








Retraction published on 2 Feb 2023, see Frontiers in Bioscience-Elite 2023, 15(1)

1 Clínica Universitaria de Navarra, Universidad de Navarra, Av. Pio XII 36, 31008 Pamplona, Spain
2 Centre for Applied Medical Research, Universidad de Navarra, Av. Pio XII 55, 31008 Pamplona, Spain
Academic Editor: Alessandro Poggi
Abstract
Stimulation of dendritic cells (DC) is considered critical in cancer
immunotherapy. BATF-3-dependent subsets, that express in humans CD141 (BDCA-3),
promote CD8 T-cell cross-priming against tumor antigens. Here, we evaluate two
clinical-grade stimuli for peripheral blood CD141
Keywords
- Myeloid dendritic cells (mDC)
- Cross-presenting CD141+ (BDCA-3+)
- RNA-seq
- Microarrays
- Immunotherapy
- Clinical trial
Dendritic cells (DCs) are professional antigen-presenting cells (APCs) and are
found in most tissues of the human body [1]. Upon activation, DCs migrate to the
lymph nodes and activate T cells [2]. Among the primary circulating blood DCs,
there are two main subsets, the myeloid DCs (mDCs) and the plasmacytoid DCs
(pDCs). The mDC subset can be subdivided into CD1c
Efficient activation is a crucial step for DC-based cancer immunotherapy [11, 12, 13, 14, 15, 16, 17]. DC activation can be measured based on different parameters. Firstly, surface expression of maturation markers like CD40, CD80, CD83, CD86, HLA-DR, or PD-L1, as well as chemokine receptors like C-C chemokine receptor type 7 (CCR7), can be measured using flow cytometry [18]. Co-stimulatory molecules like CD80 or CD40, but also co-inhibitory molecules like PD-L1, do not only represent the maturation state of DCs but also play an important role in regulating T cell stimulation. Secondly, cytokines and chemokines released by the cells indicate their viability, functionality, and maturation state. Furthermore, as indicators of the DC activation state, their ability to prime T cells or other immune cells can be measured by co-culture experiments in which T cell proliferation or phenotypic changes are used as a read-out. All these methods are based on a pre-defined set of markers and targets. On one hand, this means that those experiments are focused and comparable. On the other hand, however, important effects that are not known or expected a priori, or do not belong to the most obvious targets, can be missed.
In clinical trials, we have used CD1c
To compare the effects of these two adjuvants in an unbiased and global manner,
we applied transcriptome analysis. We employed both RNA-seq and microarray
measurements to interrogate the DC transcriptome and asked whether these
techniques would lead to similar conclusions. RNA-seq is known to have a higher
sensitivity and therefore increases the chance to observe changes in low-abundant
genes, while the microarray approach has the advantage of requiring substantially
less starting material, which can be a limiting factor for rare cell populations
like peripheral blood CD141
For RNA-seq and microarray measurements, cells were obtained from aphaeresis of
5 different donors. Due to the limited cell numbers of donor 1, only 4 donors
could be used for RNA-seq. Peripheral blood mononuclear cells (PBMCs) were
isolated by using Ficoll density centrifugation (Ficoll Paque Premium, GE
Healthcare, Chicago, IL, USA). Anti-Human Lineage Cocktail 1 in FITC (LIN1) (BD
Bioscience Pharmingen, San Jose, CA, USA) containing antibodies for CD3, CD16,
CD19, CD20, CD56 receptors, together with the anti-FITC conjugated magnetic
microbeads of Miltenyi Biotec (Bergisch-Gladbach, Germany) were used to deplete
the LIN1
pRNA complexes were made freshly before adding to the cells. Protamine (protamine hydrochloride MPH 5000 IE/mL; Meda Pharma BV Amstelveen, the Netherlands) was diluted to 0.5 mg/mL in RNase free water and mixed with 2-kbp-long single-stranded mRNA (coding for human gp100 protein) [14]. It was extensively mixed and incubated for 5–10 minutes at room temperature, before being added to the cells.
CD141
Data were transformed to log2 values for performing hierarchical clustering analysis (One minus Pearson correlation). Using the standard settings of the MORPHEUS - Versatile matrix visualization and analysis software version 0.0.2 (Broad Institute; Cambridge, MA, USA; https://clue.io/morpheus).
Data were analyzed using the R platform for statistical computing. Specifically, the package “edgeR”, version 3.16.5, in Bioconductor version 3.4 (http://bioconductor.org/, released on 31 October 2018) was used for whole-transcriptome principal coordinates analysis (using the “plotMDS” command), differential gene expression analysis, and GO term analysis. Differential expression was determined by fitting a generalized linear model using the “glmFit” command, and significance was determined using the likelihood ratio test provided by the “glmLRT” command. Data from the Clariom D assays were imported into R using the Bioconductor packages “affycoretools” [23] and “oligo” [24] and were analyzed using the empirical Bayes procedure [25] implemented in the “limma” package, version 3.30.13 (The Walter and Eliza Hall Institute of Medical Research, WEHI, Australia. Maintainer: Gordon Smyth (smyth@wehi.edu.au)). Throughout, p-values were corrected for multiplicity using the Benjamini-Hochberg method. For GO term analyses, a significance cutoff of 0.05 was used. All data analysis scripts are available as supporting information for this article.
We obtained CD141
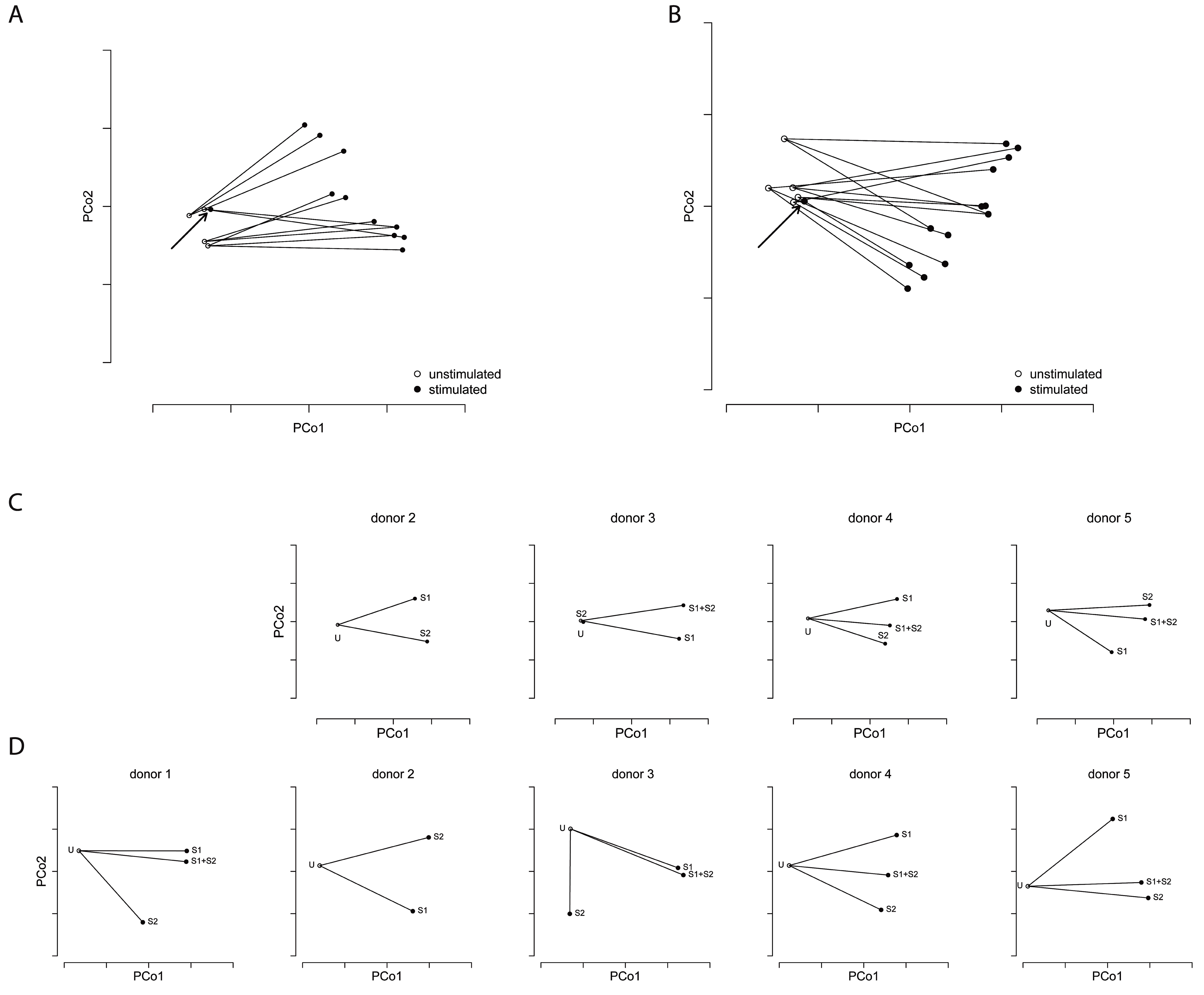 Fig. 1.
Fig. 1.Stimulation has strong effects on the transcriptomes of
CD141
Since CD141
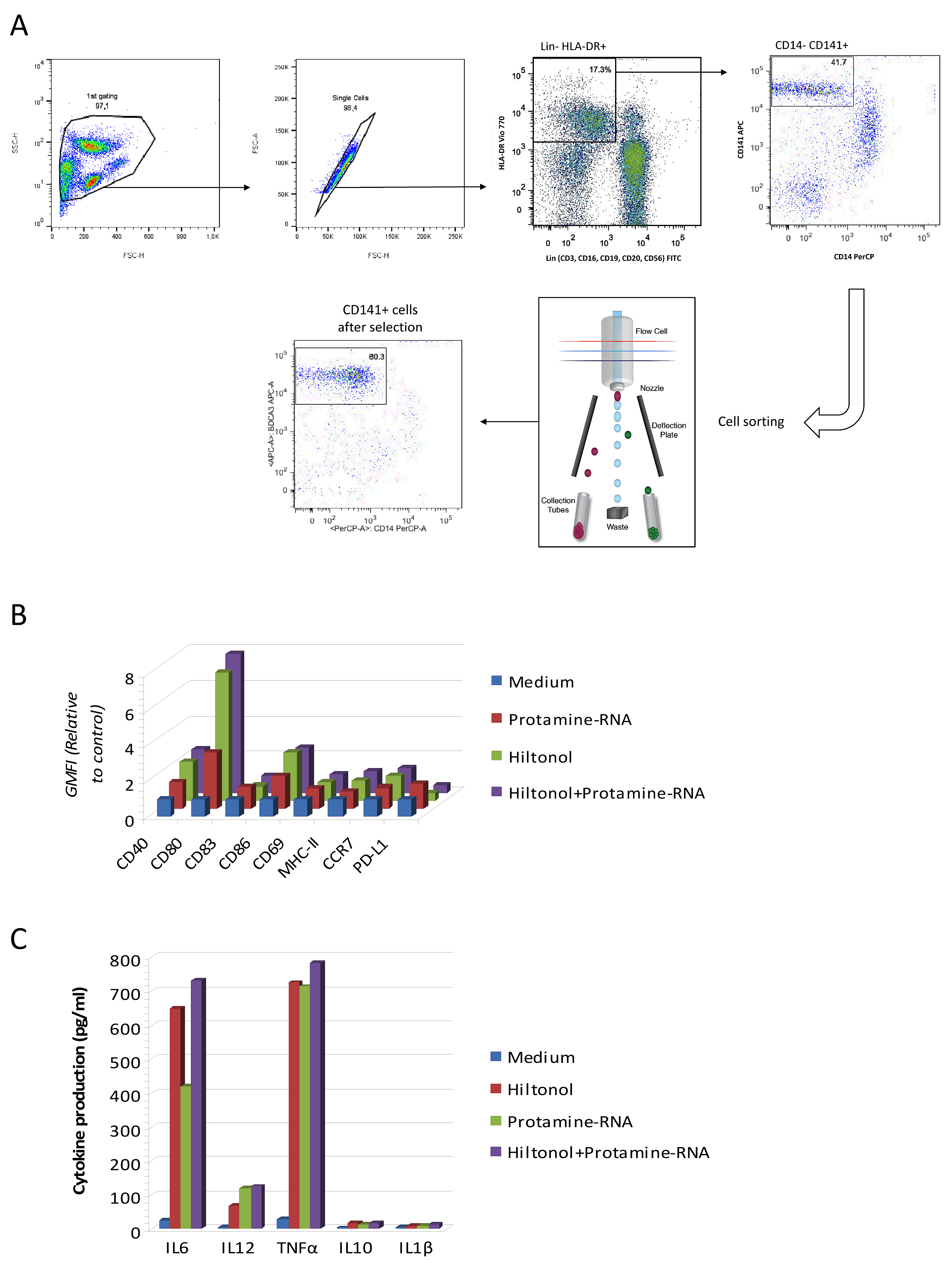 Fig. 2.
Fig. 2.Characterization of CD141
Likewise, after stimulation, we have carried out experiments to verify the effectiveness of the stimuli (Hiltonol and pRNA) checking the maturation state of cells (Fig. 2B,C). For this purpose, we have analyzed dendritic cell markers (Fig. 2B) and the production of cytokines in supernatant (Fig. 2C) previously described for this type of stimuli in DC populations [4, 9].
Significant increases in cell markers CD40, CD80, CD86, MHC-II and CCR7 were
observed (Fig. 2B). It is consistent with a strong stimulation of an APC cell
profile with an increase in co-stimulatory molecules, antigen presentation
capacity and migration to lymph nodes. Also, cells cultured in vitro
secrete IL-6, IL-12, and TNF-
Next, we directly compared the RNA-seq and microarray results for single genes, to see if the conclusions from both analyses are similar. Focusing on the set of genes that showed significant up-or downregulation upon stimulation (Fig. 3A,B), we observed that the direction of the change was the same in all cases (i.e., they were either up- or down-regulated in both datasets) with the single exception being the E2F3P1 pseudogene (Fig. 3B). The correlation between the estimated fold changes in the RNA-seq versus microarray data for the selected gene set was 0.9 and 0.91, respectively (Fig. 3A,B), largely due to the consistency in the direction of the effect. The fold-change estimated from the RNA-seq datasets was often several orders of magnitude larger than that obtained from the microarray datasets, which was expected due to the higher dynamic range of RNA-seq [22]. However, this analysis does show that both datasets are expected to yield similar qualitative conclusions, especially in downstream analyses that focus more on the direction of a change (i.e., after applying a fold-change cutoff) than on its magnitude, such as gene ontology (GO) analyses.
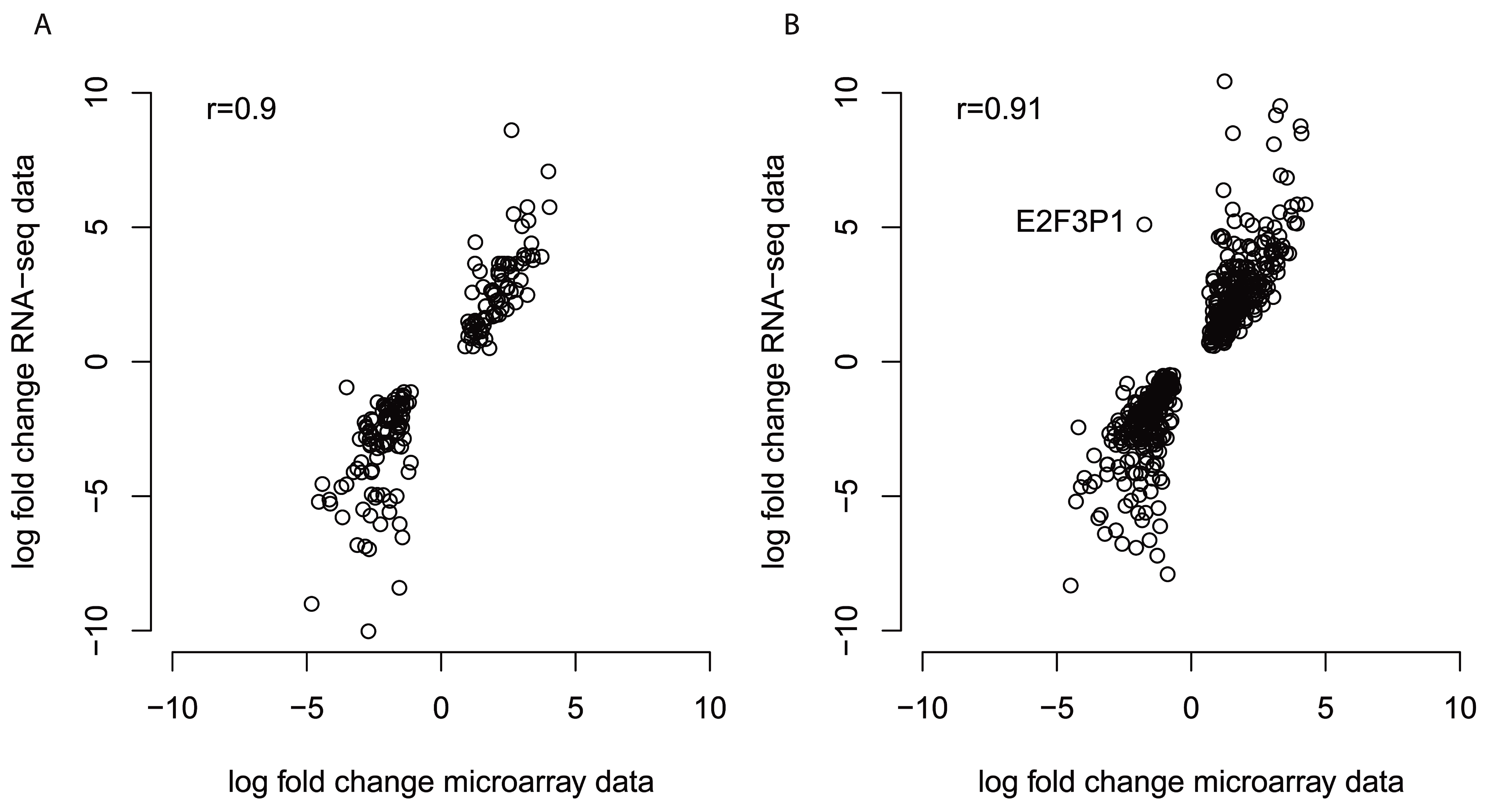 Fig. 3.
Fig. 3.RNA-seq and microarray analysis strongly agree on the effects of each stimulus. Microarray and RNA-seq fold change estimates for significantly up- or downregulated genes (applying a p-value cut-off of 0.05 after multiple testing correction) upon (A) Hiltonol and (B) pRNA were plotted against each other. The overall correlation was quantified using Spearman’s rho.
Aiming to identify similarities and differences between the two stimuli, we next
correlated the transcript fold-change values upon each stimulus (pRNA or
Hiltonol) to each other for the RNA-seq (Fig. 4A) versus the microarray (Fig. 4B)
data. This showed that (a) both stimuli upregulated more genes in CD141
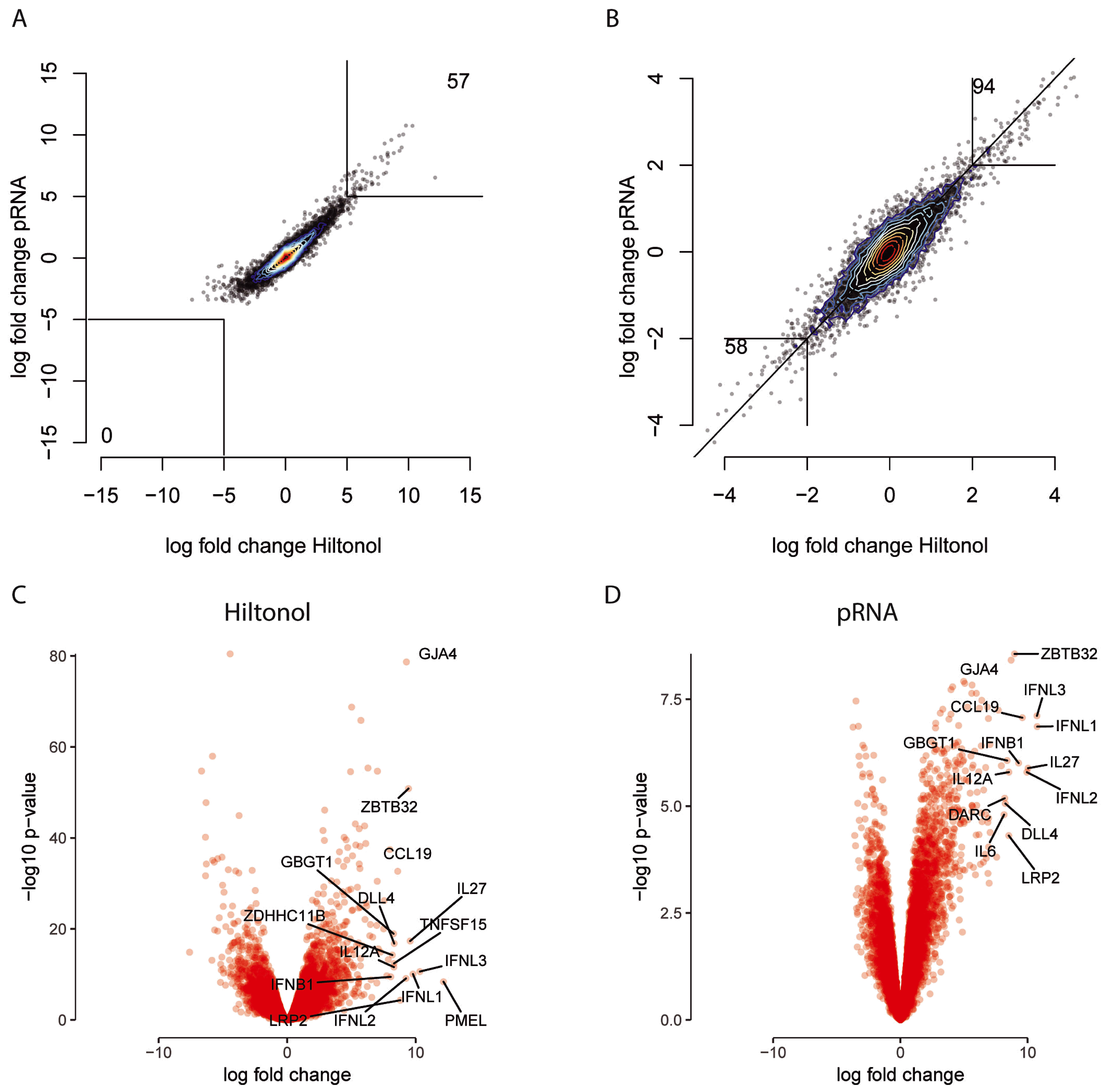 Fig. 4.
Fig. 4.Hiltonol and pRNA stimulation have similar effects on the
transcriptome of CD141
For reasons of simplicity, we therefore focus on the RNA-seq data only in the remaining of this paper.
To detect the most dominant changes upon each stimulus, we generated volcano
plots in which the genes with a log fold change of higher than 8 were labeled
with the gene names (Fig. 4C,D). Upon both stimuli, genes for IFN-
To identify specific pathways that changed in CD141
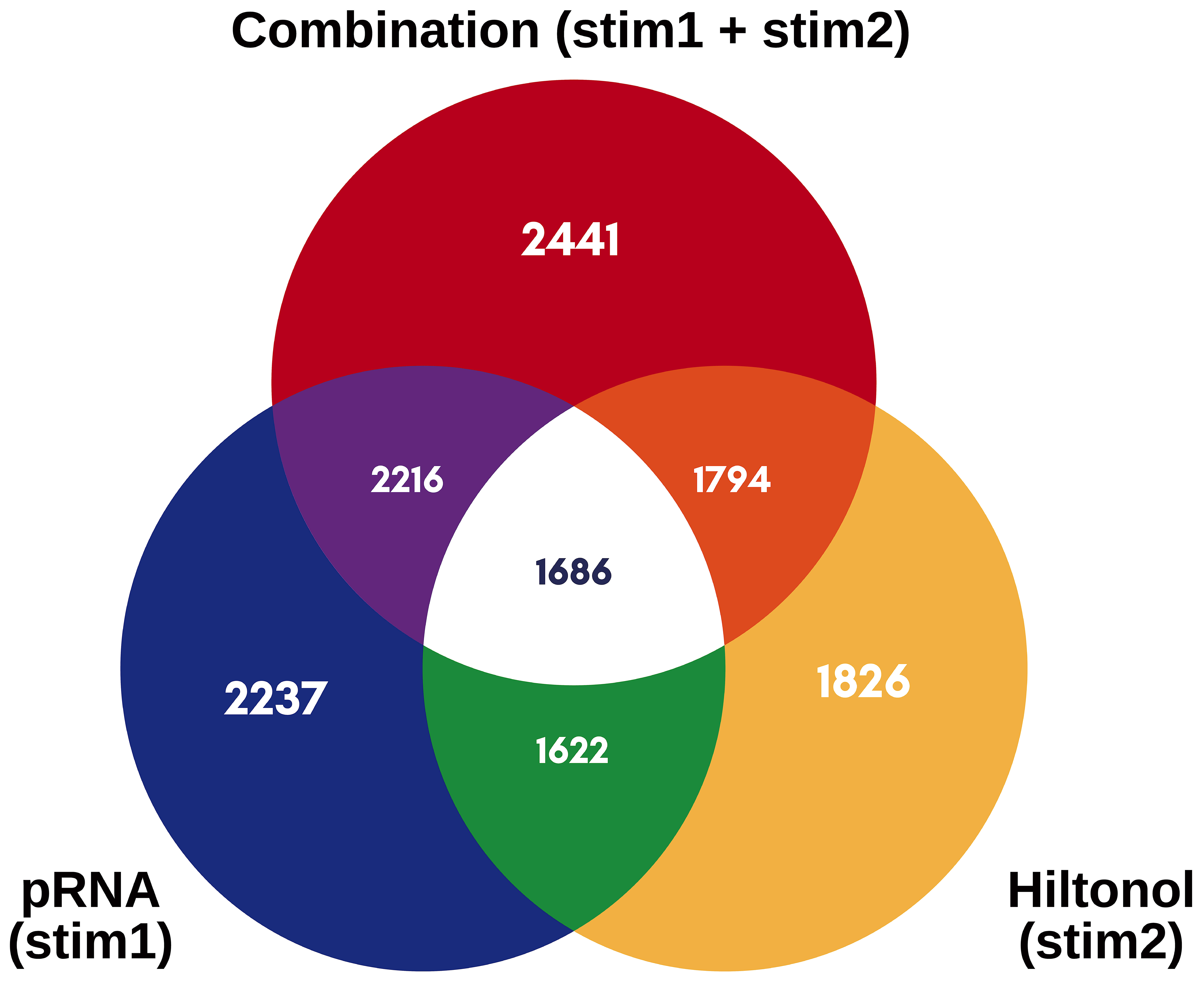 Fig. 5.
Fig. 5.Venn-diagram based on differentially expressed genes (DEGs) between different stimuli. Diagrams with overlapping genes compared the three treatments: protamine-RNA complex (stim1), Hiltonol (stim2) and combination (stim1 + stim2).
| Hiltonol | ||||
| Term | Ontology | N genes | N up/down | –log |
| Response to stress | BP | 3557 | 1028 | 28.7 |
| Cytoplasm | CC | 10,548 | 2601 | 28.2 |
| Immune system process | BP | 2348 | 717 | 26.1 |
| Viral process | BP | 942 | 344 | 25.8 |
| Regulation of response to stimulus | BP | 3503 | 1000 | 25.6 |
| Multi-organism cellular process | BP | 947 | 344 | 25.4 |
| Symbiosis, encompassing mutualism through parasitism | BP | 973 | 351 | 25.3 |
| Interspecies interaction between organisms | BP | 973 | 351 | 25.3 |
| Response to virus | BP | 304 | 147 | 24.3 |
| Binding | MF | 13,201 | 3127 | 24.1 |
| pRNA | ||||
| Term | Ontology | N genes | N up/down | –log |
| Response to virus | BP | 304 | 70 | 23.5 |
| Defense response to virus | BP | 226 | 59 | 22.9 |
| Defense response | BP | 1461 | 169 | 18.8 |
| Defense response to other organisms | BP | 475 | 80 | 17.8 |
| Immune response | BP | 1509 | 170 | 17.7 |
| Response to biotic stimulus | BP | 839 | 114 | 17.6 |
| Immune system process | BP | 2348 | 232 | 17.3 |
| Response to stress | BP | 3557 | 315 | 16.9 |
| Response to external biotic stimulus | BP | 801 | 109 | 16.9 |
| Response to other organisms | BP | 801 | 109 | 16.9 |
| Hiltonol + pRNA | ||||
| Term | Ontology | N genes | N up/down | –log |
| Immune system process | BP | 2348 | 694 | 30.4 |
| Response to stress | BP | 3557 | 975 | 30.2 |
| Cytoplasm | CC | 10,548 | 2428 | 27.3 |
| Regulation of response to stimulus | BP | 3503 | 943 | 26.0 |
| Cytoplasmic part | CC | 7919 | 1890 | 25.8 |
| Regulation of immune system process | BP | 1302 | 416 | 24.6 |
| Cell surface receptor signaling pathway | BP | 2595 | 727 | 24.3 |
| Intracellular signal transduction | BP | 2588 | 719 | 22.9 |
| Immune response | BP | 1509 | 461 | 22.7 |
| Response to cytokine | BP | 782 | 275 | 22.7 |
| Changes in clusters of transcripts were detected using a GO term analysis. The
top 10 most differential expressed gene clusters upon each stimulus and the
combination was selected. The description for each gene cluster is shown in the
columns. The GO contains three sub-ontologies: molecular function (MF), cellular
component (CC), and biological process (BP). N genes stand for the total
amount of genes contained in a cluster and N up/down denotes the number of genes
that changed their expression values upon the stimulus. The table is sorted on
the –log | ||||
Importantly, the top 100 affected GO terms contained no pathways related to cell damage or apoptosis (Supplementary Tables 1,2,3 show different pathways affected after stimulation).
Chemokines, chemokine receptors, and interferons are among the most relevant
groups of genes for the T cell activating function of DCs. Therefore, we
investigated these gene groups directly (Fig. 6). Hierarchical clustering of
samples based on a selected set of these genes mirrored the results of our
earlier PCoA analysis: unstimulated conditions and stimulated conditions formed
clear separate clusters. This confirms the clear effect of the stimuli on the
phenotype and the function of the CD141
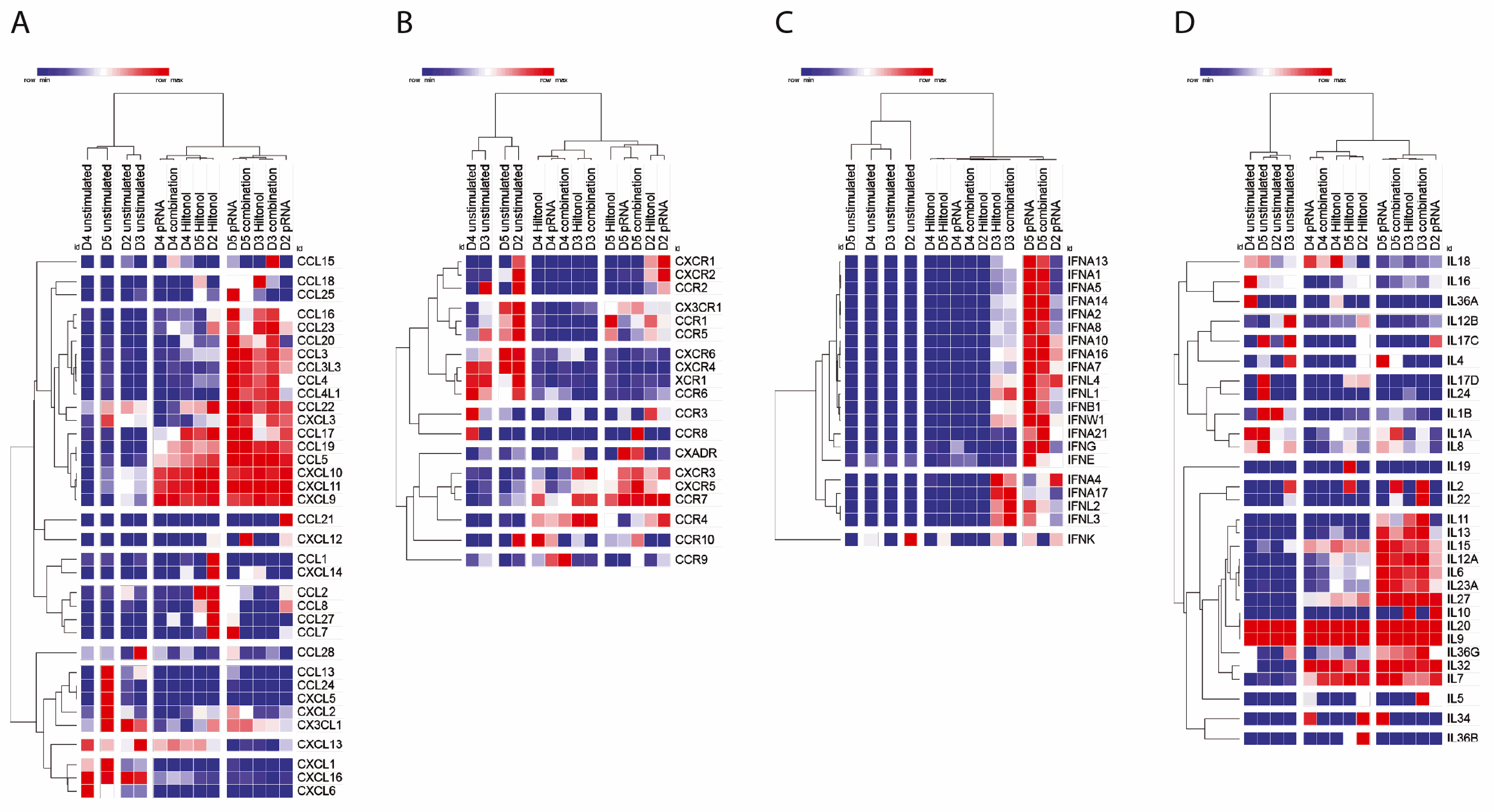 Fig. 6.
Fig. 6.Relative expression of genes belonging to chemokines, chemokine receptors, and cytokines (type I IFNs). Genes that are related to (A) chemokines, (B) chemokine receptor, (C) type I IFN, and (D) other cytokines are clustered regarding their similar expression pattern. Each column represents one condition and each row represents one gene.
Comparing the chemokine receptor genes, again unstimulated samples cluster
together whereas the samples of the different stimuli form a second, mixed
cluster (Fig. 6B). This analysis showed a cluster of 3 chemokine receptors (CCR6,
CXCR4, and CXCR6) that were all downregulated upon stimulation. Another cluster
(lower part of Fig. 6B) contained genes that were upregulated upon stimulation,
including the gene for the migration-related chemokine receptor CCR7, but also
for CXCR3, CXCR5, and CCR4. Type I/III interferons were mainly present upon pRNA
stimulation (Fig. 6C). In conclusion, a comparison of the cytokine genes revealed
one big cluster containing IL-27, IL-36
Finally, we were interested in the transcript levels of established CD141
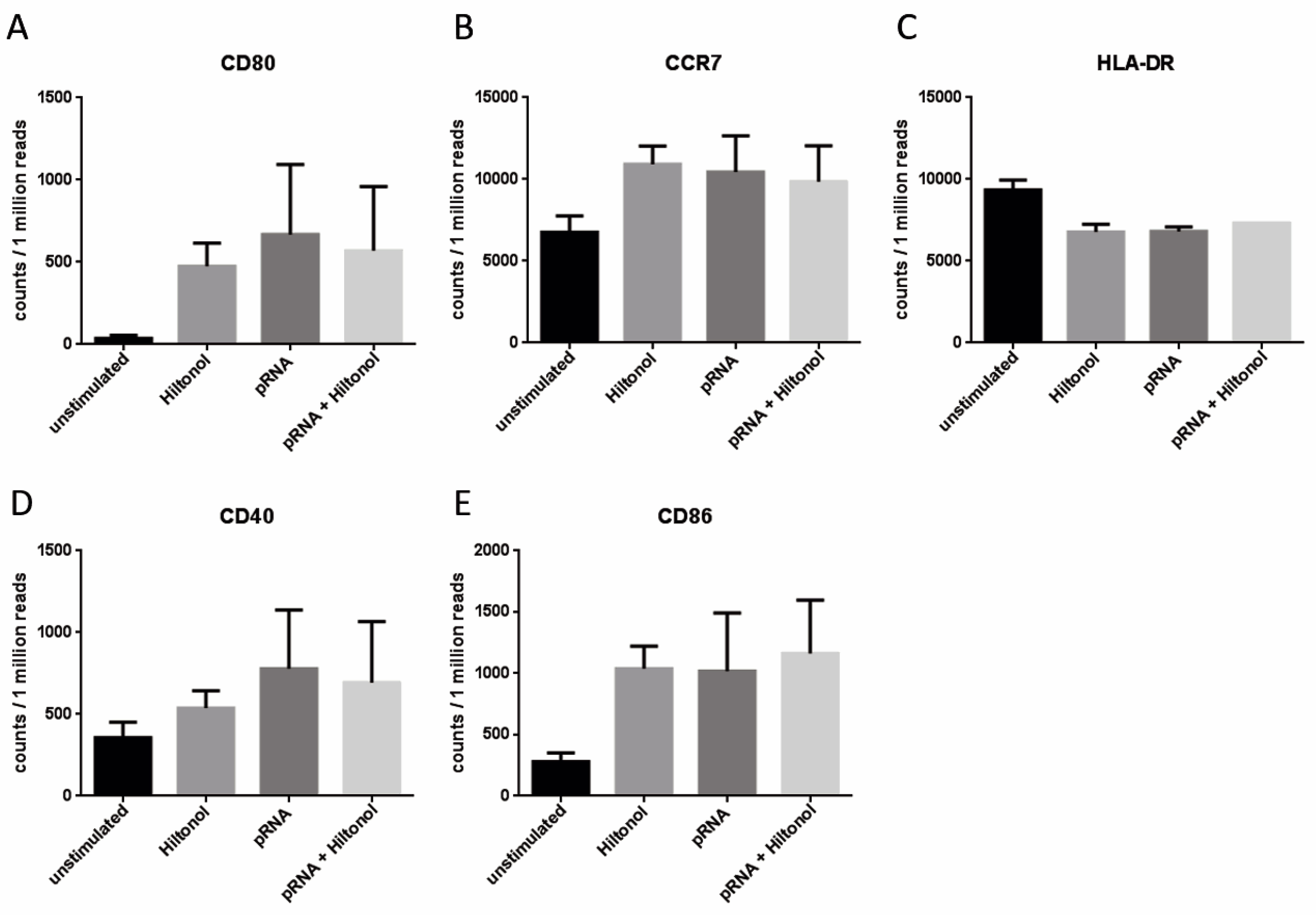 Fig. 7.
Fig. 7.Both stimuli upregulate maturation markers on CD141
To investigate the similarities and differences of CD141
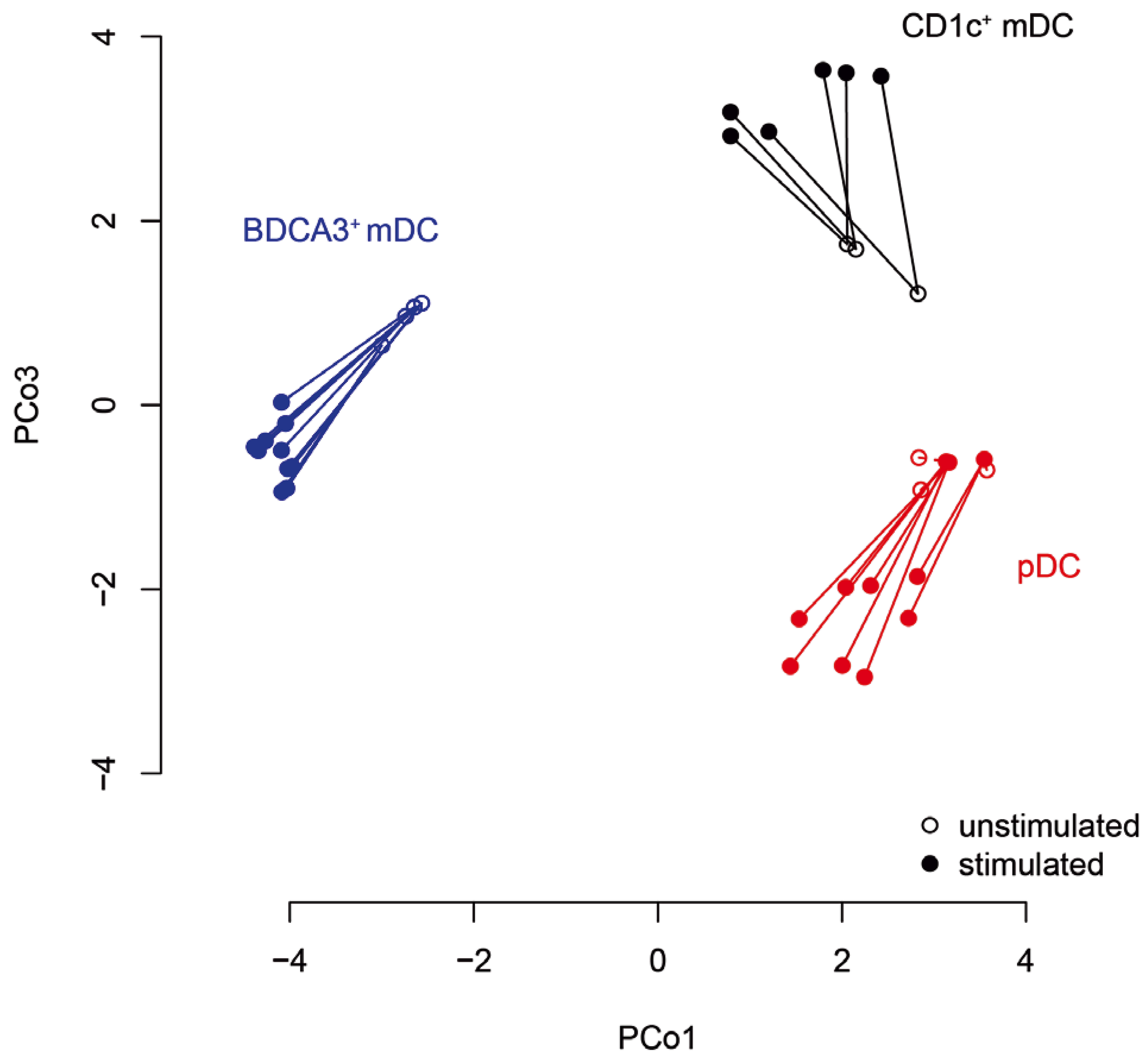 Fig. 8.
Fig. 8.Comparing CD141
We compared two clinical-grade TLR stimuli and their effect on CD141
We used RNA-seq to obtain unbiased and global overviews of the transcriptome.
Previously, we studied the effect of pRNA and other adjuvants on CD1c
Although pRNA binds most likely TLR7/8 and Hiltonol is a ligand for TLR3, their
general effects on CD141
A minor difference between pRNA and Hiltonol could be observed by comparing the upregulation of type I/III IFN related genes (Fig. 4), which indicated that the combination and predominantly more samples with a pRNA stimulation upregulated type I/III IFN genes. However, these results do not reach statistical significance in the whole-transcriptome analysis. Further functional assays measuring released interferons on the protein level should be performed to investigate this feature.
Since the cost of RNA-seq has decreased, the technique has become commercially available and the required amount of RNA has diminished, it has become a standard technique for measuring gene expression, including for some clinical applications [30, 31, 32, 33]. A major advantage of RNA-seq is its ability to quantify and detect novel transcripts, unlike microarray techniques [34, 35]; further, it directly yields RNA sequences, which simplifies data analysis. The usefulness of RNA-seq for immunotherapy was recently demonstrated by the use of this technique to detect mutations or to search for neo-antigens [36, 37, 38]. Another study performed by Van Allen et al. [39] investigated transcriptomic changes of melanoma cells upon treatment with ipilimumab, a CTLA-4 inhibitor. Such studies point to the potential for RNA-seq to become an integral component of personalized medicine.
In summary, we provide extensive and unbiased genome-wide data regarding how
CD141
We show different clinical-grade stimuli for an immune system cell called
CD141
Our analysis shows that microarrays and RNA-seq lead to similar conclusions
about the activation state of CD141
In conclusion, all these results collectively suggest that both stimuli
(Hiltonol and protamine RNA) are potent and safe as clinical-grade adjuvants for
enhancing tumoral immune responses. Currently, numerous clinical trials are being
carried out using new subtypes of dendritic cells that enhance immune responses.
In our case, CD141
mDC, myeloid dendritic cells; poly I:C, polyinosinic:polycytidylic acid; GO, genetic ontology; TLR, toll-like receptor; IFN, interferon; PCoA, Principle Coordinate Analysis.
BA and CA conceived the study. CA designed the experiments. BA and CA produced and analyzed the data, and contributed to data interpretation. BA and CA wrote the manuscript. All authors read and approved the final manuscript.
Ethical Approval and Consent to participate; Human samples material provided by healthy volunteers (Sanquin, Nijmegen, the Netherlands and Pamplona, Spain) after obtaining written informed consent per the Declaration of Helsinki and according to institutional guidelines. All experiments were conducted following government laws and under approval by the Ethics committee (Ethical approval number: 2018.012; study name “PROSPECTIVE TRANSLATIONAL STUDY OF DETERMINATION OF PREDICTIVE FACTORS OF EFFICACY AND TOXICITY IN PATIENTS WITH CANCER”). Informed consent statement was obtained from all subjects involved in the study.
The authors thank Mario Alfaro and Iñigo Alfaro for his help with supporting time.
This research received no external funding.
The authors declare no conflict of interest. Given his/her role as Editor, Carlos Alfaro had no involvement in the peer-review of this article and has no access to information regarding its peer-review. Full responsibility for the editorial process for this article was delegated to any person of Editorial Board.
Supplementary material associated with this article can be found, in the online version, at https://www.imrpress.com/journal/FBE/14/1/10.31083/j.fbe1401002.