.
Hyperphosphorylation of Drp1 Ser-637 also leads to increased mitochondrial proton
gradient energy transfer, which increases the oxidative metabolism and prevented
high-fat diet (HFD)-induced obesity [31]. Adipose tissue-specific DNA
methyltransferase 1 (DNMT1) knockout blocks the interaction between
enhancers and DRP1 by influencing DNA methylation, thereby inhibiting
mitochondrial fission and leading to adipocyte hypertrophy and impaired expansion
of adipocyte precursors. Loss of DNMT1 can lead to the damage of adipocyte
Induced by apoptosis, Drp1 is transferred to mitochondria and preferentially locates at potential sites of division [33]. Endoplasmic reticulum is reported to play a central role in Drp1 aggregation on mitochondria fission. Drp1 oligomerization can be transferred from ER to mitochondria or peroxisome by the communication between the ER and mitochondria [34]. Formin-like protein Inf2 mediates the Drp1 polymerization of ER, leading to the increased ER-mitochondrial contact. The increased endoplasmic reticulum calcium inflow to mitochondria causes the mitochondrial division [35, 36]. ER stress leads to diverse mitochondrial outcomes. Ablation of adipose tissue specific DRP1 leads to the failure of ER lipid droplets (LDs) release and shape change. Abnormal lipid droplets shape and accumulation during ER stress can further lead to mitochondrial dysfunction, lipolysis disorders, reduced thermogenesis, and finally defective systemic lipid metabolism [37]. In contrast, the deletion of hepatic DRP1 gene induces ER stress, and promotes the expression of fibroblast growth factor 21 (FGF21) in liver [38]. Increased expression of FGF21 and ER stress can lead to the increased energy expenditure in mice, thus playing a protective role in HFD-induced obesity [38, 39, 40]. Currently strong evidence shows that CerS6-derived sphingolipids promote Drp1-mediated mitochondrial fragmentation in obesity [41]. Ablation of CerS6 in a mouse model of HFD-induced insulin resistance facilitates a successful reversal of the fragmentation of hepatic mitochondrial network, rescues the insulin-sensitive phenotype and prevented HFD-induced obesity [42].
Reactive oxygen species (ROS) is considered as the mediator of mitochondrial fission in endothelial cells [43]. ER stress leads to the increased ROS production in adipocytes and decrease the catalase synthesis, thus affecting the lipid metabolism [44]. Besides, ROS acts as signal molecules to activate P38, a member of mitogen activated kinase-like protein (MAPK) family [45]. Obesity can increase the phosphorylation of P38 in the skeletal muscle, which is reversed by reduced mitochondrial fission due to DRP1 ablation [46]. Therefore, increased DRP1 expression increases the ROS level by inducing cell division, forming a vicious cycle that eventually leads to abnormal cell metabolism.
The dynamin-related GTPase Fis1 locates in the outer membrane of mitochondria and also induces the mitochondrial division [22, 47]. Fis1 recruits Drp1 from the cytoplasm to participate in the mitochondrial division, resulting in increased mitochondrial fragmentation and changed mitochondrial shape [48, 49]. Researches show that increased mitochondrial fission proteins Drp1 and Fis1 with decreased fission protein Mfn2 can lead to impaired mitochondrial function and oxidative stress [50]. Maternal obesity during pregnancy can reduce the expression of Fis1, Opa1, Mfn1, Mfn2 in offspring, resulting in reduced energy metabolism and fat utilization. Studies reveal that alteration of regulatory mitochondrial factors in offspring may lead to the impaired mitochondrial health and increased susceptibility to obesity later in life [51]. After mitochondrial fusion induced by silencing mitochondrial fission proteins including Fis1 and Drp1 in adipocytes, the content of triacylglycerol (TG) in adipocytes was reduced. It was speculated that the mitochondrial fusion may be more efficient in carrying out pyruvate dehydrogenation, oxidative phosphorylation and lipid metabolism [52]. A novel selective peptide inhibitor, P110, is reported to inhibits the Drp1/Fis1 interaction and production of ROS in cultured neurons. Thus, P110 is helpful with treatment of obesity [53].
As a component of the mitochondrial network, the dynamin-related protein Opa1, encoded by nuclear gene OPA1, plays a role in the mitochondrial aggregation and stabilization of inner membrane [54, 55, 56, 57]. Opa1 is located in the inner membrane of mitochondria and coordinates the mitochondrial cristae, which is essential in the mitochondrial respiratory chain and oxidative metabolism [58, 59, 60, 61]. Different isoforms of Opa1 play different roles. They can restore the cristae structure, mtDNA abundance and energy efficiency. The complete recovery of mitochondrial dynamic network requires the interaction of different Opa1 isoforms [62]. OPA1 gene inactivation leads to dramatic alterations in mitochondrial network, in which mitochondrial fragments are scattered, and the mitochondrial cristae is broken and disorganized [63].
Mitochondrial protease OMA1 zinc metallopeptidase (Oma1) can inactivate Opa1
under stress and inhibit mitochondrial fusion [64]. It has been proved that
Cardiolipin (CL), located in the inner membrane of mitochondria, is also important for the mitochondrial cristae stability. It has been reported that mitochondrial lipids, especially CL, play a major role in regulating the mitochondrial cristae shape and dynamics under PH changes [69, 70]. It has been discovered that carbohydrate response element-binding protein (ChREBP) KO mice are resistant to obesity, and recent research has found that is due to the combined influence of decreased CL synthesis and downregulated expression of Opa1 in brown adipose tissue (BAT). All of these two changes can cause abnormal mitochondrial cristae structure in BAT of ChREBP KO mice to display an anti-obese phenotype [71].
The stability of the mitochondrial cristae in different tissues also ultimately
affects obesity. Recently it has been proved that adequate mitochondrial fusion
and fission in neurons is important for metabolic regulation. In
proopiomelanocortin (POMC) neurons, OPA1 gene inactivation leads to
dramatic alterations in mitochondrial cristae topology, mitochondrial Ca
The mitofusins (Mfn1 and Mfn2) lead to the mitochondrial fusion and regulate the mitochondrial cristae structure [74]. Both homomorphic and heteromorphic complexes work together to promote the mitochondrial outer membrane fusion in mammals [24, 75, 76, 77]. By influencing mitochondrial fusion, Mfn1 and Mfn2 regulate obesity-related metabolic changes such as reduced glucose oxidation, mitochondrial membrane potential, cellular respiration, and lipid toxicity [78, 79]. Enhanced mitochondrial fusion is beneficial to the mitochondrial biogenesis and lipid metabolism, which is impaired in obesity [78, 80, 81]. What’s more, decreased transcription of MFN2 gene is reported in obese humans, and the expression of MFN2 is negatively correlated with body mass index (BMI) [82]. Mutations in MFN2 (p.R104W, p.R707W) are found in obese patients [83, 84]. Broken mitochondrial networks and mitochondrial dysfunction with MFN2 mutations are speculated to cause inhibition of leptin expression and increase adipocyte proliferation and survival [85]. Moreover, selective deletion of MFN1 and MFN2 in mice impairs the mitochondrial fusion (both mitochondrial size and shape) in agouti-related protein (Agrp) neurons, resulting in impaired electrical activity of Agrp neurons, with increased resistance to fat gain and reduced weight gain in mice during high-fat diet [86]. Moreover, specific ablation of MFN2 in POMC neurons in the hypothalamus leads to loss of mitochondrial endoplasmic reticulum contact, resulting in endoplasmic reticulum stress, leptin resistance, hyperphagia, and reduced energy expenditure, which eventually lead to severe obesity [87].
The expression of mitochondrial fusion genes MFN1 and MFN2 is
regulated by peroxisome proliferator-activated receptor
In addition, Mfn2 dysfunction represses nuclear-encoded subunits of oxidative
phosphorylation (OXPHOS) complexes I, II, III and V, thereby inhibiting pyruvate,
glucose, and fatty acid oxidation and reducing mitochondrial membrane potential
[81]. Mfn2 can also affect energy metabolism by regulating the interaction
between lipid droplets and mitochondria [96]. Contact between activated
mitochondria and lipid droplets will promote the transfer of fatty acids from
lipid droplets to mitochondria for
Human membrane-associated RING-CH(March)-V/March5 is a novel mitochondrial outer
membrane transmembrane protein. March5 plays a promoter role in the mitochondrial
fusion [25]. March5 can bind Mfn2 to promote the formation of long tubular
mitochondria and mitochondrial networks. It can also promote the ubiquitination
of Drp1 to inhibit the mitochondrial division. March5 is regulated by peroxisome
proliferator-activated receptor-
In general, mitochondrial fission and fusion can affect the pathophysiology of obesity in several ways: (a) the connection between organelles, (b) protein molecular modification, (c) neuronal activity, (d) mitochondrial cristae stability and OXPHOS system function, (e) adipose tissue thermogenesis.
Mitochondrial dynamics provides new approaches to treatment of obesity. Several candidates are under study in animal and in vitro models based on mitochondrial fusion/fission theory, such as P110 and Vitexin. These inhibitors may be helpful for the treatment of diseases with unbalanced mitochondrial dynamics including obesity.
Drp1, Dynamin-related protein 1; Fis1, mitochondrial fission 1 protein; Opa1, optic atrophy 1; Mfn1, mitofusin 1; Mfn2, mitofusin 2; March5, membrane-associated RING-CH-type finger; ROS, reactive oxygen species; ER, endoplasmic reticulum; LDs, lipid droplets; FAs, fatty acids; POMC, proopiomelanocortin; Agrp, agouti-related protein; BAT, brown adipose tissue.
JF and JW contributed to the conception of this manuscript. XL drafted the manuscript. JW contributed to manuscript writing and revision for intellectual content. JW, GD and NZ conducted the literature search critically. WW and KH provided constructive discussions and contributed to paper review. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This work was supported by National Key Research and Development Program of China (No. 2021YFC2701901 and 2016YFC1305301), the National Natural Science Foundation of China (No. 81570759 and 81270938), the Fundamental Research Funds for the Central Universities (2020XZZX002-22), the Research Fund of Zhejiang Major Medical and Health Science and Technology & National Ministry of Health (WKJ-ZJ-1804), Zhejiang Provincial Natural Science Foundation of China (LQ20H070003), and Zhejiang Province Natural Sciences Foundation Zhejiang Society for Mathematical Medicine (LSZ19H070001).
The authors declare no conflict of interest.