


1 Department of Neuroscience, H. Lundbeck A/S, Ottiliavej 9, DK-2500 Valby, Denmark
Abstract
Background: Tauopathies such as Alzheimer’s disease (AD) are
characterized by abnormal hyperphosphorylation of the microtubule-associated
protein tau (MAPT) aggregating into neurofibrillary tangles (NFTs). O-linked
Keywords
- Thiamet G
- rTg4510
- OGA
- hyperactivity
- tau pathology
- tauopathy
Filamentous aggregates and intracellular deposition of the microtubule-associated protein tau (MAPT) are neuropathological hallmarks in Alzheimer’s disease (AD) and other tauopathies [1]. Tau aggregates deposited as fibrils in neurons, known as neurofibrillary tangles (NFTs), are detergent-insoluble and consist of hyperphosphorylated tau. Tau is considered hyperphosphorylated when phosphorylated at seven or more sites, while under normal conditions tau is phosphorylated at two to three sites [2]. Dynamic tau phosphorylation and dephosphorylation are critical for microtubule stability and axonal transport [3]. Hyperphosphorylated tau does not perform these or other physiological functions [4]. Disease progression and degree of cognitive impairment in AD correlates with the spatiotemporal distribution of NFTs [5], suggesting that pathological tau is involved in neurodegeneration [6]. Point mutations in the MAPT gene are associated with familial tauopathies termed MAPT-related frontotemporal dementia (MAPT-FTD) [7]. These mutations can enhance tau oligomerization and aggregation [8, 9], indicating that pathological tau can trigger neurodegeneration in animal models harboring MAPT-FTD mutant transgenes, such as the tau P301L mutation [10, 11]. Findings from tau transgenic mouse models suggest that oligomeric tau species, not NFTs, are responsible for neuronal dysfunction and neurodegeneration [11, 12, 13]. Oligomerization and aggregation of tau involves hyperphosphorylation, altered conformation, and assembly into fibrils and NFTs [14, 15]. In addition to phosphorylation, tau can undergo other post-translational modifications that are also believed to be involved in tau oligomerization and aggregation [16].
The addition of N-acetylglucosamine (GlcNAc) via an O-glycosidic linkage (O-GlcNAc) to the hydroxyl side chains of serine and threonine residues is a physiological post-translational modification of proteins [17]. O-GlcNAcylation is a ubiquitous and dynamic process which is regulated by O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) catalyzing the addition and removal of GlcNAc to and from target proteins [18]. O-GlcNAcylation depends on the availability of uridine diphosphate glucose (UDP)-GlcNAc, which is generated in the hexosamine biosynthetic pathway from glucose [19]. Consequently, O-GlcNAcylation of proteins is thought to be regulated by glucose metabolism. Impairment in glucose metabolism has been reported as an early feature of AD [20], suggesting that dysregulation of O-GlcNAcylation could occur early in the disease. Further, protein O-GlcNAcylation is decreased in AD brains and linked to tau hyperphosphorylation [21]. O-GlcNAcylation can occur on residues of proteins that are known to be phosphorylated. Tau is believed to be O-GlcNAcylated on or close to residues hyperphosphorylated in AD brains [22, 23, 24, 25]. The loss of O-GlcNAc from these residues could be prevented by inhibiting the removal enzyme OGA and thereby preventing the accumulation of hyperphosphorylated tau. Accordingly, an acute increase in O-GlcNAcylation with a potent OGA inhibitor Thiamet G reduced tau phosphorylation in rodent brain at several tau residues [26, 27]. On the other hand, chronic increase in O-GlcNAcylation by Thiamet G decreased tau aggregation without changing normal tau phosphorylation, leading to protective effects in different tau transgenic mouse models [28, 29, 30, 31].
Here, we report that acute and chronic treatment with the potent OGA inhibitor Thiamet G [26] resulted in sufficient brain exposure and robust increase in O-GlcNAcylation in rTg4510 mice. The rTg4510 mouse model express inducible human four-repeat mutant P301L tau driven by the calcium/calmodulin dependent kinase II promoter under control of the tetracycline transactivator, mainly in the forebrain, and display an age-dependent progression of tau pathology including hyperphosphorylated tau species, NFTs, neuronal loss, and behavioral abnormalities [11, 32]. In this mouse model, tau pathology is characterized histologically by pre-tangle tau and NFTs and biochemically by oligomeric and fibrillar hyperphosphorylated tau species separated by differential centrifugation in soluble and sarkosyl-insoluble fractions [11, 13]. Increase in O-GlcNAcylation levels by chronic Thiamet G treatment reduced soluble and insoluble hyperphosphorylated tau species in rTg4510 mice. We and others have shown that rTg4510 mice exhibit a non-mnemonic behavioral defect [33, 34] characterized by hyperactivity, which is dependent on transgene P301L tau expression and correlates with the progression of tau pathology [34]. Concomitant with the reduction of hyperphosphorylated tau species the number of hyperactive rTg4510 mice was significantly reduced by chronic Thiamet G treatment. We demonstrate here that a sustained increase in brain O-GlcNAcylation levels can decrease hyperphosphorylated tau species and rescue behavioral impairments in the rTg4510 mouse tauopathy model.
All reagents and antibodies were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. Thiamet G was synthesized at the Department of Medicinal Chemistry, H. Lundbeck A/S, Valby, Denmark according to published methods [26]. Thiamet G at 10 µM was tested in an in vitro diversity profile panel composed of 71 binding and 27 enzyme assays broadly defined with roughly an equal number of selective, central, and peripheral therapeutically relevant targets (Eurofins Cerep, Celle-Lévescault, France).
Murine cortical neurons were isolated from day E14–16 non-transgenic or rTg4510 embryos and cultured in Neurobasal medium supplemented with 2% B-27 supplement with antioxidants, 0.5 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (all solutions from Invitrogen, Waltham, MA, USA) as described [35]. Embryos were genotyped using primer pairs as described [11]. Cortical neurons were treated with 1 µM cytosine arabinoside at days in vitro (DIV) 4 to halt proliferating cells and incubated with Thiamet G dissolved in phosphate buffered saline (PBS) at DIV 7. Culture medium was supplemented with 20 mM glucose before addition of Thiamet G.
For detection of cell viability following Thiamet G addition, the percentage of viable cells in the neuronal cultures was quantified by their capacity to reduce 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) following incubation with 0.5 mg/mL MTT for 60 min.
Cells were lysed in RIPA-buffer (50 mM Tris HCl, 1% NP-40/IGEPAL CA630, 0.25%
Na-deoxycholate, 150 mM NaCl and 1 mM egtazic acid (EGTA)) supplemented with a 1% protease and
phosphatase inhibitors mix (Roche, Basel, Switzerland) and 200 nM hexokinase
inhibitor PUGNAc [O-(2-Acetamido-2-deoxy-D-glucopyranosylidenamino) N-phenylcarbamate] (Tocris, Bristol, UK). Following 30 min of lysis on ice, cell
lysis was spun down (15,000
The generation of rTg4510 mice was described previously [11]. In our colony, the
tau transgene responder was expressed in the FVB/N mouse strain (Taconic,
Silkeborg, Denmark), and the tTA activator system was maintained in the 129S6
mouse strain (Taconic, Silkeborg, Denmark). Their F1 progeny carried responder
and activator transgenes in the rTg4510 mice along with non-transgenic (FVB/129)
littermates. Only F1 mice were used in this study. All mice were bred in
specific-pathogen-free conditions at Taconic (Silkeborg, Denmark) and genotyped
from tail DNA using the primer pairs as described [11]. Mice were group-housed,
where water and food (Brogaarden, Lynge, Denmark) were available ad
libitum along with enrichment materials. The light/dark cycle was 12 h; room
temperature was 21
For single dosing experiments, non-transgenic mice were dosed subcutaneously 75 mg/kg Thiamet G and rTg4510 and non-transgenic littermate mice were dosed per os 200 mg/kg Thiamet G or vehicle (10% HP-beta-CD, pH 4). Mice were euthanized 0.5, 1, 2, 4, 6, or 24 hours (h) following dosing and blood and brain tissue samples were collected. For subchronic and chronic dosing experiments, mice were dosed by drinking water for 1 week or 18 weeks, respectively, where 3.75 mg/mL Thiamet G was included in the drinking water. Water consumption was 5 mL/day per mouse and the average weight of the rTg4510 mice was 30 grams. Accordingly, a daily Thiamet G dose of approximately 600 mg/kg was given. Thiamet G was found to be stable in drinking water at room temperature for 1 week and the water supply was changed weekly. To suppress transgene expression, doxycycline (200 mg/kg) enriched chow (Bio-Serv, Flemington, NJ, USA) was administered to 6 week old rTg4510 mice in the control group ad libitum for 18 weeks during the study.
Mice were euthanized by cervical dislocation. Immediately following
decapitation, blood was collected in ethylenediaminetetraacetic acid (EDTA)-coated tubes. Samples were kept on ice
before being centrifuged at 20,000
Forebrain homogenates were separated by differential centrifugation into
Tris-buffered saline (TBS)-extractable (S1), S1 precipitate (S1p), and
sarkosyl-insoluble pellet (P3) fractions as established previously [13] for tau
biochemistry performed as described [36]. Briefly, forebrain homogenates were
separated by centrifugation at 27,000
Western blotting, analysis and quantitation of immunoreactivity was performed as described previously [36]. Total protein O-GlcNAcylation was detected using mouse monoclonal CTD110.6 [37] or RL2 (ThermoFisher Scientific, Waltham, MA, USA) antibody. Total tau was detected using mouse monoclonal Tau5 (Abcam, Cambridge, UK) and rabbit polyclonal tau E1 antibodies (raised against amino acids 19–33 (GLGDRKDQGGYTMHQ) of the longest isoform of human tau 2N4R) [38]. Tau phosphorylation was detected using rabbit polyclonal antibodies pS199/202 (Invitrogen, Waltham, MA, USA), pS400 (Invitrogen, Waltham, MA, USA), and mouse monoclonal pS396 antibody D1.2 [35]. Tau5 is a pan tau antibody and recognizes human and murine tau at non-phosphorylated and phosphorylated epitopes. The E1 antibody recognizes exclusively human tau at non-phosphorylated and phosphorylated epitopes, while pS199/202, pS396 and pS400 antibodies recognize the phosphorylated epitopes of murine and human tau. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization and detected with mouse monoclonal (Meridian Life Science, Memphis, TN, USA) or rabbit polyclonal (Abcam, Cambridge, UK) antibody.
Forebrain homogenates were analyzed using a Crosslink immunoprecipitation kit from Pierce (ThermoFisher Scientific, Waltham, MA, USA) in accordance with the manufacturer’s protocols. A set of 2 samples containing homogenates from a vehicle and a Thiamet G treated rTg4510 mouse were processed in parallel. The A/G agarose resin was crosslinked to 10 µg mouse CTD110.6 and 300 µg pre-cleared brain lysate was incubated with the resin-coupled O-GlcNAc antibody. Samples were eluted, collected by centrifugation, dissolved in SDS-sample buffer containing DTT (100 mM), and the immunoprecipitated O-GlcNAcylated proteins were probed with the E1 tau and O-GlcNAc RL2 antibodies by western blot.
Concentrations of Thiamet G were determined in plasma and brain homogenate using
ultra performance liquid chromatography (UPLC) followed by tandem mass
spectrometry (MS/MS) detection. Brain samples were homogenized by adding four
times the volume of dimethyl sulfoxide (DMSO):2-propanol:H20 (20:30:50). The mobile phases A and B,
which consisted of water/acentonitrile (50:50) containing 10 nM NH
The free fraction of Thiamet G in plasma and brain homogenates was measured by
equilibrium dialysis and liquid chromatography (LC), followed by tandem mass
spectrometry (MS/MS) analysis. Thiamet G dissolved in DMSO and diluted in 50%
acetonitrile (ACN)/H
A locomotor activity assay was performed in activity cages equipped with
infrared light sources and photocells as described previously [34]. The frequency
with which the mouse crossed the photo beams was used as a measure of activity in
a 3-hour locomotor activity test. Hyperactivity was defined as total activity
counts in the non-transgenic littermate population higher than the mean + 3
All data were analyzed with GraphPad Prism version 9 (GraphPad Software, San
Diego, CA, USA). Paired samples t tests using a 95% confidence interval
and a two-tailed p value was used to analyze the immunoprecipitation
(IP) data sets. All other data comparing two groups were analyzed using unpaired
t tests, with a Welch’s correction factor, 95% confidence interval, and
two-tailed p value. The difference in the number of hyperactive mice
between the groups was analyzed with a chi-square test, with p values of
Thiamet G is reported as selective inhibitor of the eukaryotic OGA [26]. We
confirmed that 10 µM Thiamet G did not show binding or activity on
98 other central and peripheral therapeutically relevant targets in a Cerep
diversity profile screen (data not shown). We investigated consequences of OGA
inhibition by Thiamet G in primary cortical neurons isolated from rTg4510 mouse
embryos over-expressing human 0N4R P301L tau. Overexpression of the transgene tau
did not lead to tau hyperphosphorylation, aggregation, or toxicity in rTg4510
primary neurons. Neuronal functions and viability were unchanged compared to
cortical cultures isolated from non-transgenic littermates (data not shown).
Neurons were treated with Thiamet G in concentrations ranging from 0.0001 to 100
µM for 6 and 24 h. No cell toxicity was observed in neurons
following Thiamet G treatment, measured by a MTT reduction test (data not shown).
A substantial increase in global O-GlcNAc levels of numerous proteins at the
range from 30 kDa to 220 kDa or greater was detected 6 h after Thiamet G
treatment by western blotting using the CTD110.6 antibody (Fig. 1A). We observed
approximately 100% increased overall O-GlcNAc signal in neurons treated with 100
nM Thiamet G compared to buffer control (Fig. 1B). By densitometry quantification
and normalization to GAPDH signal, the half-maximal effective concentration
(EC
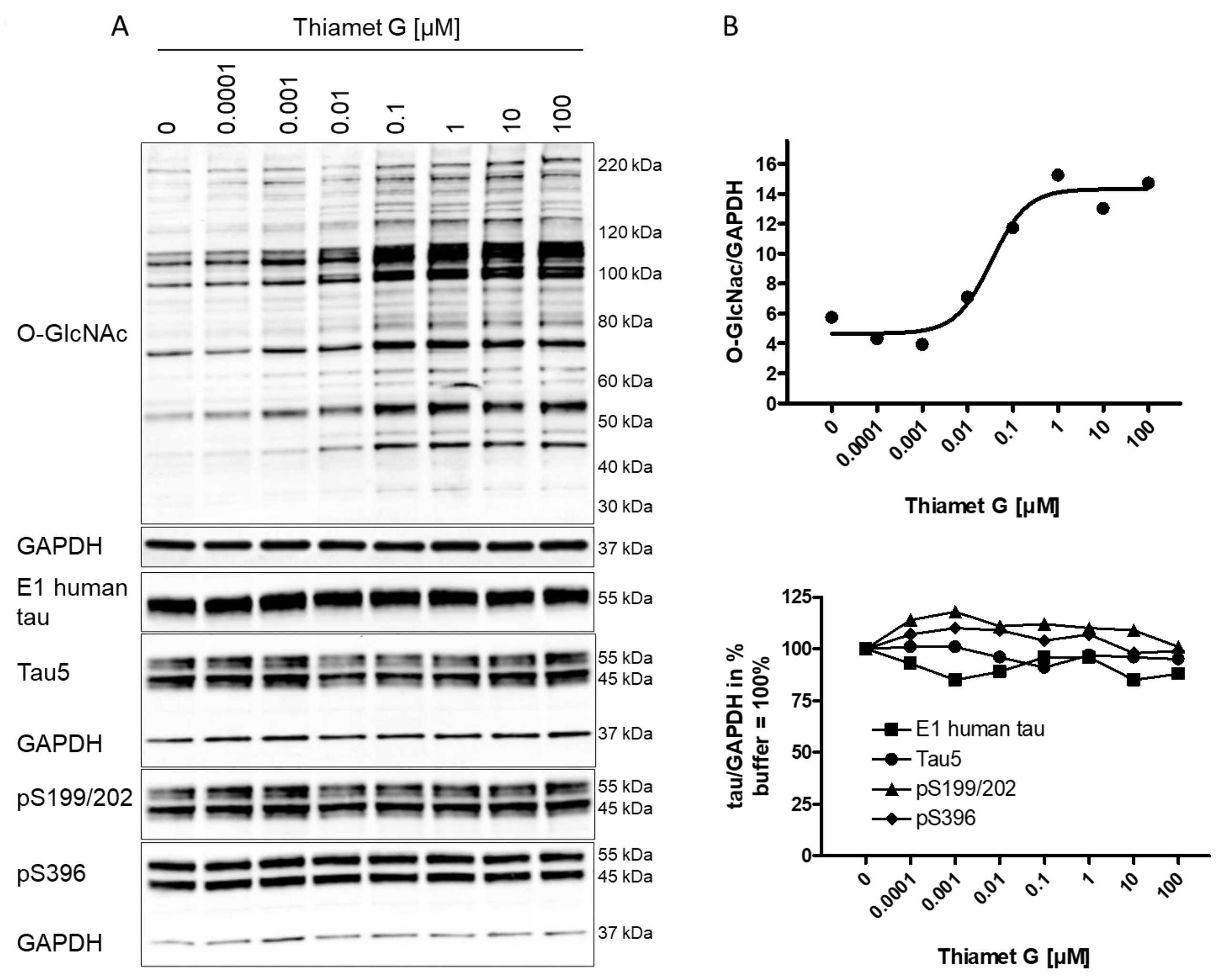
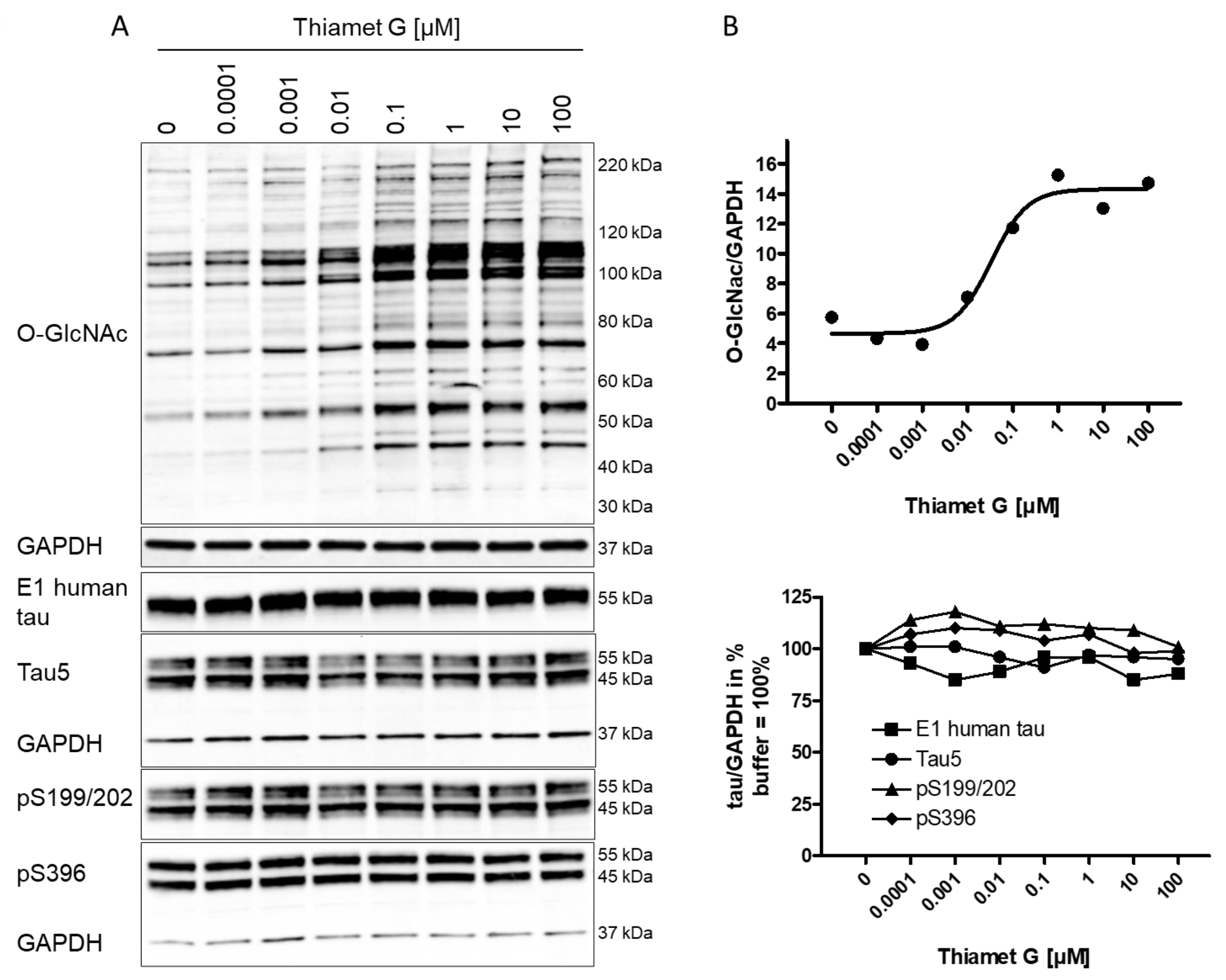 Fig. 1.
Fig. 1.Concentration-dependent increase in protein O-GlcNAc levels in
neurons from rTg4510 mice treated with Thiamet G. Cortical neurons isolated from
rTg4510 mouse embryos were incubated at days in vitro
(DIV) 7 with Thiamet G at concentrations as indicated. After 6
hours, neurons were lysed and analyzed by western blot for global protein
O-GlcNAcylation (O-GlcNAc), human tau (E1), total tau (Tau5), and phosphorylated
tau at S199/202 (pS199/202) and S396 (pS396) epitopes. For normalization, the
O-GlcNAC blot was re-probed with GAPDH. The E1 blot was re-probed with pS396 and
GAPDH, and the pS199/202 blot was re-probed with Tau5 and GAPDH. Phosphorylated
and non-phosphorylated human 0N4R transgene tau was displayed at 55 kDa with the
E1 antibody. Murine and human 0N4R transgene tau species are displayed at 45 and
55 kDa, respectively, with the Tau5 antibody. (A) Representative western blots
from n = 3 experiments. (B) Quantification of western blots is shown as graphs in
absolute units O-GlcNAc/GAPDH and in % tau/GAPDH with buffer control (0) set to
100%. Individual samples are presented as filled shapes. Thiamet G EC
Next, the impact of OGA inhibition by Thiamet G on the phosphorylation of mouse and human transgene tau was assessed in rTg4510 neuronal cultures. Normally phosphorylated human 0N4R P301L tau runs as 55 kDa band on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Hyperphosphorylated 0N4R P301L tau mobility is shifted and runs as 64 kDa band on SDS-PAGE. We detected transgene human 0N4R P301L tau displayed at 55 kDa with the E1 antibody specific for human tau by western blotting (Fig. 1A). Additionally, neurons expressed endogenous murine tau isoforms which were displayed at 45 and 55 kDa on SDS-PAGE and detected together with the human transgene tau with the phosphorylation-independent Tau5 antibody. Human and murine tau were phosphorylated at S199/202 and S396 epitopes as detected with the pS199/202 and pS396 tau antibodies by western blotting. When treated with Thiamet G, we observed a marked increase in global O-GlcNAc levels in the 0.1–100 µM concentration range (Fig. 1A), and no changes in the normal total human and murine tau levels detected with the E1 and Tau5 antibodies (Fig. 1B). No changes of tau phosphorylation at the S199/202 and S396 epitopes of murine and human tau were observed. Using densitometry quantification and normalization to GAPDH signaling, we confirmed that Thiamet G treatment did not lead to significant changes in phosphorylation at S199/202 and S396 epitopes of human or mouse tau in rTg4510 primary neurons (Fig. 1B).
Initially, we tested whether Thiamet G can cross the blood brain barrier after a
single subcutaneous dose of 75 mg/kg and calculated that the half-life of Thiamet
G was approximately 1.5 and 3.5 h in plasma and brain, respectively (Table 1).
Next, we investigated oral administration and dosed 24 week old rTg4510 and
non-transgenic littermate mice with a single dose of 200 mg/kg acutely for 6 or
24 h or subchronically for 1 week with 600 mg/kg/day Thiamet G in drinking water.
At this age rTg4510 mice exhibited prominent tau pathology including NFTs [34, 36] and hyperphosphorylated tau (Supplementary Fig. 1). We analyzed
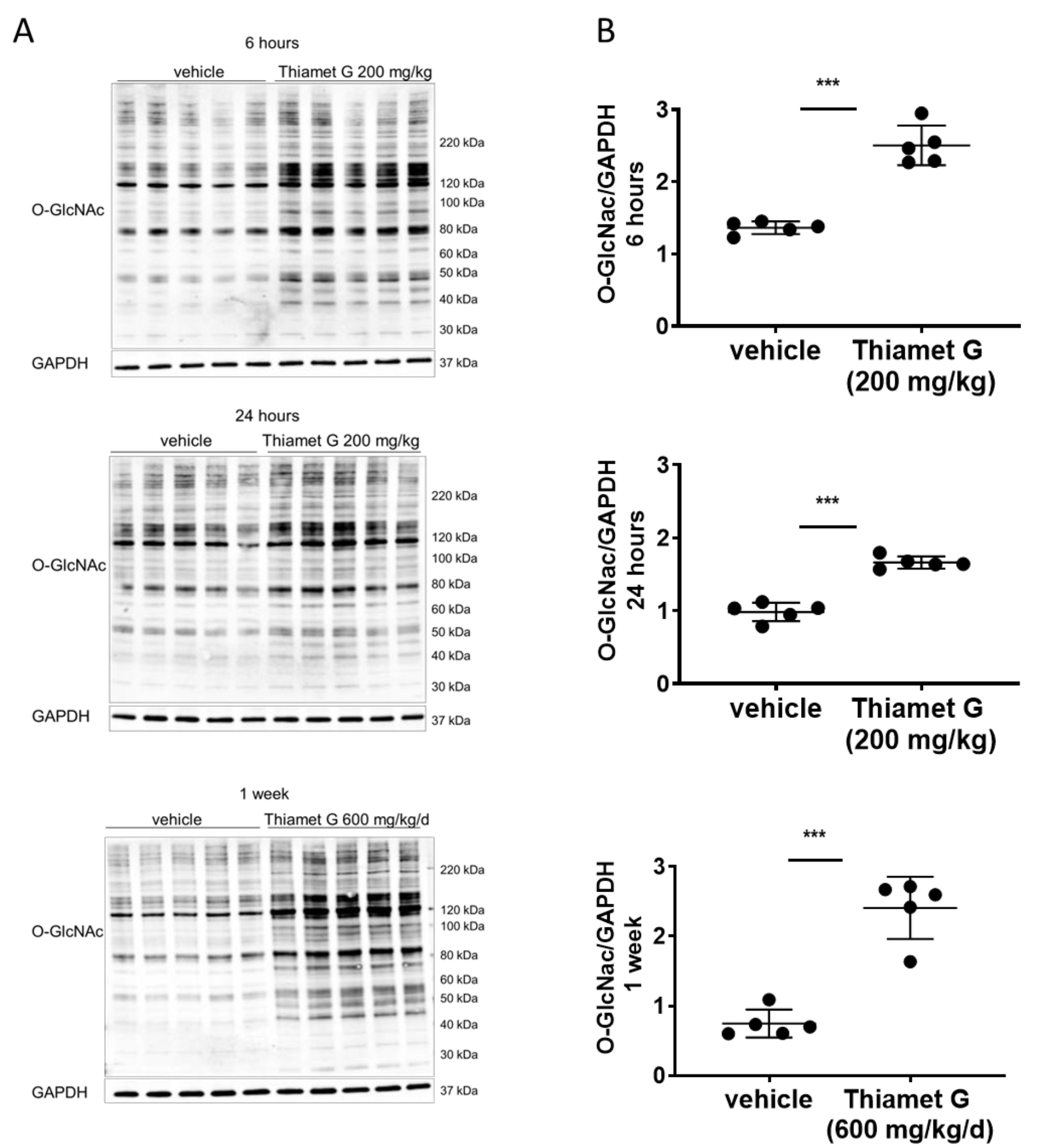
Thiamet G concentrations in plasma and brain tissue (Table 2), and total O-GlcNAc
levels in brain tissue (Fig. 2). Non-transgenic littermate (data not shown) and
rTg4510 (Fig. 2) mice displayed similar Thiamet G concentrations in plasma and
brain tissue, and a comparable increase in global O-GlcNAc levels. After 6 h, 24
h, and 1 week of dosing we measured 0.5, 0.2, and 1.1 µM Thiamet G,
respectively, in brain samples from rTg4510 mice (Table 2). The free fraction of
Thiamet G in the brain was determined to be 67%, leading to approximately 360,
120, and 720 nM unbound Thiamet G after acute 6 h, 24 h, and 1 week dosing in the
drinking water, respectively (Table 2). These concentrations were approximately
11-, 4-, and 22-fold higher than the measured EC
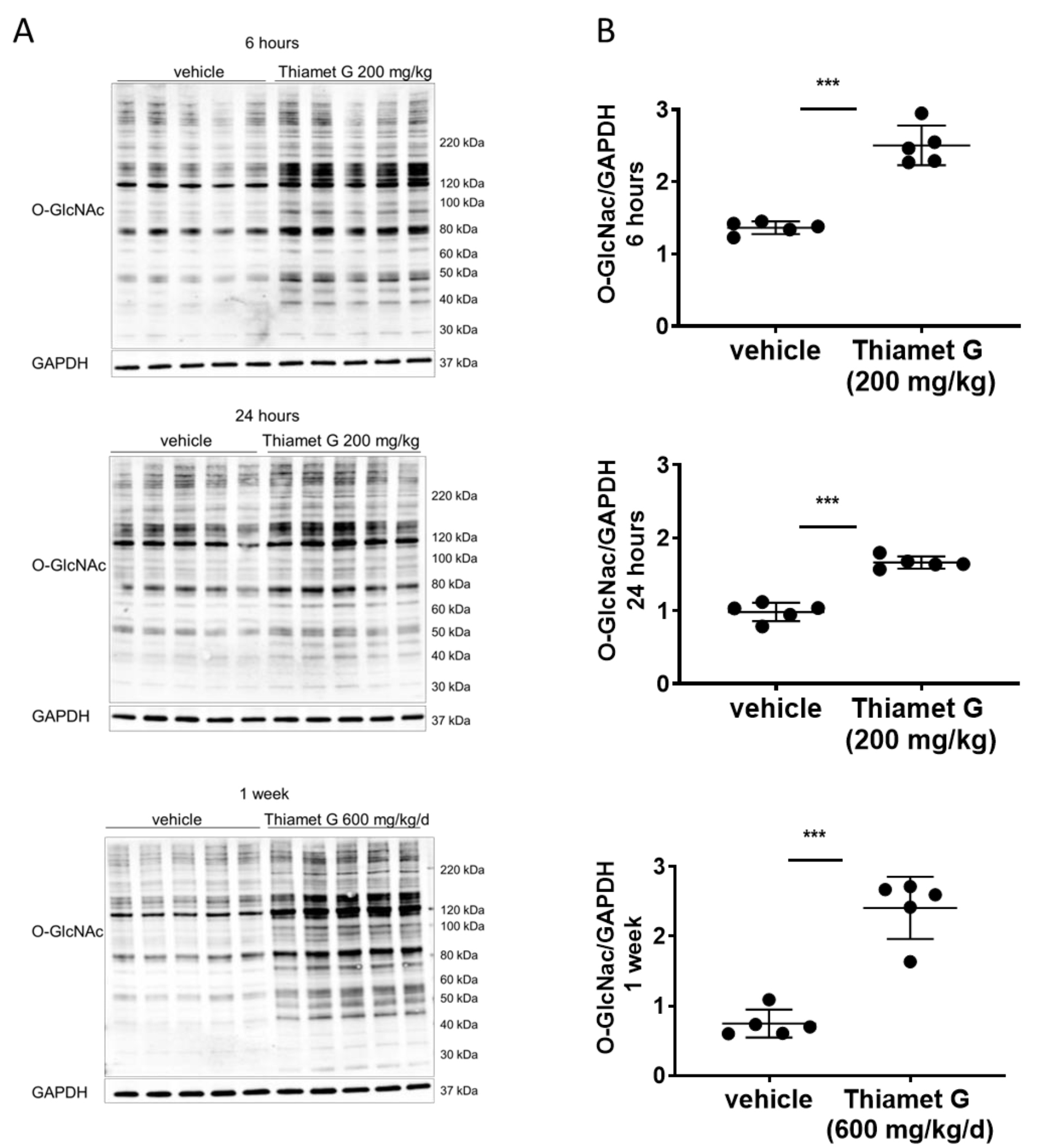 Fig. 2.
Fig. 2.Acute and subchronic Thiamet G dosing increases protein O-GlcNAc
levels. Soluble fraction of brain homogenates from 24 week old rTg4510 mice
treated either with vehicle (n = 5) or Thiamet G (n = 5) acutely per os
(200 mg/kg) for 6 or 24 hours, or subchronically for 1 week (600 mg/kg/day), were
analyzed by western blot for global protein O-GlcNAcylation (O-GlcNAc). For
normalization, blots were re-probed for GAPDH. (A) Representative western blots
from n = 3 experiments. (B) Quantification of western blots is shown as graphs in
absolute units O-GlcNAc/GAPDH, individual samples are presented as filled circles
and in lines as means
| Time [hours] | Plasma mean |
Brain mean |
Brain/plasma ratio |
| 0.5 | 17.7 |
3.6 |
0.2 |
| 1 | 13.7 |
4.5 |
0.3 |
| 2 | 4.4 |
3.2 |
0.7 |
| 4 | 1.0 |
1.7 |
1.8 |
| 6 | 0.5 |
1.0 |
2.1 |
| 24 | 0.05 |
0.11 |
2.3 |
Brain and plasma samples from 12 week old non-transgenic mice (n = 3) treated with Thiamet G subcutaneously (single dose 75 mg/kg) at time points indicated were analyzed for Thiamet G concentrations and brain/plasma ratio calculated. Half-life of Thiamet G in plasma and brain tissue was calculated as 1.5 and 3.5 hours, respectively. SEM, Standard error of the mean.
| Treatment time | Plasma mean |
Brain mean |
Brain/plasma ratio | Brain free fraction [µM] |
| 6 hours acute | 2 |
0.5 |
0.3 | 0.4 |
| 24 hours acute | 0.1 |
0.2 |
1 | 0.1 |
| 1 week subchronic | 6 |
1.1 |
0.18 | 0.7 |
| 18 weeks chronic | 9 |
1.1 |
0.12 | 0.7 |
Brain and plasma samples from 24 week old rTg4510 mice (n = 5) treated with Thiamet G per os (single dose 200 mg/kg) for 6 and 24 hours or in the drinking water (600 mg/kg/day) for 1 week were analyzed for Thiamet G concentrations and brain/plasma ratio calculated. Brain and plasma samples from 6 week old rTg4510 mice (n = 5) treated with Thiamet G in the drinking water (600 mg/kg/day) for 18 weeks until 24 weeks of age were analyzed for Thiamet G concentrations and brain/plasma ratio calculated. Thiamet G unbound fraction in the brain was measured as 67% and brain free fractions calculated accordingly.
Next, we investigated whether tau phosphorylation and hyperphosphorylation were modulated by OGA inhibition with Thiamet G under the acute treatment regimen in aged rTg4510 mice. As previously characterized by others [11, 32] and us [34, 36], rTg4510 mice develop an age-dependent neuropathology displaying pre-tangle tau and NFTs corresponding to oligomeric and fibrillar hyperphosphorylated tau [13] at 24 weeks of age (Supplementary Fig. 1).
Tau pathology in rTg4510 mice, which overexpress human 0N4R P301L tau, can be analyzed biochemically by differential centrifugation and sarkosyl fractionation of brain homogenates (Supplementary Fig. 2). On SDS-PAGE, normally phosphorylated human 0N4R P301L tau is displayed as a 55 kDa band. Hyperphosphorylated 0N4R P301L tau’s mobility is shifted and displayed as 64 and 70 kDa bands. The majority of hyperphosphorylated tau species run at 64 kDa (Supplementary Fig. 1). Hyperphosphorylated 64 kDa and 70 kDa tau species isolated in the sarkosyl-insoluble pellet fraction (P3) consist of fibrillary tau aggregates, as characterized by immunoelectron microscopy [13], and are regarded as the biochemical equivalent of NFTs [11, 32]. Hyperphosphorylated 64 kDa tau species in the soluble fraction (S1) precipitate under high speed centrifugation into the precipitate fraction of S1 (S1p). These low speed soluble and high speed precipitable hyperphosphorylated 64 kDa tau species consist of tau oligomers, as characterized by immunoelectron microscopy, and are regarded as the biochemical equivalent of pre-tangle tau [13].
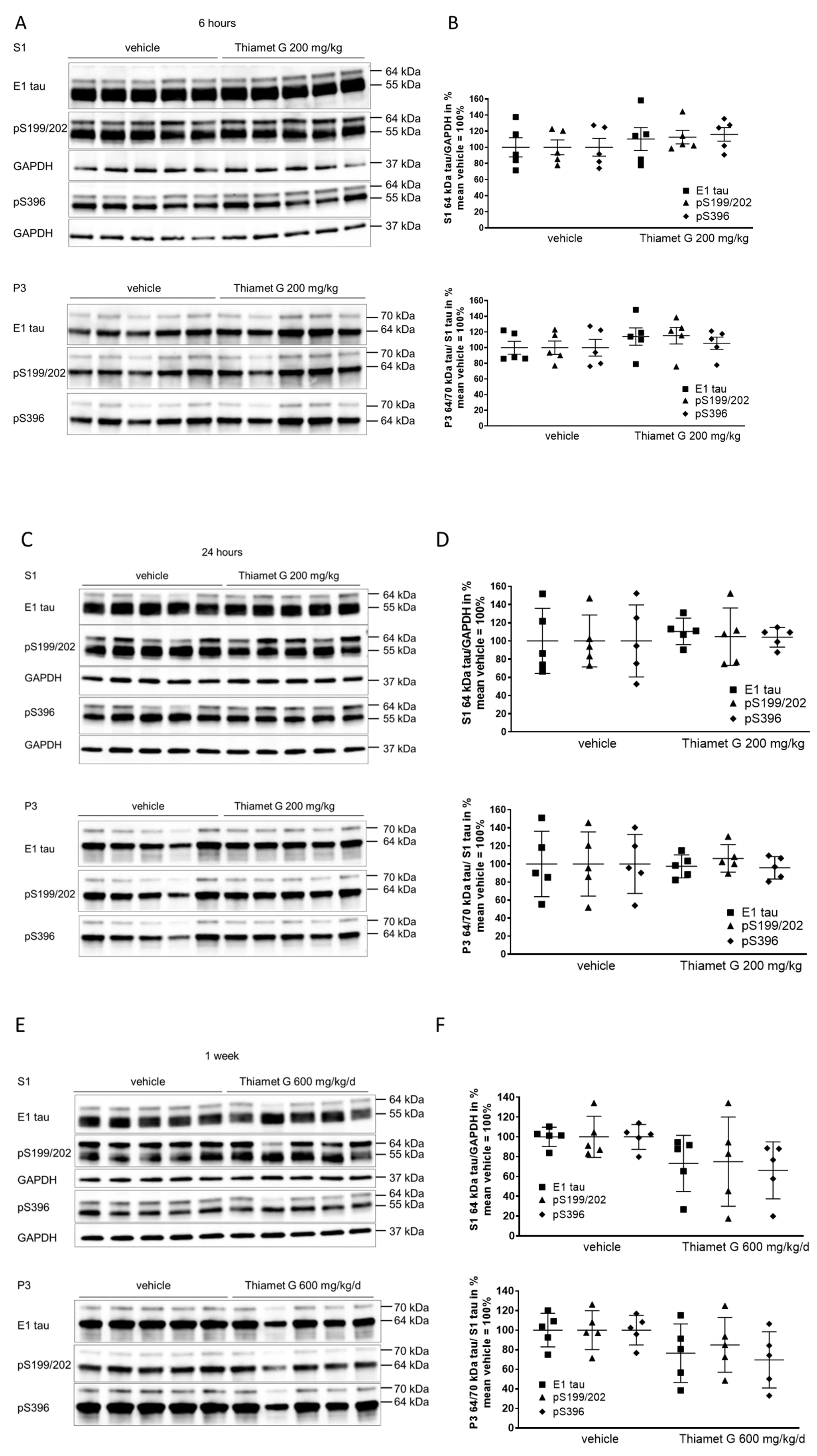
We isolated forebrain homogenates into soluble (S1) and sarkosyl-insoluble pellet (P3) fractions by differential centrifugation (Supplementary Fig. 2) as previously characterized [13]. To investigate the different tau species, we used the human tau specific E1 antibody and phospho tau antibodies that detect phosphorylation at the S199/202 and S396 epitopes. Normal human 0N4R P301L tau was displayed as 55 kDa in S1 detected with the E1 and phospho tau pS199/202 and pS396 antibodies (Fig. 3A,C). Hyperphosphorylated tau species were displayed as mobility shifted tau of 64 kDa in S1, and 64 kDa and 70 kDa in P3 (Fig. 3A–D). Levels of soluble 55 kDa human tau were not changed in brain samples 6 and 24 h after acute treatment with Thiamet G (S1, Fig. 3A,C). Levels of soluble hyperphosphorylated 64 kDa tau in S1 were not altered after acute Thiamet G treatment. Additionally, neither insoluble hyperphosphorylated 64 kDa or 70 kDa tau species in P3 were altered (Fig. 3A–D).
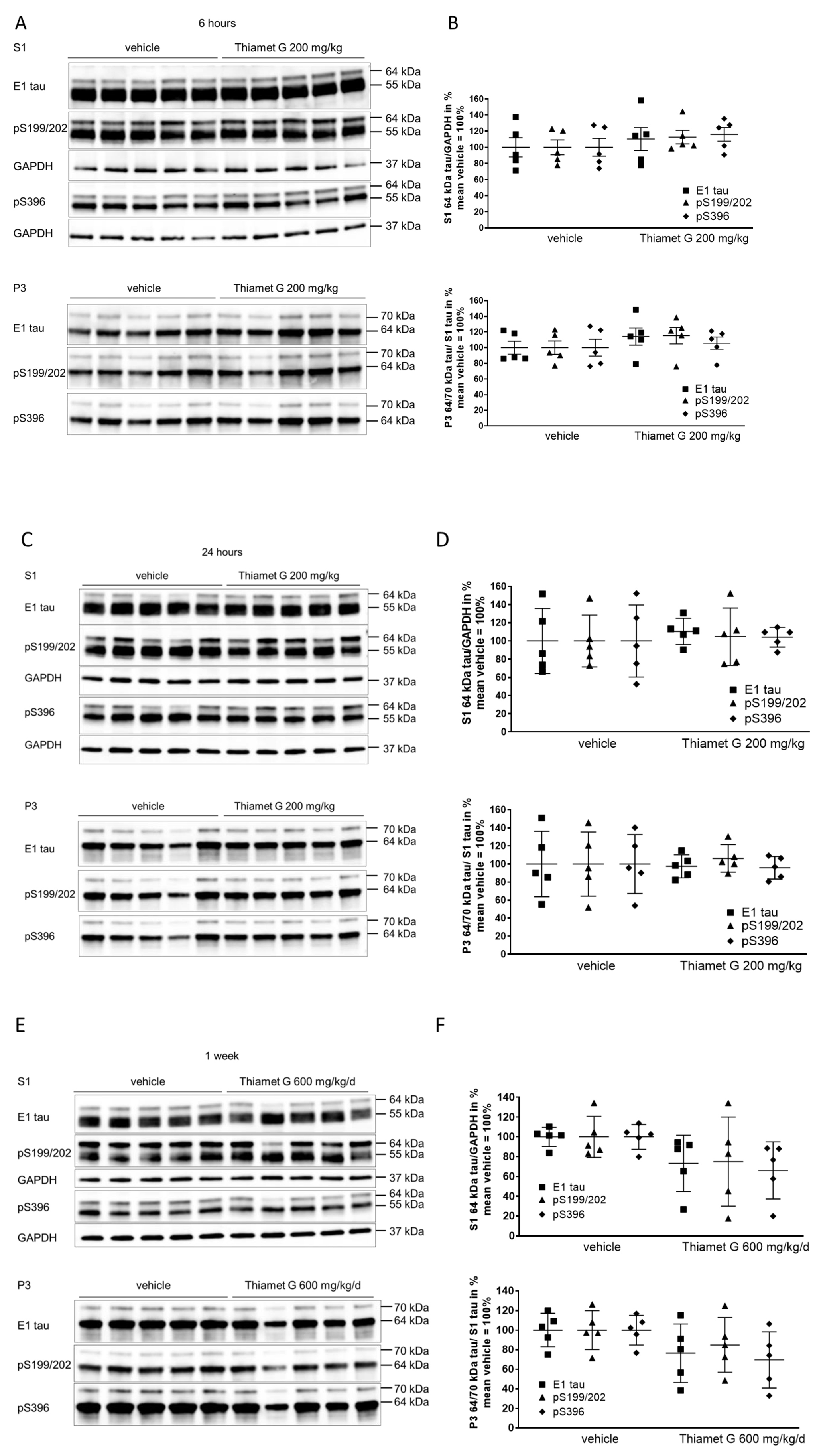 Fig. 3.
Fig. 3.Acute and subchronic Thiamet G dosing and effects on
hyperphosphorylated tau species. Brain homogenates from 24 week old rTg4510 mice
treated either with vehicle (n = 5) or Thiamet G (n = 5) acutely per os
(200 mg/kg) for 6 and 24 hours or for 1 week subchronically in the drinking water
(600 mg/kg/day) were fractioned into soluble (S1) and sarkosyl-insoluble (P3)
fractions and analyzed by western blot for total human tau (E1) and
phosphorylated tau at S199/202 (pS199/202) and S396 (pS396) epitopes. For normalization of S1 blots GAPDH was used. The E1 blot was
re-probed with pS396 and GAPDH and the pS199/202 blot was re-probed with GAPDH.
The P3 E1 blot was re-probed with pS396. Phosphorylated and non-phosphorylated
human 0N4R tau transgene is displayed at 55 kDa in S1 fraction,
hyperphosphorylated tau is mobility-shifted and displayed at 64 and 70 kDa in S1
and P3 fractions. (A,C,E) Representative western blots from n = 3 experiments.
(B,D,F) Quantification of western blots is shown as graphs in S1 % tau/GAPDH and
P3 % tau/S1 tau with vehicle set to 100%. Individual samples are presented as
filled shapes and in lines as means
Soluble 55 kDa tau was not reduced by subchronic Thiamet G treatment for 1 week and phosphorylation levels remained unchanged (S1, Fig. 3E). On the other hand, subchronic Thiamet G treatment led to a 20–30% decrease in soluble 64 kDa and insoluble 64 and 70 kDa hyperphosphorylated tau species (Fig. 3E,F), although this difference did not reach statistical significance. From these results, we concluded that Thiamet-G in acute and subchronic oral doses reaches sufficient brain concentrations, increases the total protein O-GlcNAc levels in brains, but has no effect on phosphorylation levels of soluble 55 kDa human 0N4R P301L tau. In subchronic 1 week oral dosing, Thiamet G showed a trend to reduce soluble and insoluble hyperphosphorylated tau species in 24 week old rTg4510 mice.
Based on the trend observed in aged rTg4510 mice, we initiated chronic Thiamet G
treatment in young rTg4510 mice to investigate the long-term effect of OGA
inhibition on O-GlcNAc levels and accumulation of tau pathology over time. 6 week
old rTg4510 mice, which did not present with hyperphosphorylated tau
(Supplementary Fig. 1) or tau pathology as we reported previously [34, 36], were continually treated for 18 weeks with 600 mg/kg/day Thiamet G in the
drinking water. Biochemical analysis was performed on brain homogenates isolated
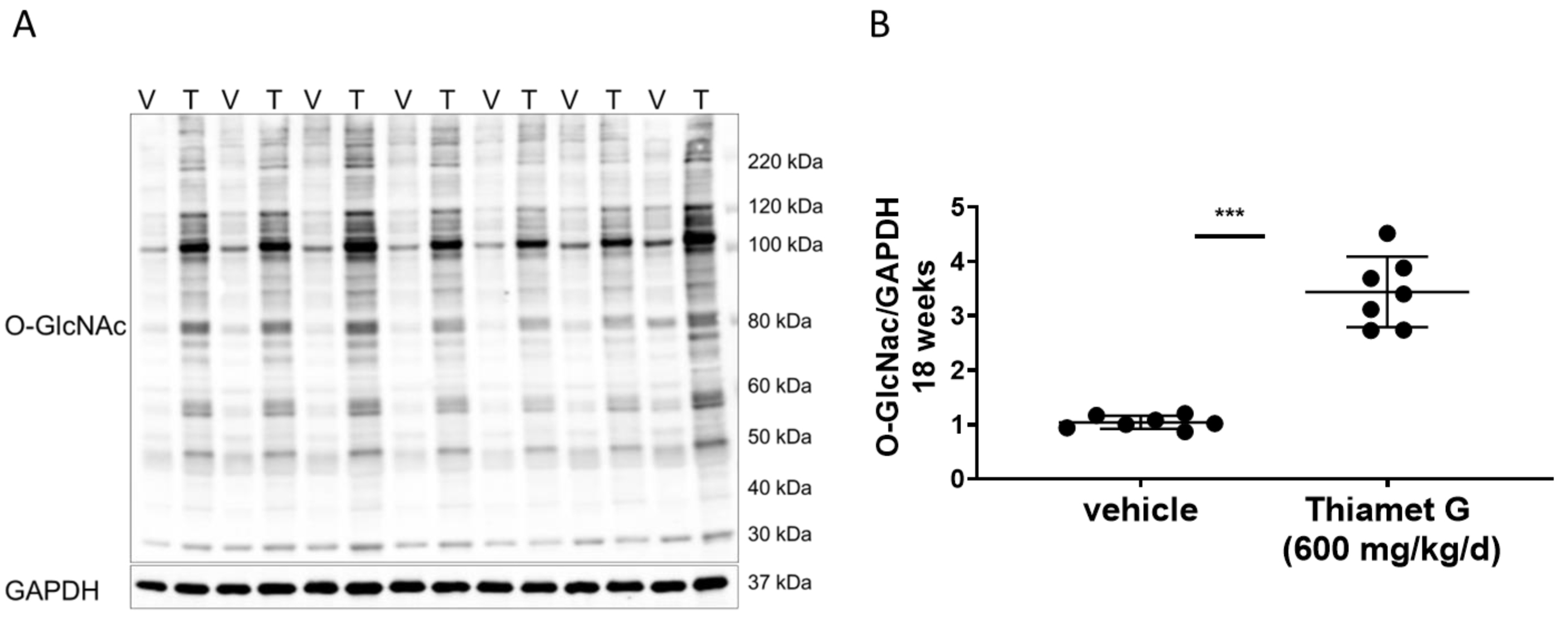
from 24 week old rTg4510 and non-transgenic littermate mice, where we found
similar brain concentrations. We measured 1.1 µM Thiamet G in brain
samples from rTg4510 mice, which were similar to the brain sample concentrations
of Thiamet G found in rTg4510 mice treated subchronically for 1 week (Table 2).
The unbound Thiamet G concentration in the brain amounted to 740 nM, which was
22-fold higher than the EC
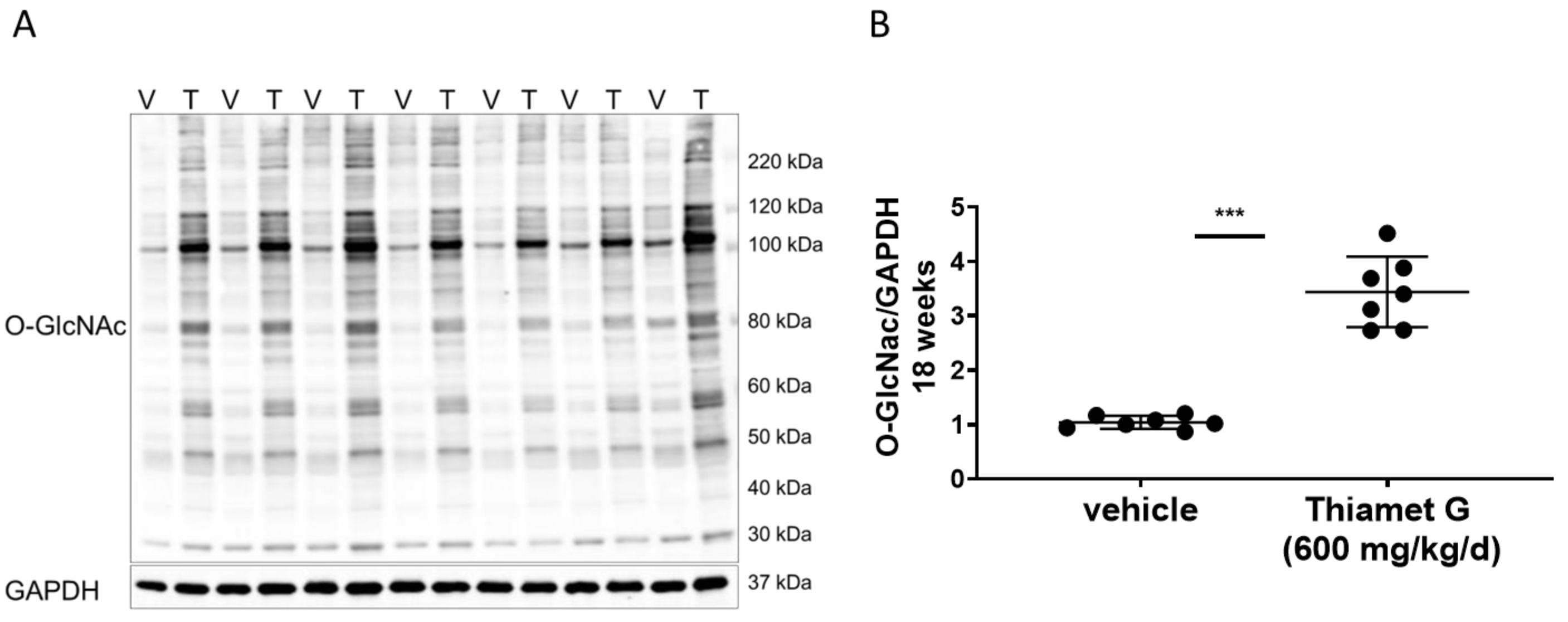 Fig. 4.
Fig. 4.Chronic Thiamet G treatment increases protein
O-GlcNAcylation in aged rTg4510 mice. Soluble fraction of brain
homogenates from 24 week old rTg4510 mice treated either with vehicle (V, n = 7)
or Thiamet G (T, n = 7) in the drinking water (600 mg/kg/day) from 6 weeks of age
were analyzed by western blot for global protein O-GlcNAcylation (O-GlcNAc). For
normalization, the blot was re-probed for GAPDH. (A) Representative western blot
from n = 3 experiments. (B) Quantification of western blot is shown as a graph in
absolute units O-GlcNAc/GAPDH. Individual samples are presented as filled circles
and in lines as means
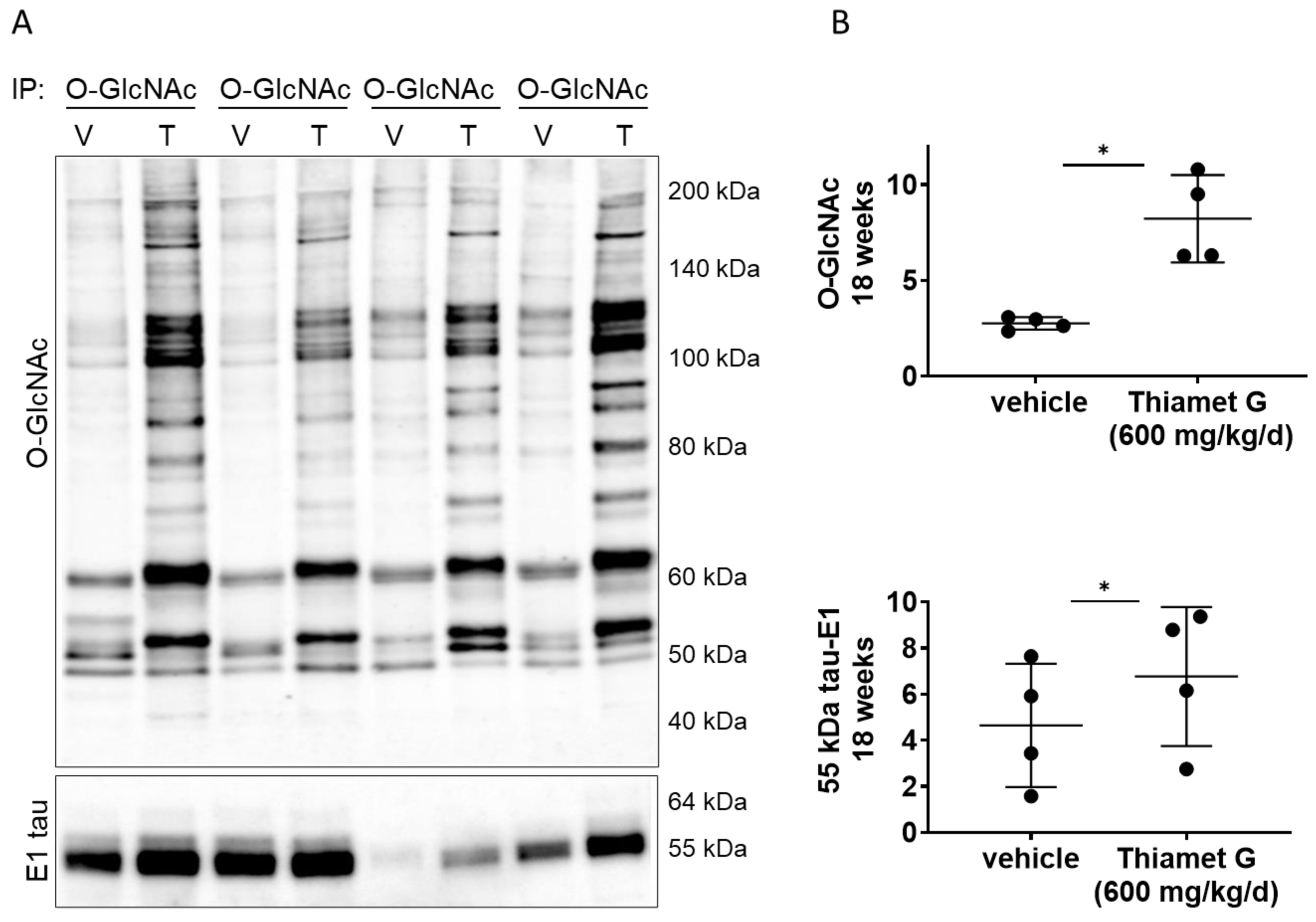
Next, we investigated whether chronic Thiamet G treatment for 18 weeks in rTg4510 mice could affect O-GlcNAcylation of human P301L tau. We performed immunoprecipitation (IP) experiments with forebrain homogenates of rTg4510 mice treated with vehicle or Thiamet G, and immunoprecipitated O-GlcNAc modified proteins with the CTD110.6 antibody, which selectively recognizes O-GlcNAcylated proteins (in pairs, n = 4 for each treatment group; Fig. 5A). As expected, we detected a significant 3-fold increase in protein O-GlcNAcylation in the immunoprecipitates from rTg4510 mice treated with Thiamet G compared to vehicle (Fig. 5B). We then probed the O-GlcNAc immunoprecipitates with the human tau specific E1 antibody and detected mostly 55 kDa tau. 64 kDa tau bands were almost undetectable in the O-GlcNAc immunoprecipitates. We repeatedly observed considerable variations in the 55 kDa tau levels between the individual immunoprecipitates (Fig. 5A). However, we detected a mild but significant 1.5-fold increase of 55 kDa tau species in O-GlcNAc immunoprecipitates from rTg4510 mice treated with Thiamet G (Fig. 5B), suggesting that chronic Thiamet C treatment for 18 weeks could increase tau O-GlcNAcylation.
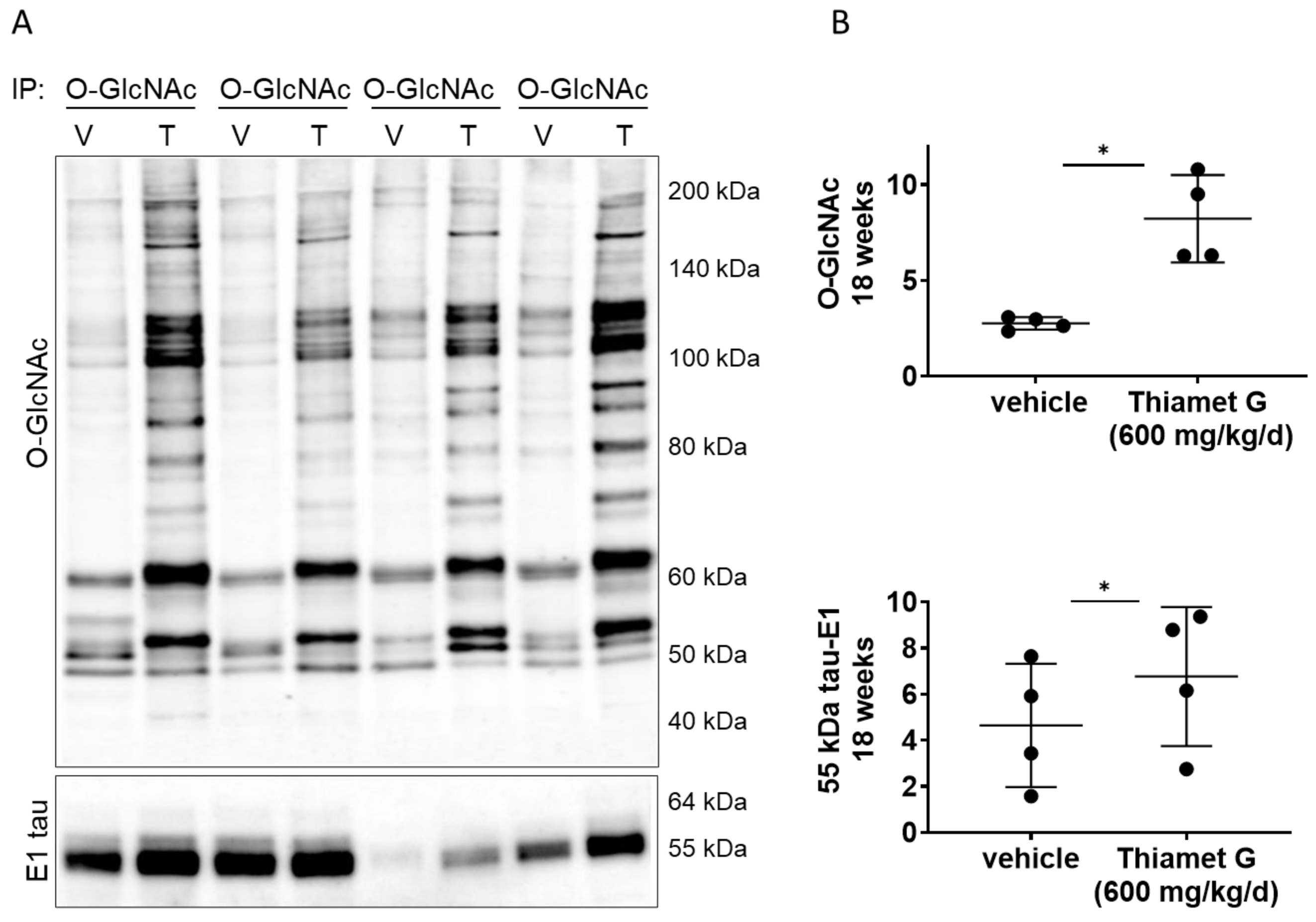 Fig. 5.
Fig. 5.Chronic Thiamet G treatment increases tau O-GlcNAcylation in
aged rTg4510 mice. Pairs of brain homogenate from 24 week old rTg4510 mice
treated either with vehicle (V) or Thiamet G (T) in the drinking water (600
mg/kg/day) from 6 weeks of age were prepared individually for immunoprecipitation
of O-GlcNAcylated proteins with the CTD110.6 antibody. Immunoprecipitates (n = 4
pairs) were analyzed by western blot for O-GlcNAc and human tau with the E1
antibody. (A) Representative western blots from n = 3 experiments. (B)
Quantification of western blots is shown as graphs in absolute units O-GlcNAc and
human 55 kDa tau. Individual samples are presented as filled circles and in lines
as means
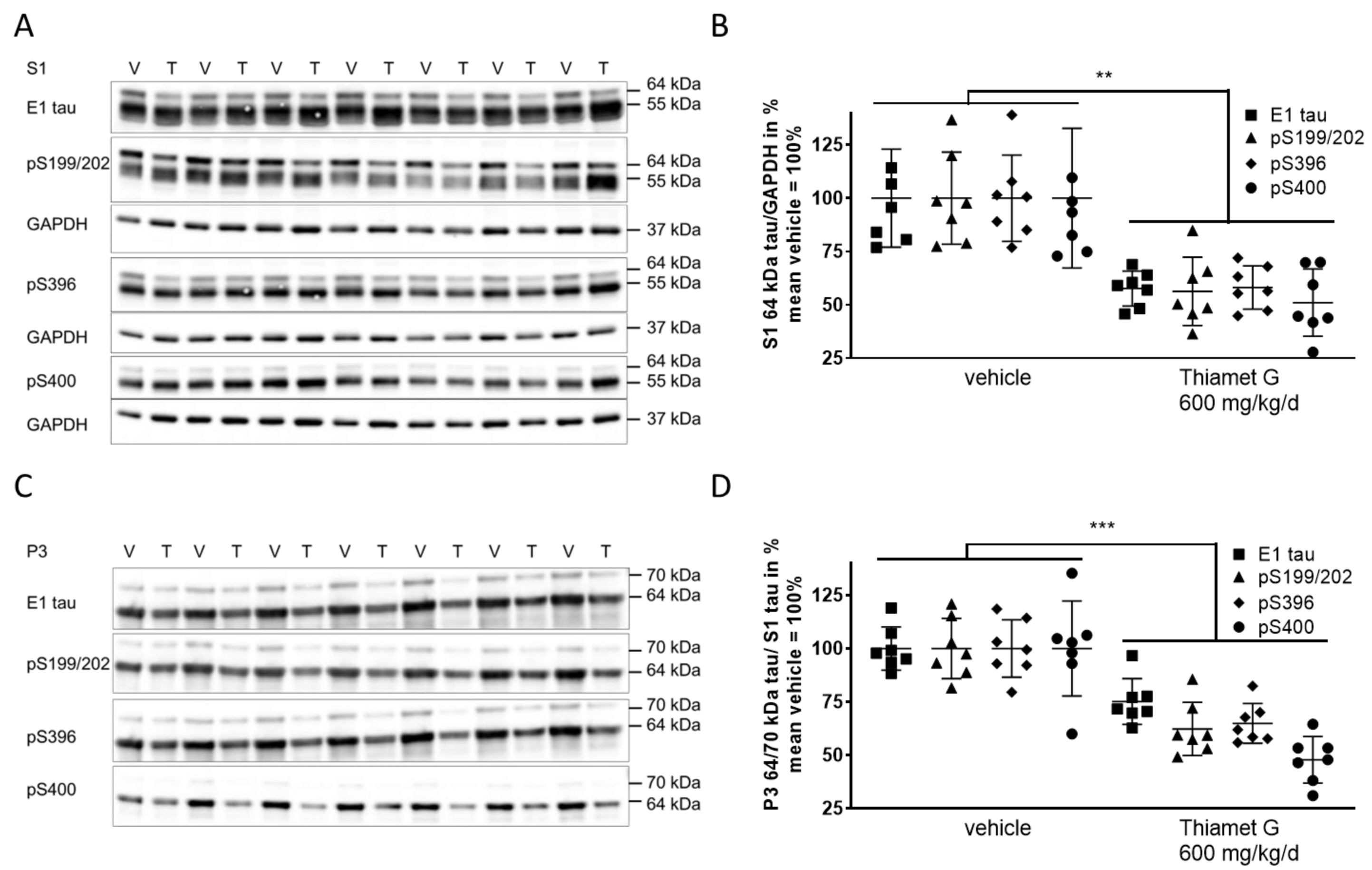
Next, we investigated whether chronic Thiamet G treatment prevented accumulation of soluble and insoluble hyperphosphorylated tau in 24 week old rTg4510 mice. S1 and P3 were isolated from forebrain homogenates and different human tau species were measured using E1 and phospho tau pS199/202, pS396, and pS400 antibodies, respectively. At 24 weeks of age, vehicle treated rTg4510 mice displayed considerable levels of soluble hyperphosphorylated 64 kDa tau in S1 (Fig. 6A,B) as well as insoluble hyperphosphorylated 64 and 70 kDa tau species in P3 (Fig. 6C,D), in agreement with our biochemical characterization (Supplementary Fig. 1) reported previously [34, 36]. In Thiamet G treated rTg4510 mice we observed a significant reduction of soluble hyperphosphorylated 64 kDa tau by 45% in S1 based on detection with the phospho tau antibodies pS199/202, pS396, and pS400 (Fig. 6B). Insoluble hyperphosphorylated 64 and 70 kDa tau in P3 was also significantly reduced by 42% (Fig. 6D). The highest reduction was observed with the pS400 tau antibody showing 52% and 49% reduction of hyperphosphorylated tau species in S1 and P3, respectively. We did not observe any changes in the total levels or phosphorylation of soluble 55 kDa tau in S1 (Fig. 6A). These results suggest that long-term OGA inhibition by Thiamet G reduced accumulation of soluble and insoluble hyperphosphorylated tau when treatment was initiated before rTg4510 mice exhibited tau pathology.
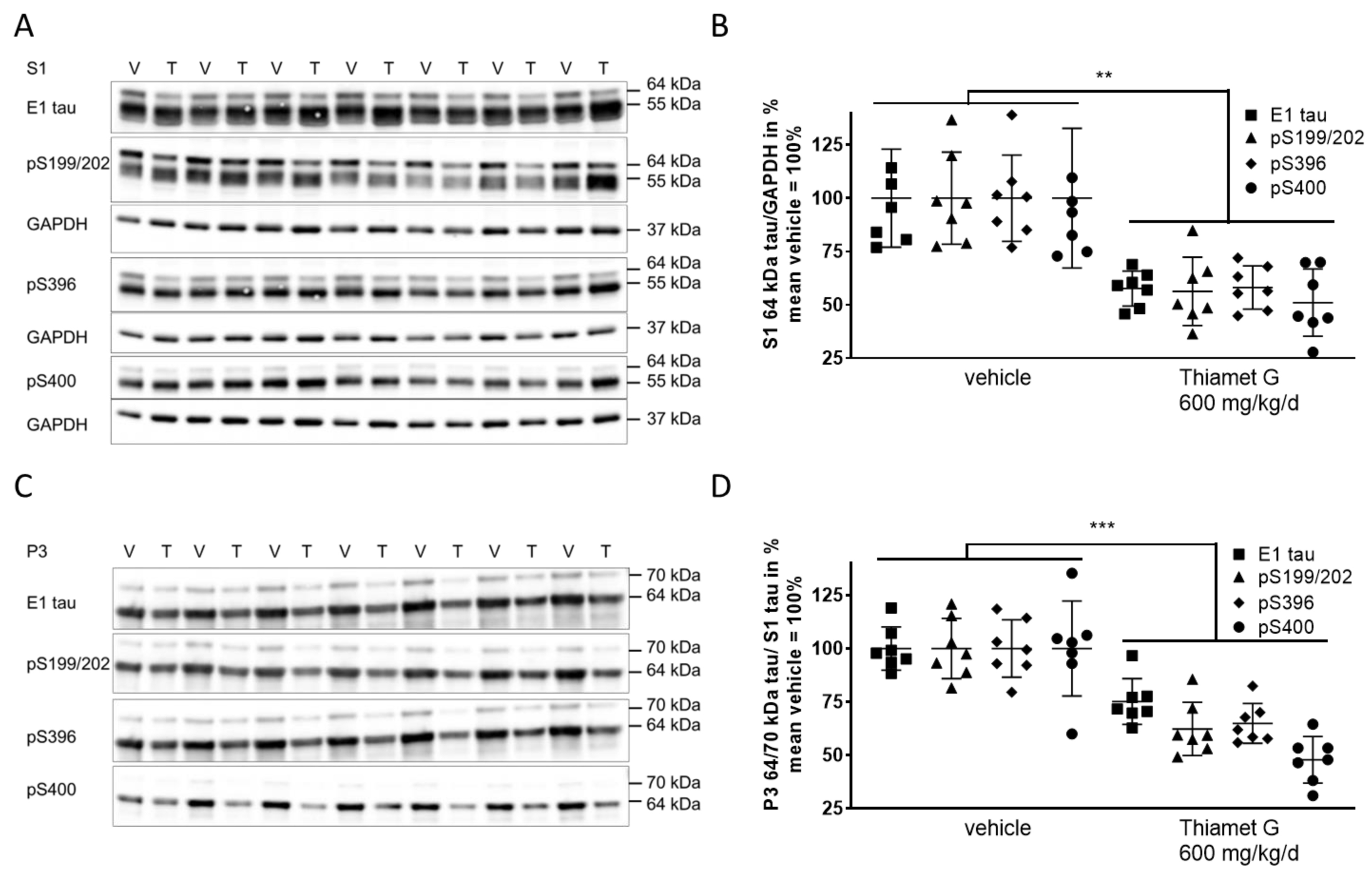 Fig. 6.
Fig. 6.Chronic Thiamet G treatment reduces accumulation of
hyperphosphorylated tau in aged rTg4510 mice. Brain homogenates from 24 week old
rTg4510 mice treated either with vehicle (V, n = 7) or Thiamet G (T, n = 7) in
the drinking water (600 mg/kg/day) from 6 weeks of age were fractioned into
soluble (S1) and sarkosyl-insoluble (P3) fractions and analyzed by western blot
for total human tau (E1) and phosphorylated tau at S199/202, S396, and S400
epitopes. For normalization of S1 blots GAPDH was used. The E1 blot was re-probed
with pS396 and GAPDH. pS199/202 and pS400 blots were re-probed with GAPDH. The P3
E1 blot was re-probed with pS396. Phosphorylated and non-phosphorylated human
0N4R tau was displayed at 55 kDa in S1 fractions. Hyperphosphorylated tau was
mobility-shifted and displayed at 64 and 70 kDa in S1 and P3 fractions. (A,C)
Representative western blots from n = 3 experiments. (B,D) Quantification of
western blots is shown in graphs in S1 64 kDa tau/GAPDH in % and P3 64 and 70
kDa tau/S1 tau in %. Individual samples are presented as filled shapes and in
lines as means
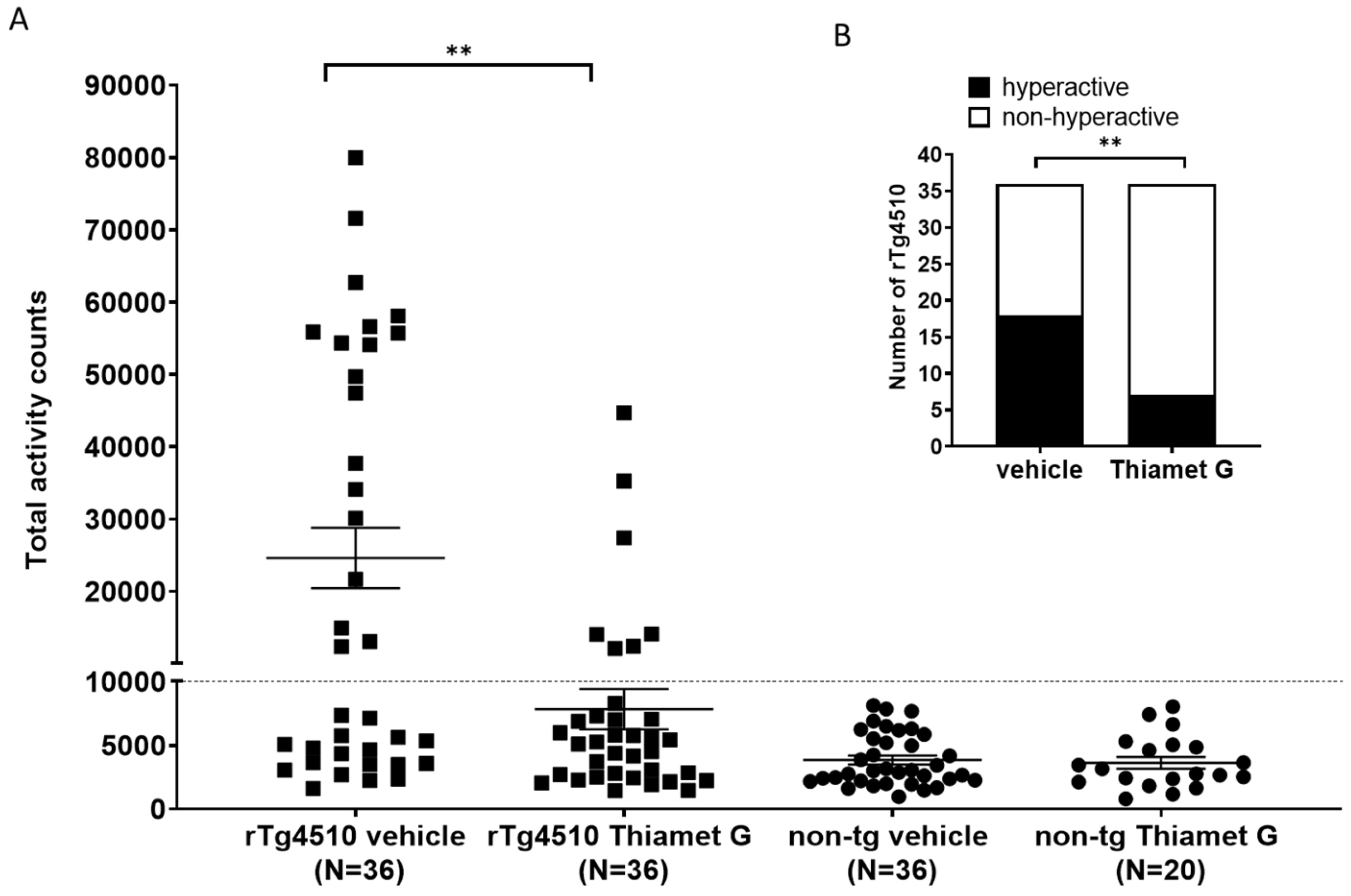
rTg4510 mice develop a non-mnemonic behavioral defect, characterized by a
hyperactive phenotype that increases with the progression of tau pathology [33, 34]. We reported previously that between 46–59% of the rTg4510 mice in our
colony were hyperactive when they reach 24 weeks of age. Suppression of transgene
tau expression with doxycycline from 6 weeks of age reduced the number of rTg4510
mice displaying the hyperactive phenotype at 24 weeks of age, suggesting that
hyperactivity is dependent on tau pathology [34]. Here, we investigated whether
long-term treatment with Thiamet G influence hyperactivity in the rTg4510 mice.
56 non-transgenic littermates (n = 36 treated with vehicle and n = 20 treated
with Thiamet G) and 72 rTg4510 mice (n = 36 treated with vehicle and n = 36
treated with Thiamet G) were dosed from 6 weeks of age with 600 mg/kg/day Thiamet
G in the drinking water and tested for increased locomotor activity at 24 weeks
of age. Non-transgenic littermates displayed normal locomotor activity between
963 and 8124 total activity counts with an average of 3848
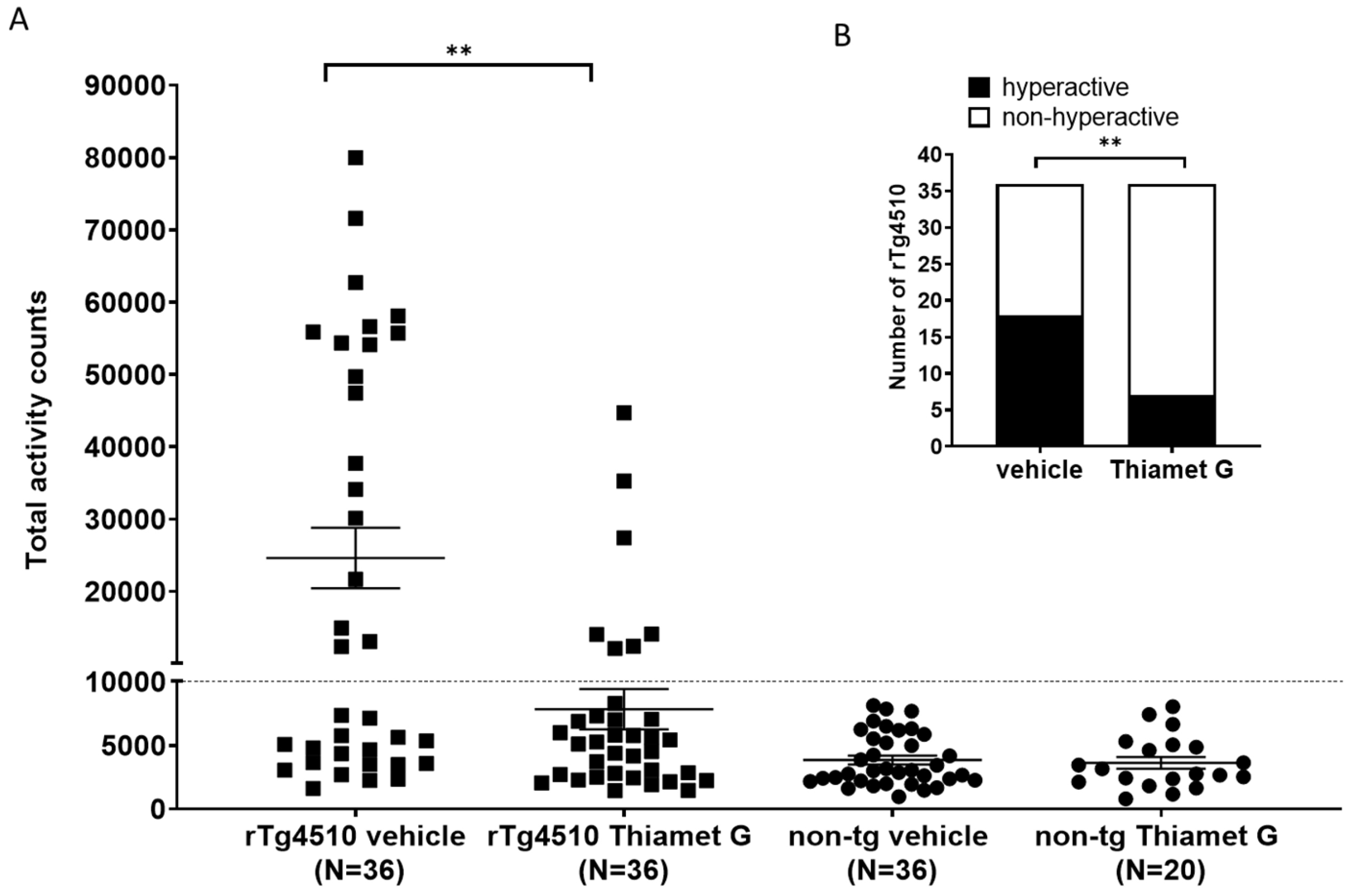 Fig. 7.
Fig. 7.Chronic Thiamet G reduces hyperactivity in aged rTg4510 mice.
Non-transgenic littermates (non-tg, n = 56) and rTg4510 mice (n = 72) treated
with vehicle (non-tg, n = 36; rTg4510, n = 36) or Thiamet G (non-tg, n = 20;
rTg4510, n = 36) in the drinking water (600 mg/kg/day) from 6 weeks of age were
tested in a 3-hour locomotor activity test in a novel cage environment at 24
weeks of age. (A) Data are presented as total activity counts. Hyperactivity was
defined as
Taken together, these in vivo results show that Thiamet G can cross the
blood brain barrier in rTg4510 mice and non-transgenic littermates following oral
dosing. The free fraction of Thiamet G in the brain of rTg4510 mice after chronic
dosing was 740 nM, which is 22-fold above the EC
Here we show that chronic treatment with the selective and potent OGA inhibitor Thiamet G results in a significant reduction of soluble and insoluble hyperphosphorylated tau species, and attenuation of the hyperactive phenotype in rTg4510 mice.
Thiamet G is a potent inhibitor of OGA; in neurons we determined an EC
Previous reports from different tauopathy mouse models harboring the P301L mutation in human tau such as JNPL3 [26, 28], Tau.P301L [39], and rTg4510 [30, 31, 40, 41] suggest that chronic OGA inhibition is beneficial. Several studies in the transgenic mouse strains JNPL3 and rTg4510 concluded that chronic treatment with Thiamet G increased tau O-GlcNAcylation and reduced tau aggregation and NFTs [28, 30, 31]. Another study showed beneficial effects of chronic Thiamet G treatment on a behavioral defect that was dependent on pathological tau [40].
Our findings are in agreement with previous reports in tauopathy models. Chronic
treatment with 600 mg/kg Thiamet G for 18 weeks resulted in a significant
reduction of tau pathology in 24 week old rTg4510 mice. The 600 mg/kg dose was
chosen to ensure complete inhibition of OGA and to obtain a significant increase
in brain O-GlcNAcylation in rTg4510 mice. This dose of Thiamet G achieved a free
Thiamet G concentration in the brain of 740 nM, which was 22-fold above the
EC
We observed a significant reduction in tau pathology in forebrain homogenates from rTg4510 mice treated chronically with Thiamet G. We investigated hyperphosphorylated 64 kDa tau species from the low speed soluble (S1) and the sarkosyl-insoluble pellet (P3) fractions as characterized before [13]. The sarkosyl-insoluble hyperphosphorylated 64 and 70 kDa tau species from P3 consists mainly of aggregated fibrillar tau and are regarded as the biochemical NFTs equivalent [11, 32]. The hyperphosphorylated 64 kDa tau species from S1 represent low speed soluble, high speed precipitable tau species, consisting mainly of tau oligomers and are regarded as the biochemical pre-tangle tau equivalent [13]. Oligomeric tau aggregates are believed to precede NFTs formation and are more correlated with neuronal dysfunction and degeneration than the sarkosyl-insoluble 64 and 70 kDa tau species in rTg4510 mouse brains [11, 13, 32]. In AD it is unclear whether NFTs are the primary neurotoxic trigger, as the number of degenerated neurons is much higher than neurons containing NFTs in some brain areas [42]. Tau aggregates, with structural similarity to the oligomeric hyperphosphorylated 64 kDa tau species from rTg4510, were found in AD brains [43]. We demonstrate here that soluble and insoluble hyperphosphorylated 64 and 70 kDa tau species were significantly reduced by chronic treatment with Thiamet G. We found a marked 42–52% reduction of pathological tau in rTg4510 mice. Our findings agree with other reports in rTg4510 mice [30, 31, 40], highlighting the effectiveness of OGA inhibition at attenuating tau pathology in mouse tauopathy models.
A reciprocal interplay between phosphorylation and O-GlcNAcylation of specific tau residues, and reduction of tau phosphorylation after acute, but not chronic, OGA inhibition has been reported previously [26]. We observed no effect of OGA inhibition with Thiamet G on phosphorylation levels of normal tau in neuronal cultures or in brain homogenates from rTg4510 mice. In neuronal cultures we observed a marked increase in global O-GlcNAc levels with Thiamet G, but phosphorylation levels of human transgene 0N4R P301L and murine tau were unchanged. In vivo, we observed a clear increase in global O-GlcNAc levels in brain homogenates from 24 week old rTg4510 mice treated acutely with Thiamet G for 6 or 24 h. However, no effect on tau levels or phosphorylation was observed on normally phosphorylated 55 kDa tau or hyperphosphorylated 64 and 70 kDa tau species following acute Thiamet G treatment. Conversely, we demonstrated a significant reduction in hyperphosphorylated 64 and 70 kDa tau species in brain homogenates from 24 week old rTg4510 mice following chronic Thiamet G treatment. Under this condition, levels of normally phosphorylated 55 kDa tau species were not altered. In our hands OGA inhibition was not reducing phosphorylation of normal tau, which is consistent with findings by others [28, 29, 30, 31]. Our study supports that chronic OGA inhibition reduced pathological tau measured biochemically as soluble and insoluble hyperphosphorylated tau species.
Studies in JNPL3 and rTg4510 mice reported that Thiamet G increases tau O-GlcNAcylation [28, 30, 31]. We show a significant 1.5-fold increase of tau in O-GlcNAc immunoprecipitates from Thiamet G treated rTg4510 mice, suggesting that chronic Thiamet G treatment for 18 weeks can increase tau O-GlcNAcylation. Interestingly, we predominately detected 55 kDa tau on immunoprecipitated O-GlcNAcyled proteins, suggesting that hyperphosphorylated 64 kDa tau might be modified less by O-GlcNAcylation. Similar observations have been reported previously that O-GlcNAcylation of tau was mainly associated with soluble 55 kDa and not with aggregated 64 kDa tau in rTg4510 mice [30, 31]. The question arises as to how OGA inhibition could reduce tau pathology in these models when pathological tau was less O-GlcNAcylated. A possible explanation could be that O-GlcNAcylation of tau could reduce the oligomerization susceptibility of tau as shown in vitro experiments [24, 28]. In this way tau could be kept in a monomeric state with less posttranslational modifications and aggregation could be intervened. We speculate that a reduction of tau’s aggregation propensity might be a possible mechanism by which OGA inhibition could reduce accumulation of pathological tau.
This study mainly explored the efficacy of Thiamet G on reducing accumulation of tau pathology by treatment initiated in young rTg4510 mice preceding tau pathology. Interestingly, subchronic Thiamet G treatment showed limited efficacy in aged rTg4510 mice with existing tau pathology. In 24 week old rTg4510 mice treated for 1 week with Thiamet G, we observed a 3.5-fold increase in global O-GlcNAc levels and 20–30% reduction of tau pathology, however effects on soluble and insoluble hyperphosphorylated tau species were not significant. Our findings may suggest that Thiamet G could not only reduce formation of tau pathology in a preventive manner but also reduce already existing tau pathology. Based on this initial observation, future studies should investigate further whether OGA inhibitor treatment can also attenuate existing tau pathology in aged rTg4510 mice.
We previously identified a non-mnemonic behavioral defect in the rTg4510 mice, which was characterized by abnormally increased locomotor activity classified as hyperactivity. The hyperactive phenotype appeared in a subset of rTg4510 mice and was associated with a disrupted day/night cycle. This phenotype became more severe with progression of tau pathology [34]. This hyperactive phenotype models some of the behavioral disturbances observed in dementia patients. Some AD patients show abnormal wandering and shifted day/night activities [44, 45, 46] and some frontotemporal dementia patients show hyperactivity [47]. These behavioral disturbances, especially abnormal wandering in dementia patients, are linked to higher morbidity and mortality [46, 48]. They are also linked to impaired frontotemporal glucose consumption [49], suggestive of neuronal degeneration in frontotemporal regions. Chronic Thiamet G treatment of young 6 week old rTg4510 mice for 18 weeks reduced hyperphosphorylated tau accumulation and this was concurrent with a significant attenuation of hyperactivity in 24 week old rTg4510 mice. Our findings are consistent with a previous study investigating the effects of Thiamet G on increased locomotor activity in rTg4510 mice [40]. In our hands, the number of hyperactive rTg4510 mice decreased significantly to 19% in the Thiamet G treated group compared to 50% hyperactive rTg4510 mice in the vehicle treated group. Hyperactivity was significantly improved in rTg4510 mice by Thiamet G treatment but not completely reversed to non-transgenic littermate levels. Since Thiamet G treatment led to a robust 42–52% reduction, but not complete removal of pathological tau measured biochemically as hyperphosphorylated tau species, we anticipated that Thiamet G treatment could reduce but not completely prevent hyperactivity in rTg4510. Overall, our results suggest that OGA inhibition could lead to a decrease in tau pathology and associated behavioral disturbances in tauopathies.
Clinical trials to inhibit tau phosphorylation by glycogen synthase kinase 3 inhibitor [50] or tau aggregation by leuco-methylthioninium bis [51] were not successful and targeting O-GlcNAcylation presents a new attempt to treat AD. Based on the positive preclinical results with increasing O-GlcNAcylation, three OGA inhibitors (ASN-120290 from Asceneuron, LY3372689 from Eli Lilly, and MK-8719 from Merck/Alectos) advanced into clinical trials to test the potential to treat AD and tauopathies [52]. The beneficial effects of OGA inhibition are not limited to tau. Recent preclinical findings show that O-GlcNAcylated alpha-synuclein also exhibited reduced aggregation and toxicity [53, 54, 55, 56], suggesting that OGA inhibition could also be a new therapeutic approach for alpha-synucleinopathies.
Chronic increase of O-GlcNAcylation with the selective and potent OGA inhibitor Thiamet G reduced tau pathology measured as accumulation of hyperphosphorylated tau species and this was paralleled by attenuation of hyperactivity in rTg4510 mice. These data provide additional preclinical evidence that pharmacological inhibition of OGA may offer a therapeutic approach for slowing the progression and behavioral disturbances in tauopathies, such as AD.
Data generated or analyzed during this study are included in this published article. Datasets referred to as “data not shown” may be requested from the corresponding author.
Conceived and designed the experiments: CV, NR. Performed the experiments: NR, CV, PHJ, MG. Analyzed the data: CV, NR. Wrote the paper: CV, NR. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiments were performed in accordance with the European Communities Council Directive no. 86/609, the directives of the Danish National Committee on Animal Research Ethics, and Danish legislation on experimental animals (license no. 2014-15-0201-00339).
The excellent technical assistance of A. Bredal Christiansen and A. Ravn Neve is gratefully acknowledged. We thank M. Carnerup and L. Badolo for providing valuable advice regarding exposure and free fraction measurements.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.