

 , Xijing Zhang 1,*
, Xijing Zhang 1,*1 Department of Anaesthesiology and Perioperative Medicine and Department of Intensive Care Unit, Xijing Hospital, The Fourth Military Medical University, 710032 Xi’an, Shaanxi, China
2 College of Life Sciences, Northwest University, 710069 Xi’an, Shaanxi, China
†These authors contributed equally.
Academic Editor: Drozdstoy Stoyanov
Abstract
Ferroptosis is distinct from other apoptotic forms of programmed cell death and is characterized by the accumulation of iron and lipid peroxidation. Iron plays a crucial role in the oxidation of lipids via the Fenton reaction with oxygen. Hence, iron accumulation causes phospholipid peroxidation which induces ferroptosis. Moreover, detoxification by glutathione is disrupted during ferroptosis. A growing number of studies have implicated ferroptosis in nervous system disorders such as depression, neurodegenerative disease, stroke, traumatic brain injury, and sepsis-associated encephalopathy. This review summarizes the pathogenesis of ferroptosis and its relationship with various nervous system disorders.
Keywords
- ferroptosis
- iron
- lipid peroxidation
- glutathione
- nervous system disorders
Nervous system disorders include depression, neurodegenerative disease, stroke, traumatic brain injury (TBI), and sepsis-associated encephalopathy (SAE). These conditions seriously affect the quality of life and significantly increase the economic and social burden [1, 2, 3, 4]. A common feature of nervous system disorders is the abnormal activation of cell death [5, 6]. A better understanding of the mechanism of neuronal death should therefore lead to new approaches for the prevention and treatment of nervous system disorders.
Programmed cell death is classified into apoptosis, pyroptosis, autophagy, necroptosis, and ferroptosis according to the different molecular mechanisms and morphological features (Table 1) [7, 8, 9]. Ferroptosis was first proposed in 2012 as a novel form of programmed cell death that results from the accumulation of ferrous ions and the reduction of Glutathione (GSH) synthesis [10, 11, 12]. GSH is comprised of glutamate, cysteine, and glycine. This sulfhydryl-containing tripeptide is essential for the protection of the brain from oxidative stress through its action as a free radical scavenger and lipid peroxidation inhibitor [13]. Transferrin-bound iron is transported into the cell by transferrin receptor 1 (TFR1) and converted into ferrous ions. The accumulation of ferrous ions leads to the production of lipid-reactive oxygen species and eventually to ferroptosis [14]. Additionally, impaired generation of GSH reduces the synthesis of glutathione peroxidase 4 (GPX4), thereby blocking the conversion of lipid hydroperoxides into non-toxic lipid alcohols and leading to the accumulation of phospholipid hydroperoxides (PLOOHs) [15, 16]. PLOOHs are lipid-based forms of reactive oxygen species (ROS) that are essential for the inhibition of ferroptosis [17]. The accumulation of PLOOHs causes rapid and irreparable damage to the membrane and directly catalyzes the chain reaction of peroxidation to induce lipid peroxide-dependent cell death [18].
| Cell death forms | Cell membrane | Cytoplasm | Cell nucleus | Biochemical characteristics | Immune characteristics |
|---|---|---|---|---|---|
| Ferroptosis | Breakage and bubble | Increase mitochondrial membrane density | Normal | The increase of iron-dependent lipid peroxides | Promote inflammation |
| Apoptosis | Integrity | No significant change | Nuclear shrinkage chromatin condensation | DNA fragmentation decreases the mitochondrial membrane potential | Inhibit inflammation |
| Autophagy | Integrity | Lysosomal vacuoles are double membranes | Normal | LC3 changed from LC3I to LC3II | Promote inflammation |
| Necroptosis | Membrane rupture | Swelling of organelles | Partial condensation of chromatin | Enrichment of kinase and decrease of ATP level | Promote inflammation |
| Pyroptosis | Loss of membrane integrity | Loss of organelles | DNA condensation and fragmentation | Formation of inflammasomes activation of caspase-1 and release of a large number of pro-inflammatory factors | Promote inflammation |
The link between ferroptosis and a wide range of disorders including cardiovascular disease, cancer, and metabolic disease has been studied extensively [19, 20, 21]. For example, cardiomyopathy was improved following the upregulation of GPX4. Furthermore, an iron chelating agent was able to reduce the level of the ferrous ion, ameliorate the side effects of cardiomyopathy, and prevent ferroptosis [22]. Recent studies have also shown a close relation between ferroptosis and various nervous system disorders. The aim of this review is to summarize the development and mechanism of ferroptosis and its role in nervous system disorders, thereby suggesting possible new options for the treatment of nervous system disorders.
Ferroptosis is a form of programmed cell death caused by the accumulation of iron-dependent lipid peroxide [23]. In 2003, Dolma et al. [24] reported that Erastin and RSL3 (RAS-selective lethal agents) could induce cell death in RAS mutated cells. In 2007, Yagoda et al. [25] found that Erastin-induced cell death was distinct from apoptosis, autophagy, necroptosis, and other forms of cell death. The morphology of this new type of cell death was characterized by a loss of mitochondrial integrity. The following year, two compounds similar to Erastin (RSL5 and RSL3) were identified that could induce iron-dependent, non-apoptotic cell death in tumor cells [26]. This form of cell death could be inhibited by iron-chelating agents (DFOM) and antioxidants (vitamin E) [27]. In 2012, Dixon et al. [10] formally named this as “ferroptosis”. In contrast to other types of cell death, the cell membrane is broken during ferroptosis while the mitochondrial membrane density is increased. The concept of “ferroptosis” was further developed in 2012 [28].
Iron is the most abundant transition metal element in the Earth’s crust and plays a critical role in oxygen transport, cellular energy metabolism, and many enzymatic reactions [29, 30]. Under normal conditions, iron is absorbed from food in the duodenum and mucosa of the upper jejunum [31]. The ferrous ions released from the ferric transporter on the outer surface of the capillary endothelial cell are oxidized by ceruloplasmin into ferric ions, which are then captured by transferrin (Tf) in the intercellular fluid and cerebrospinal fluid. Tf binds the ferric ions and forms a Tf-TFR1 complex with transferrin receptor 1 (TFR1), which is then endocytosed via clathrin-coated pits at the cell surface [32, 33]. In the acidic endosomes, ferric ions are reduced into ferrous ions and transported by divalent metal transporter 1 (DMT1) to labile iron pools in the cytoplasm. Here, the ferrous ions are stored in ferritin or imported into mitochondria via mitotic ferritin. Non-transferrin-bound iron at the cell surface is directly introduced into the labile iron pool by DMT1 [34, 35, 36]. The accumulation of intracellular ferrous ions promotes lipid peroxidation, leading to the production of ROS and ultimately to ferroptosis (Fig. 1) [37].
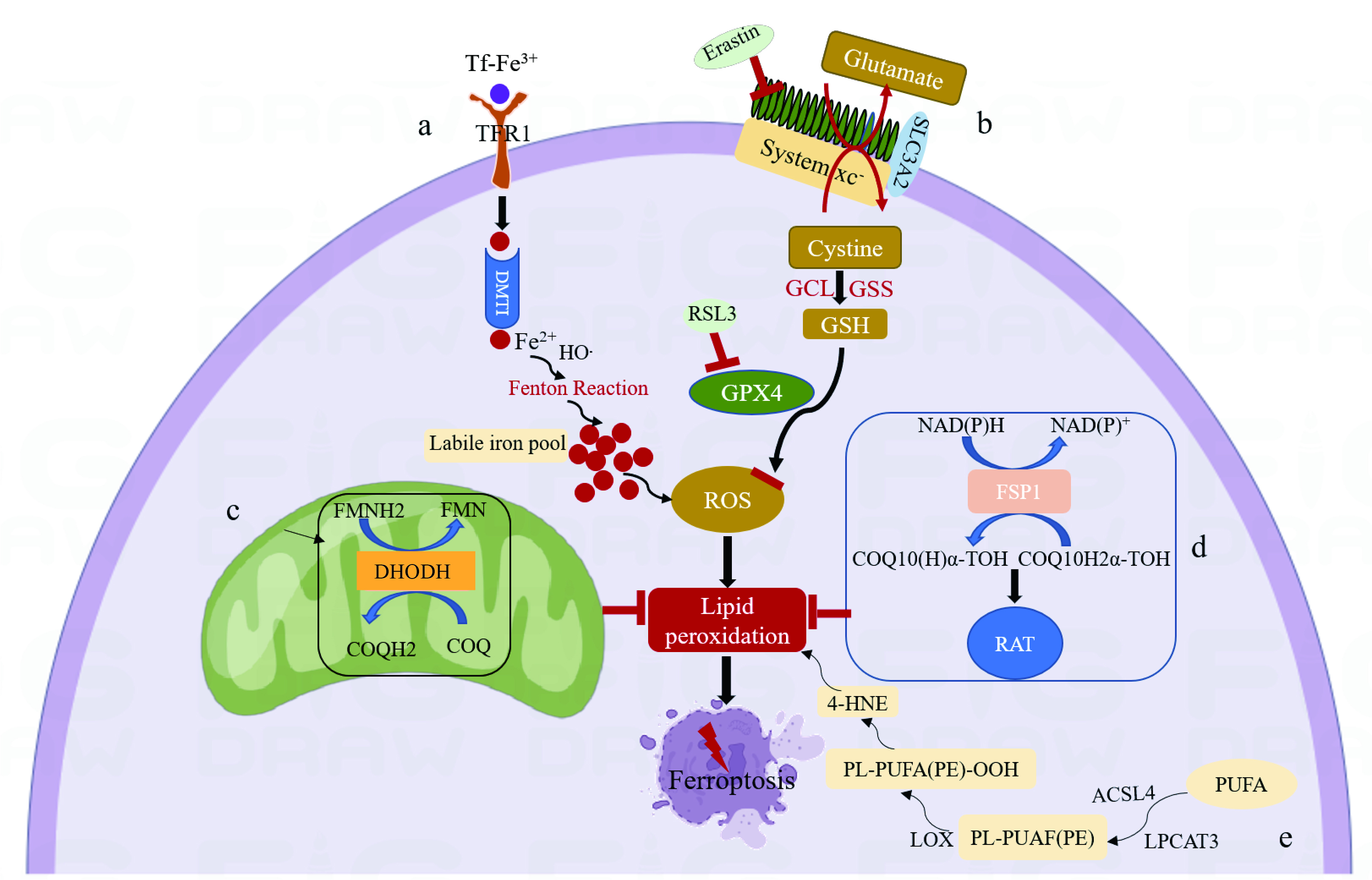 Fig. 1.
Fig. 1.Mechanism of ferroptosis. (a) Tf binds ferric ions and forms a Tf-TFR1 complex with TFR1. The following endocytosis into acidic endosomes, ferric ions are reduced into ferrous ions and transported by DMT1 to labile iron pools in the cytoplasm. The accumulation of intracellular ferrous ions promotes lipid peroxidation, leading to the production of ROS and ultimately to ferroptosis. (b) System xc-exchanges intracellular glutamate for extracellular cystine in a 1:1 ratio, which is then synthesized into GSH by GCL and GSS. GSH is a reducing cofactor for glutathione peroxidase (GPX4), a membrane lipid repair enzyme that eliminates lipid hydroperoxides to protect against ferroptosis. (c) Dihydroorotate Dehydrogenase (DHODH) in the mitochondrial membrane resists ferroptosis by oxidizing dihydroorotate (DHO) to orotic acid (OA) and reducing ubiquinone (CoQ10) to Dihydroubiquinone (CoQH2). (d) Ferroptosis suppressor protein 1 (FSP1) converts ubiquinone into ubiquinol and generates a lipophilic, radical-trapping antioxidant (RTA) that prevents lipid peroxide formation and inhibits ferroptosis. (e) Polyunsaturated fatty acids (PUFA) are substrates for lipid peroxidation and react with acyl-CoA synthetase long-chain family member-4 (ACSL4) to generate Malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), eventually leading to ferroptosis.
Lipid peroxidation is the hallmark of ferroptosis. In the
initial stage, lipid peroxidation can occur through both enzymatic and
non-enzymatic reaction processes [38, 39]. However, which of these two is
essential for ferroptosis remains controversial. Several studies
have suggested that non-enzymatic lipid peroxidation is responsible for
ferroptosis [40]. This is driven by the Fenton reaction, which uses iron and
oxygen to catalyze a chain reaction that leads to the production of phospholipid
hydroperoxides (PLOOHs) [40]. Polyunsaturated fatty acid (PUFA) phospholipids are
substrates for lipid peroxidation. Enzymes such as acyl-CoA synthetase long-chain
family member-4 (ACSL4) inhibit ferroptosis by reducing PUFA phospholipid
synthesis. ACSL4 is therefore a marker of sensitivity to ferroptosis [41].
Mechanistically, ACSL4 enriches cellular membranes with long polyunsaturated
System xc
Previous studies on ferroptosis defense mechanisms have focused on GPX4 [54]. Recently, two articles published in 2019 reported on a novel ferroptosis suppressor, FSP1, that was independent of GSH [55, 56]. FSP1 was originally named AIFM2 and has homology to apoptosis-inducing factor (AIF) [55]. However, it lacks the N-terminal mitochondrial targeting sequence present in AIF and does not promote apoptosis. AIFM2 was therefore renamed FSP1 [55]. It is located in lipid droplets (LDs) and the plasma membrane [57]. It can convert ubiquinone on cell membranes into its reduced form, panthenol, which inhibits peroxidation and prevents ferroptosis [55]. By employing a synthetic lethal CRISPR/Cas9 screen, Doll et al. [55] found that FSP1 was an oxidoreductase that can reduce coenzyme Q10 (CoQ) to generate a lipophilic, radical trapping antioxidant (RTA) that prevents lipid peroxide formation (Fig. 1). Venkatesh et al. [58] reported that inhibition of MDM2 and MDMX resulted in increased levels of FSP1 protein, which in turn led to increased levels of the endogenous lipophilic antioxidant CoQ, thereby inhibiting ferroptosis. Song et al. [59] showed that miR-4443 regulates the expression of FSP1 through METLL3 in an m6A manner, thus regulating ferroptosis.
DHODH is a flavin-dependent enzyme located in the mitochondrial membrane [60]. By selectively inhibiting dihydroorotate dehydrogenase, thereby inhibiting pyrimidine synthesis [61]. This novel, GSH-independent ferroptosis defense system was first reported by Boyi’s team in 2021 [61]. They demonstrated that DHODH inhibits ferroptosis by oxidizing dihydroorotate (DHO) to orotic acid (OA) and reducing ubiquinone (CoQ10) to Dihydroubiquinone (CoQH2) in the mitochondrial membrane (Fig. 1). To our knowledge, there have been no further advances regarding the DHODH and ferroptosis mechanism. The molecular mechanisms that link ferroptosis with various diseases also warrant further exploration.
Depression is a major global health issue that affects about 300 million people worldwide [62]. It is characterized by persistent mood depression and restlessness and includes psychomotor disorders and cognitive impairments [63]. It also has the characteristics of being universal, chronic, and recurrent [64]. Unfortunately, completely effective treatment for depression has yet to be discovered. Currently, treatment for depression mainly involves drug therapy and psychotherapy. However, their efficacy is quite limited and drug therapy requires long-term medication, while psychotherapy is slow [64, 65, 66]. Therefore, it is very important to explore the pathophysiological mechanism of depression. Studies have shown that hippocampal volume is significantly reduced in both adults and adolescents with depression [67]. Patients who have recovered from depression also show a decreased hippocampal volume [67]. The destruction and atrophy of neurons during depression may be the cause of this reduced volume [68, 69]. Previous studies on depression and the death of hippocampal neurons have focused mainly on apoptosis [70]. For example, Zhao et al. [71] found that repetitive transcranial magnetic stimulation (RTMS) reduces depression-like behavior by inhibiting the apoptosis of hippocampal neurons. Dexmedetomidine can also reduce the apoptosis of hippocampal neurons in Sprague Dawley rats and decrease depressive behavior by inhibiting the p38 MAPK/c-Myc /CLIC4 signaling pathway [72]. As described above, ferroptosis is a newly discovered form of programmed cell death that is caused mainly by the accumulation of iron and lipid peroxides [73]. Ferroptosis offers a new treatment strategy for depression. Cao et al. [74] confirmed that mice with chronic unpredictable mild stress (CUMS) could be used as an animal model for depression. Jiao et al. [75] found that GPX4 expression was decreased in this model whereas that of FTH1, ACSL4, and COX2 was increased, indicating that ferroptosis was activated. Wang et al. [76] found that sodium hydride (NaHS) can reduce iron deposition and oxidative stress and increase the protein level of GPX4 and SLC7A11 in the prefrontal cortex of type 1 diabetic mice, thereby reducing ferroptosis and alleviating anxiety-depressive behavior (Table 2, Ref. [76, 77, 78, 79, 80, 81, 82, 83, 84, 85]). Using the chronic social defeat stress (CSDS) mouse model, Dang et al. [77] found that edravone (EDA) could upregulate GPX4 expression to alleviate depressive behavior, but that EX527 and ML385 can reduce Nrf2, HO-1, and Gpx4 protein levels and thereby attenuate the antidepressant effects of EDA (Table 2). In conclusion, these results confirm that ferroptosis occurs in mouse models of depression. Although the underlying molecular mechanism remains unknown, the upregulation of GPX4 can inhibit ferroptosis and thereby reduce depressive behavior in mice.
| Nervous system disorder | Reagents | Functions | References | |
| Depression | Sodium hydride | GPX4 and SLC7A11. | Wang Y, et al. [76] | |
| Edravone | GPX4 | Dang R, et al. [77] | ||
| EX527 and ML385 | Nrf2, HO-1, and Gpx4 | |||
| Neurodegenerative diseases | ||||
| Alzheimer’s disease | Lip-1 | Trapping free radical | Hambright WS, et al. [79] | |
| Parkinson’s disease | Iron ion chelating agents and ferroptosis inhibitor Fer-1 | Iron and GPX4 | Mahoney-Sanchez L, et al. [78] | |
| ferric ammonium citrate | Iron | Zhang P, et al. [80] | ||
| Stroke | ||||
| Ischemic stroke | Carvacrol | GPX4 | Guan X, et al. [81] | |
| Cerebral hemorrhage | Iron ion chelating agent Deferoxamine | Iron | Okauchi M, et al. [85] | |
| Traumatic Brain Injury | Ferrostatin-1 (Fer‐1) | Iron deposition | Xie BS, et al. [82] | |
| Sepsis-associated encephalopathy | Iron ion chelating agents | Iron | Yao P, et al. [83] | |
| miR-9-5p | Transferrin receptor | Wei XB, et al. [84] | ||
In conclusion, ferroptosis is a potential target for the treatment of depression, but there is still no clinical evidence to support the use of ferroptosis inhibitors in humans.
Neurodegenerative diseases are caused by the accumulation of oligomers and the aggregation of misfolded proteins [86, 87]. Due to the aging of society, neurodegenerative diseases have a considerable socio-economic impact. They include Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) [88, 89], and are characterized by a loss of function of specific neuronal groups and of their connections [88]. Emerging evidence has shown that ferroptosis can disrupt neuronal viability and is involved in the pathological mechanism of neurodegenerative diseases [78, 90].
AD is a complex neurodegenerative disease characterized by
extracellular accumulation of the proteins amyloid-
PD is the most common
severe movement disorder in the world and one of the most disabling diseases of
the central nervous system [48]. Attributed to a selective loss of neurons in the
substantia nigra, the symptoms of PD include tremors, stiffness, slow movement,
postural instability, and difficulty in walking [108, 109]. Previous studies on
PD have focused on dopamine-based therapies, but these are only effective during
the early course of the disease [78]. Further exploration of novel treatment
methods is therefore needed. The traditionally recognized pathological features
of PD are dopaminergic neuronal death and intracellular
The main focus to date for the clinical management of PD and AD is the iron chelator deferiprone, which inhibits ferroptosis in vitro [115, 116]. Deferiprone significantly reduced brain iron levels in phase II clinical trials of PD, with phase III and IV trials still ongoing. Phase II clinical trials of deferiprone are also currently underway for AD [117, 118].
Stroke is an acute disorder of blood circulation in the brain tissue caused by arteriovenous stenosis, occlusion, or rupture due to various inducing factors [119, 120]. It is also one of the most important causes of disability [121, 122]. The clinical symptoms of a stroke are language disorder and cognitive impairment [123]. Stroke can be divided into ischemic stroke and cerebral hemorrhage (ICH) stroke [124]. It has been reported that ferroptosis plays an important role in the pathophysiological processes of various cancer types [125, 126]. In recent years there has also been substantial evidence that ferroptosis is associated with the progression of stroke [127].
Ischemic stroke is characterized by the inadequate blood supply to specific areas of the brain, leading to permanent disability [128]. Insufficiency of blood in the brain activates the ischemic cascade reaction of neurons, which involves excitatory toxicity, oxidative stress, post-ischemic inflammation, and eventually cell death. Recently, ferroptosis has been found to play an important role in the development of ischemic stroke by influencing iron metabolism and lipid peroxidation, while the inhibition of ferroptosis successfully reverses ischemic damage [129]. During acute ischemia, the blood-brain barrier is destroyed and a large number of ferrous ions enter the brain parenchyma, resulting in the Fenton reaction and ferroptosis [130]. Moreover, Cui et al. [41] found that ACSL4 plays a key role in stroke. Knockdown of ACSL4 protected mice from ischemic injury by inhibiting proinflammatory cytokine production in the microglia, whereas the overexpression of ACSL4 exacerbated ischemic brain injury by promoting lipid peroxidation and facilitating neuronal death. ACSL4 is a regulator of neural death and neuroinflammation, and interfering with its expression may be a novel therapeutic approach for ischemic stroke [41]. Guan et al. [81] showed that carvacrol inhibits ferroptosis by increasing the expression of GPX4, thereby providing protection against cognitive dysfunction in ischemic-reperfusion gerbils (Table 2). These findings suggest that the inhibition of ferroptosis improves the outcome after stroke, thus offering a potentially novel treatment strategy.
ICH refers to bleeding caused by the rupture or leakage of blood vessel in the brain, the compression of brain tissue, and damage to neurons [131]. Bao et al. [132] reported that excess iron is present in the brain tissue following ICH and this iron accumulation can lead to oxidative stress and neuronal damage. The iron chelating agent DFX mitigates brain atrophy and the neurological deficits caused by ICH without causing any detectable side effects [85] (Table 2). This provides useful information for DFX-ICH clinical trials. Chen et al. [133] found that PTGS2, MDA, and COX-2 expression levels all increased post-ICH in mice and that Fer-1 treatment reduced neurological deficits, memory impairment, and brain atrophy.
The above results indicate that ferroptosis may be a potential target for the treatment of ICH, but as yet there is no clinical evidence to support the treatment of stroke with ferroptosis inhibitors.
TBI is a series of pathological changes to normal brain function caused by external physical forces [134]. It often leads to death and disability and can be divided into primary and secondary impairments [135]. Several types of programmed cell death occur during TBI pathophysiology, including necrosis, apoptosis, necroptosis, pyroptosis, and ferroptosis [136, 137]. Research has shown that TBI is followed by several characteristics of ferroptosis during the secondary impairment stage, including iron accumulation, dysfunction of iron metabolism, and up-regulation of ferroptosis-related genes [135, 82]. GPX4 activity decreases significantly at 6 h, 1 d, and 3 d after TBI, while the MDA concentration and lipid ROS levels increase significantly at 6 h, peak at 3 d, and return to baseline levels at 7 d [134, 138]. Mitochondrial shrinking was observed by transmission electron microscopy three days after TBI. Ferrostatin-1 (Fer-1) injection through the ventricle significantly reduced iron deposition and neurodegeneration, while also alleviating injury damage and improving long-term motor and cognitive performance [82] (Table 2). This strongly suggests the involvement of ferroptosis after TBI, but the precise mechanism underlying ferroptosis in TBI remains unknown [139]. Reduction of iron accumulation and inhibition of lipid peroxidation may therefore be potential treatments for TBI.
Sepsis-associated encephalopathy (SAE) can be defined as a diffuse brain dysfunction caused by an inflammatory response but without an accompanying infection of the central nervous system (CNS) [140]. Research suggests that ferroptosis is induced during sepsis and is involved in the pathogenesis of SAE [141]. In support of this, the iron concentration and GPX4 expression level are increased in the hippocampal neurons of SAE mice. Furthermore, iron chelating agents can alleviate SAE-associated cognitive dysfunction [83] (Table 2). Wei et al. [84] found that inhibition of miR-9-5p led to increased expression of transferrin receptor (TFRC) and exacerbation of SAE (Table 2). More recent research has also shown that inhibiting ferroptosis after SAE can reduce glutamatergic excitability [142]. Hence, ferroptosis may offer a new strategy for the treatment of SAE. In summary, an imbalance of the antioxidant system results in a large accumulation of lipid peroxidation. By reducing the iron concentration and the expression level of GPX4, ferroptosis can be inhibited in SAE mice, thereby improving cognitive dysfunction.
This review has outlined the development and mechanism of ferroptosis. We have also highlighted the potential role of ferroptosis in various disorders of the nervous system.
Many studies now support the contention that ferroptosis represents a potential
target for the treatment of nervous system disorders. Inhibiting ferroptosis can
effectively alleviate depression, neurodegenerative diseases, stroke, TBI, and
SAE [41, 76, 84]. For example, EDA can improve depression in CSDS mice by
upregulating GPX4 protein expression [77]. GPX4-knockout mice showed AD-like
hippocampal neurodegeneration and increased levels of lipid peroxidation, which
is a marker of ferroptosis. This could be attenuated by the ferroptosis inhibitor
Liproxstatin-1 [79]. 6-hydroxydopamine -induced ferroptosis is FTH1-mediated,
and that overexpression of FTH1 in a cell model of PD can attenuate ferroptosis
in the rat [114]. In a mouse model of stroke, ACSL4 knockout protects against
ischemic injury by inhibiting the production of microglial pro-inflammatory
cytokines [41]. In a TBI mouse model, the injection of Fer-1, a ferroptosis
inhibitor, effectively reduced iron deposition and neurodegeneration while
alleviating injury and improving long-term motor and cognitive abilities [82].
The use of an iron chelator in SAE mice reduced iron accumulation and improved
cognitive dysfunction in SAE mice [83]. Therefore, regulation of the ferroptosis
defense System xc
SAE, sepsis-associated encephalopathy; TFR1, transferrin receptors 1; GPX4, Glutathione peroxidase 4; DMT1, divalent metal transporter 1; PLOOHs, phosphColipid peroxidation; PUFA, Polyunsaturated fatty acid; ACSL4, acyl-CoA synthetase long-chain family member-4; MDA, malondialdehyde; 4-HNE, 4-hydroxynonenal; GCL, glutamate-cysteine ligase; GSS, glutathione synthetase; CoQ10, Coenzyme Q10; DHODH, Mitochondrial Dihydroorotate Dehydrogenase; CSDS, chronic social defeat stress; CNS, central nervous system; TBI, Traumatic brain injury; Fer-1, Ferrostatin-1; AD, Alzheimer’s disease; PD, Parkinson’s disease.
XZ designed an overview. ZFang provided help and advice on writing. LD wrote the manuscript. YW contributed to editorial changes in the manuscript. ZFan and XG searched a great deal of literature. YL modified the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
The authors would like to thank our mentor and colleagues.
This study was supported by National Nature Science Foundation Grant (No. 81871603) and Discipline Boost Program of the First Affiliated Hospital of Air Force Military Medical University (No. XJZT21J08).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.