
- Academic Editor
Disorders of mitochondrial function are responsible for many inherited neuromuscular and metabolic diseases. Their combination of high mortality, multi-systemic involvement, and economic burden cause devastating effects on patients and their families. Molecular diagnostic tools are becoming increasingly important in providing earlier diagnoses and guiding more precise therapeutic treatments for patients suffering from mitochondrial disorders. This review addresses fundamental molecular concepts relating to the pathogenesis of mitochondrial dysfunction and disorders. A series of short cases highlights the various clinical presentations, inheritance patterns, and pathogenic mutations in nuclear and mitochondrial genes that cause mitochondrial diseases. Graphical and tabular representations of the results are presented to guide the understanding of the important concepts related to mitochondrial molecular genetics and pathology. Emerging technology is incorporating preimplantation genetic testing for mtDNA disorders, while mitochondrial replacement shows promise in significantly decreasing the transfer of diseased mitochondrial DNA (mtDNA) to embryos. Medical professionals must maintain an in-depth understanding of the gene mutations and molecular mechanisms underlying mitochondrial disorders. Continued diagnostic advances and comprehensive management of patients with mitochondrial disorders are essential to achieve robust clinical impacts from comprehensive genomic testing. This is especially true when supported by non-genetic tests such as biochemical analysis, histochemical stains, and imaging studies. Such a multi-pronged investigation should improve the management of mitochondrial disorders by providing accurate and timely diagnoses to reduce disease burden and improve the lives of patients and their families.
Mitochondrial disorders are incurable and lead to clinical sequelae that debilitate patients and devastate families. Medical professionals must maintain an in-depth understanding of gene mutations and molecular mechanisms that cause mitochondrial disorders.
When studying molecular mechanisms of mitochondrial disorders, it is important to understand the roles of both mitochondrial and nuclear genomes in coding for mitochondrial proteins. The mitochondrial genome, composed of 16,569 double-stranded base pairs organized in a circular configuration, codes for 13 polypeptides, 22 tRNA, and 2 rRNA, totaling 37 genes [1]. About 1100 nuclear DNA (nDNA) and 37 mitochondrial DNA (mtDNA) genes code for mitochondrial components, including those in the electron transport chain (ETC), which are responsible for oxidative phosphorylation (OXPHOS). mtDNA variants represent about 15%, and nDNA variants can account for up to 85% of nDNA mitochondrial-related disorders, with incidences ranging between 1:1000 and 1:5000 [2, 3]. About one-third of mitochondrial diseases are caused by single, large-scale mtDNA deletions [4].
Fig. 1 illustrates the mitochondrial genome components, which include various
functional regions and selected locations where mutations cause
well-characterized mitochondrial disorders. The 1.1 kb D-loop contains three
mitochondrial promoters (light-strand promoter: LSP; two heavy-strand
promoters—O
 Fig. 1.
Fig. 1.Human mtDNA illustrating the coding and non-coding components in
a 16,569 bp circular DNA. The outer and inner rings comprise heavy and light
strands, respectively. The protein-coding genes encode for the complexes required
for oxidative phosphorylation complexes and are labeled Complex I–V with their
respective coding genes. Complex II is not present because it is encoded by
nuclear DNA. The promoters in the non-coding D-Loop are labeled for the L- and
H-strands (LSP and P
Fig. 2 illustrates a cloverleaf structure representing 1 of the 22 mitochondrial
tRNAs that translate the OXPHOS complexes. The tRNA acceptor stem and anticodon
bases encode the amino acid sizes and polarity, respectively. These amino acid
properties guide how the aminoacyl-tRNA synthetase acceptor site recognizes the
tRNA bases. The D-loop mainly functions as a reorganization site for
aminoacyl-tRNA synthetase, whose purpose is tRNA aminoacylation. As indicated in
Fig. 2, mutations altering tRNA bases have been implicated in various
mitochondrial disorders. For example, m.3243A
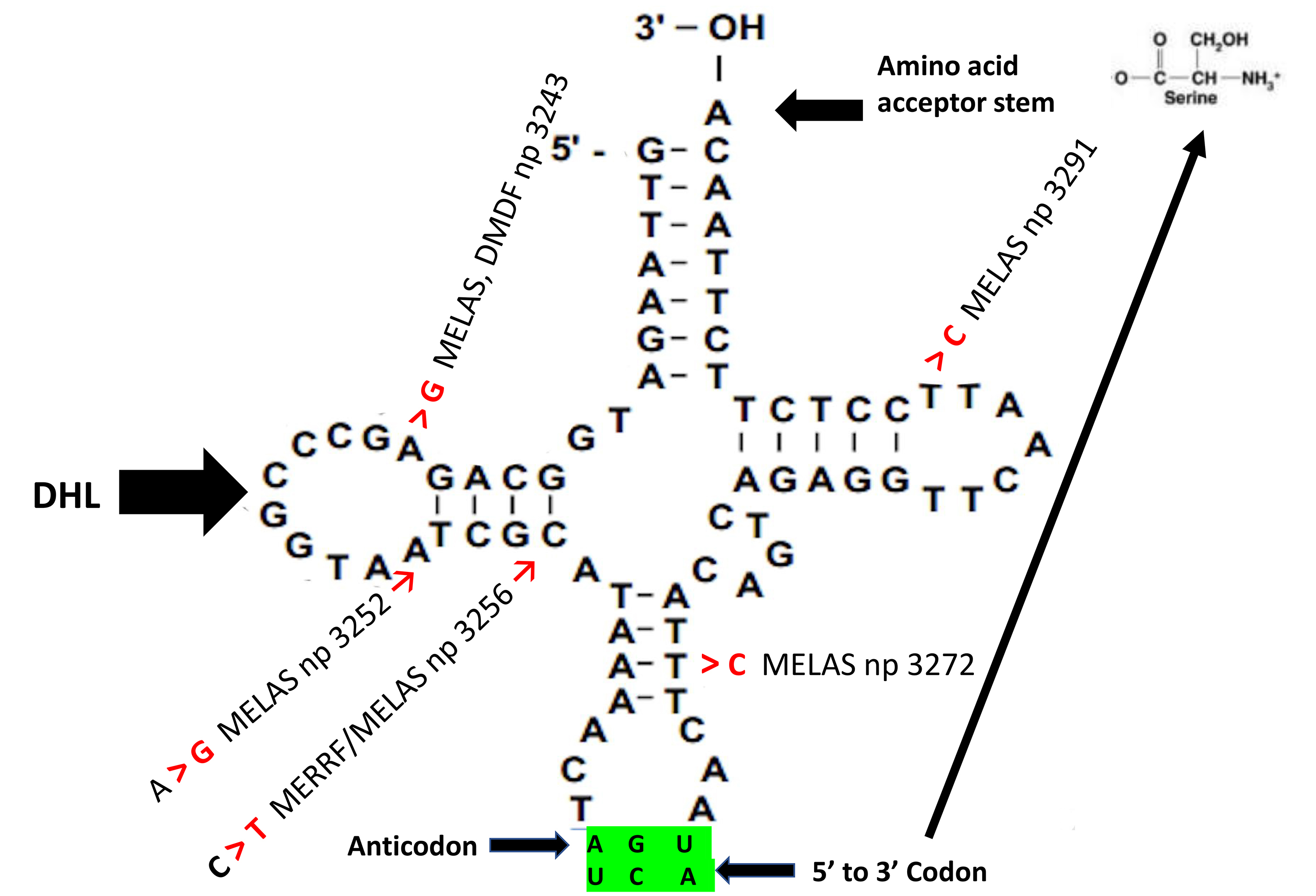 Fig. 2.
Fig. 2.Cloverleaf structure of mitochondrial tRNA
Unlike nDNA, mtDNA is genetically varied (heteroplasmic) and exists in multiple
copies per cell (2–10 mtDNA/mitochondria nucleoids and 10
Fig. 3 illustrates how heteroplasmy reflects the coexistence of wild-type and
mutant mtDNA of varying proportions in cell populations. While most mitochondrial
disorders are heteroplasmic, homoplasmy can occur when all inherited mtDNA
populations harbor a pathogenic variant. Levels of heteroplasmy required for
biochemical instability and phenotypic expression vary by disease and depend on
mutation type. For example, a heteroplasmic ratio
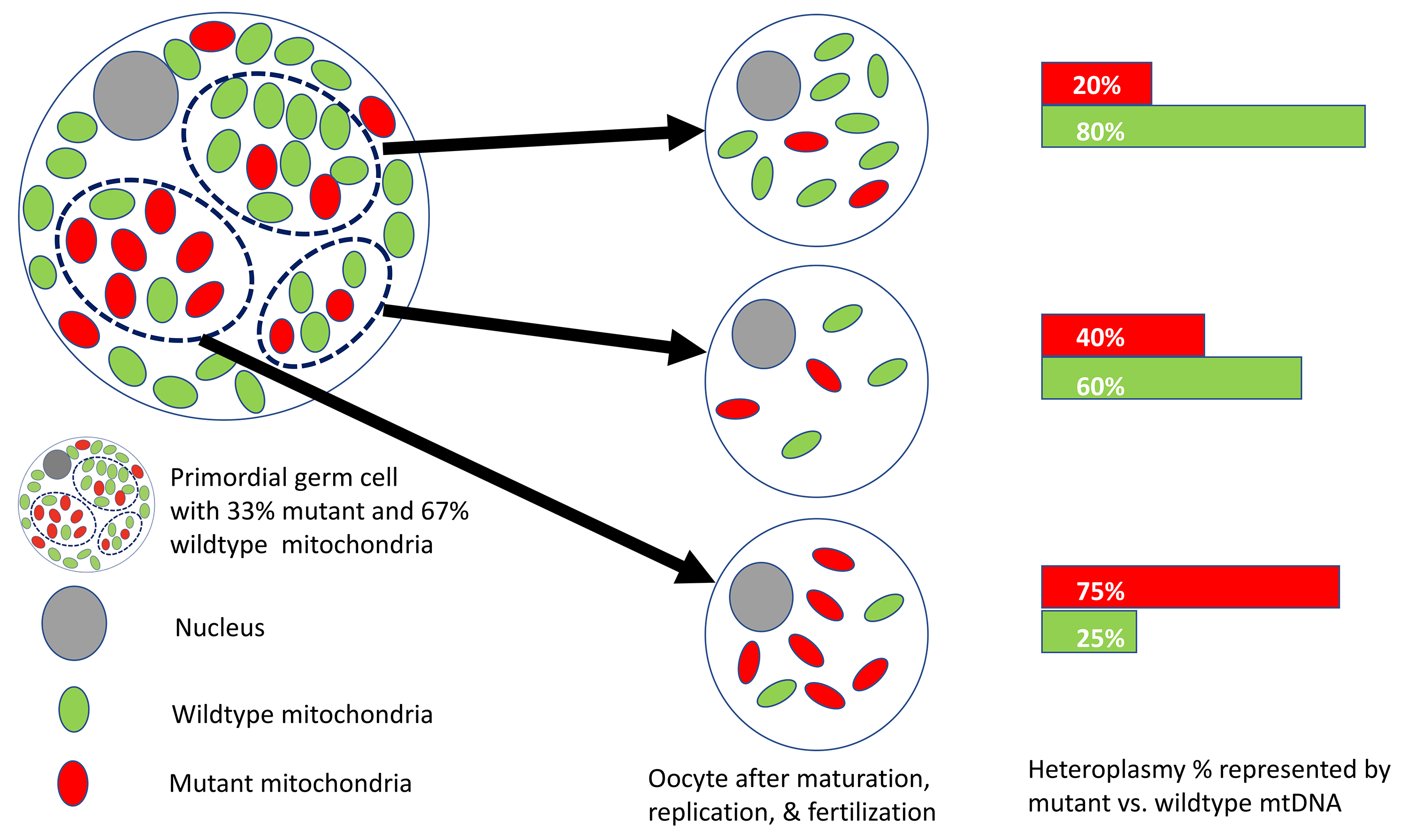 Fig. 3.
Fig. 3.Heteroplasmy illustrated at 20%, 40%, and 75% in three mature oocytes. The primordial germ cell shown is 33% heteroplasmic and can produce mature oocytes of varying heteroplasmy. Green symbols represent wild-type mitochondria, and red symbols represent mutant mitochondria in each cell. The heteroplasmic proportion is calculated as the number of mutants divided by wild-type mitochondria.
Different mitochondrial disorders may result from the same mutation, depending
on the degree of heteroplasmy. This is illustrated by heteroplasmic m.3697G
The mitochondria genetic bottleneck phenomenon can explain variable expressivity among siblings with the same mtDNA mutation [10]. This bottleneck is associated with low levels of oocyte mtDNA during early oogenesis. The mtDNA mutation profile, in a bottleneck of reduced mtDNA, is a snapshot in time that is randomly amplified after fertilization and subsequently segregated into cells representing different tissue lineages (Fig. 3). This random process lends to variable expressivity among siblings and can explain why an apparently unaffected heteroplasmic female may have affected offspring. Furthermore, interpreting heteroplasmy, in conjunction with nDNA and mtDNA variants related to mitochondrial protein disruption, often creates diagnostic odysseys in processes that include identifying molecular lesions, determining recurrence risk, and diagnosing mitochondrial disorders.
Due to the complexities distinguishing nDNA and mtDNA in health and disease, diagnosing mitochondrial disorders often involves multi-pronged approaches using molecular techniques. Whole-genome long-read mtDNA sequencing could yield additive coverage provided by next-generation sequencing, uncovering a combination of large rearrangements, point mutations, structural variants, short tandem repeat variants, and epigenetic mutations [11]. Advances in RNA sequencing techniques show promise in elucidating the etiology of mitochondrial disorders. Furthermore, RNA sequencing can elucidate the effects of tRNA genes, quantify tRNA molecules in a single muscle fiber, identify epigenetic alterations, and detect the phase of tRNA excision from gene transcripts [11].
mtDNA maintenance defects have been documented in
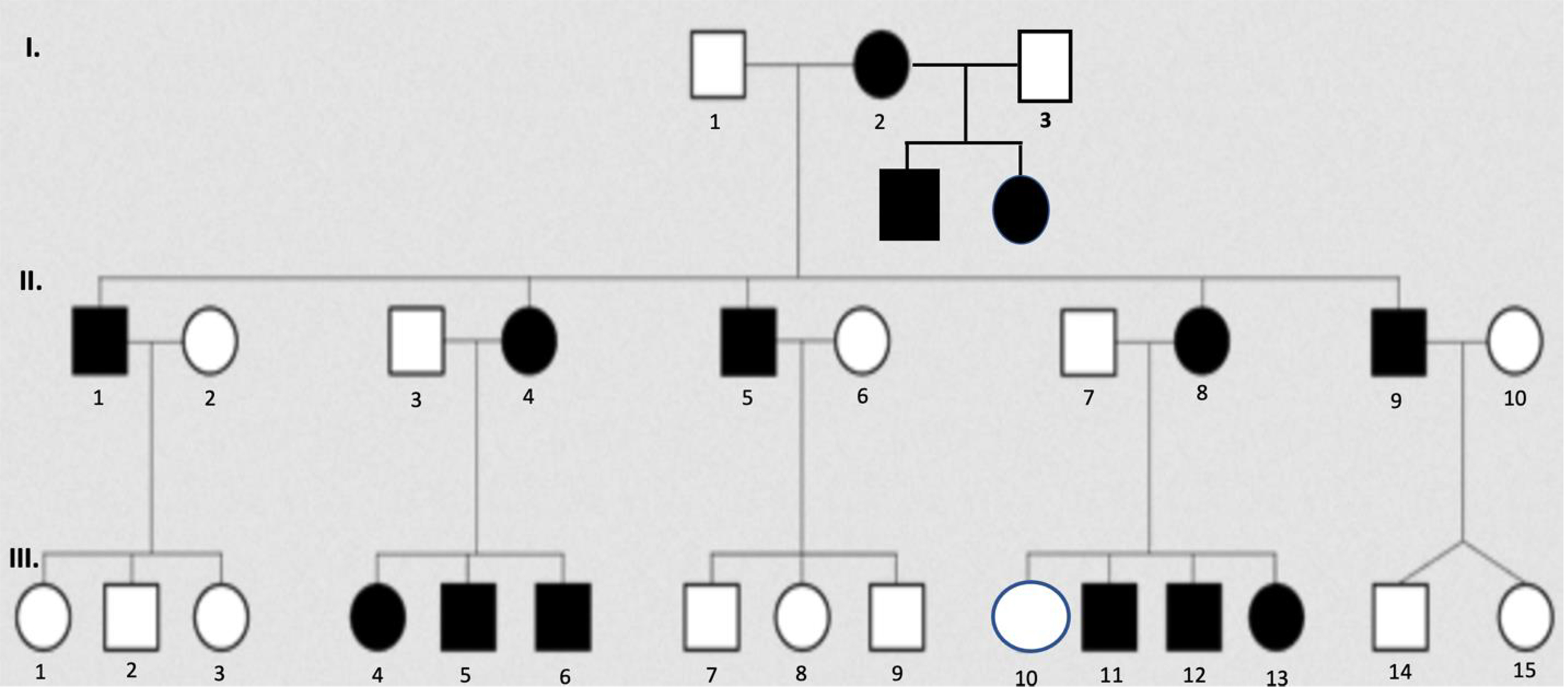 Fig. 4.
Fig. 4.Pedigree illustrates the inheritance pattern of maternal mtDNA transmission to offspring. Closed symbols represent individuals with any phenotypic feature of the disorder, and open symbols represent unaffected individuals. Squares represent males; circles represent females. Phenotypic presentations may vary depending on the degree of heteroplasmy and the nature of the mtDNA mutations. Individual III.10 may have a low mutant heteroplasmic ratio that does not cross the threshold liability for disease expression; however, she may have yet to express the phenotype, and/or her children may be affected. Note that males and females are equally affected, and transmission only occurs maternally.
More than 350 nDNA and mtDNA mutations represent multi-system disease spectrums,
sharing central themes of inadequate ATP production due to defective mitochondria
[14]. MitoCarda3.0 dataset lists
mtDNA maintenance disorders can present from infancy to early adulthood and often include phenotypes related to encephalopathy, neuropathy, myopathy, epilepsy, ataxia, ophthalmoplegia, and gastrointestinal dysmotility. Pathogenic mutations in nDNA genes coding for proteins required for mitochondrial maintenance result in disorders due to defective mtDNA synthesis, which can be quantitative (mtDNA depletion) and qualitative (mtDNA deletions) [13]. Molecular diagnostic testing identifying mtDNA depletion and/or deletions often suggests mtDNA maintenance defects. Therefore, nDNA mutations disrupting mitochondrial function alter mtDNA nucleoside transport, synthesis, expression, and/or copy number. mtDNA polymerase gamma (POLG1 and POLGA) and p55 (POLG2) control mtDNA replication [16]. Mitochondrial topoisomerase, in conjunction with nDNA TWNK, RNASEH1, and MGME1, encodes helicase, RNA polymerase, and exonuclease, respectively [17]. nDNA encodes the mitochondrial replication apparatus that directs cell cycle mtDNA replication. Specifically, in nDNA, only S-phase de novo or salvage pathways synthesize nucleotides, while mtDNA nucleotides replicate continuously and are independent of cell cycle phases.
Single gene testing is most informative when there is a pre-test probability for clinical findings and family history. In those cases, sequence analysis can identify mutation types (e.g., indels, missense, nonsense, and splice site); however, duplication analysis may be required to identify exon or whole gene deletions. mtDNA maintenance gene panels are important in limiting variants of unknown significance (VUS) that do not correlate with the phenotype. In such targeted approaches, it is imperative to investigate genes with a high likelihood of actionable findings. It is important to remember that while comprehensive genomic testing, including exome sequencing, is less stringent in selecting specific gene panels, such approaches are often diagnostically informative.
Multi-pronged approaches include history (patient and family), physical (neurologic exam), routine lab testing, biochemical studies, imaging studies (MRI, echocardiogram, and ultrasound), and molecular testing. These investigative approaches are all essential in identifying nDNA or mtDNA variant causes of mitochondrial disorders.
Cases 1–8 illustrate the most salient features of the described mitochondrial disorder. Each case follows a diagnosis, prognosis, and molecular summary (DPMS). Comprehensive discussions of gene variants are not included because they can cause various phenotypes in related mitochondrial disorders and often represent very few cases.
Case 1: A 36-year-old previously healthy female presented with painless, bilateral, pupil sparing ptosis, ophthalmoplegia, proximal myopathy, and exercise intolerance progression during the past 8 years. She demonstrated deliberate head movements to compensate for reduced ocular motility. Genetic testing revealed a 1.5 kb mtDNA deletion confined to skeletal muscle, revealing ragged red fibers (RRFs) upon biopsy (Fig. 5). She has four unaffected biological siblings.
 Fig. 5.
Fig. 5.High magnification Gomori trichrome stain of skeletal muscle micrograph showing ragged red fibers (encircled) in mitochondrial myopathy. The characteristic appearance is caused by abnormal mitochondria that progressively accumulate in the muscle fiber plasma membrane. The ragged appearance is caused by mitochondrial aggregates. Figure reference: Reproduced with permission from Wikimedia Commons; https://commons.wikimedia.org/wiki/File:Ragged_red_fibres_-_gtc_-_very_high_mag.jpg.
DPMS 1: Diagnosis: Chronic progressive ophthalmoplegia (CEPO). CEPO has
a normal life expectancy with supportive symptomatic care. Large mtDNA deletions
in oogenesis or early embryogenesis are often molecular lesions in isolated
de novo cases. Other causes of CEPO, while less common, have been
attributed to single mitochondrial rearrangements, inheritance of single
nucleotide MT-TL1 variants, as well as autosomal dominant and recessive
inheritance patterns from mutations in nDNA genes, such as POLG,
TWNK, and RRM2B. These nDNA mutations result in mtDNA deletions
in myocytes, leading to defective OXPHOS [18]. Levels of mtDNA gene variants such
as MT-TL1 m.3243A
Case 2: A 17-year-old female presented with intellectual disability, delayed puberty, progressive external ophthalmoplegia, pigmentary retinopathy, ptosis, sensory neural hearing loss (SNHL), cardiomyopathy, limb weakness, mild cerebellar ataxia, and lactic acidosis. Cerebrospinal fluid (CSF) lactate was elevated (125 mg/dL), serum creatine kinase increased, and hypoparathyroidism caused hypocalcemia. Skeletal muscle biopsy showed RRF and mitochondrial abnormalities. At 4 feet 11 inches, she is short compared to her familial constellation. However, her 12-year-old brother is in the fifth percentile for height and had unspecified eye abnormalities and developmental delays at 5 years old.
DPMS 2: Diagnosis: Kearns–Sayre syndrome (KSS). KSS usually results in
early adulthood death precipitated by cardiac conduction abnormalities. KSS
encephalomyopathy displays symptoms similar to CEPO. KSS onset is usually
Case 3: A 22-year-old male presented with acute right eye vision loss
and, 3 months later, with central vision loss due to focal retinal ganglion cell
degeneration combined with optic nerve atrophy. mtDNA analysis revealed a 70%
heteroplasmic m.11778G
DPMS 3: Diagnosis: LHON. LHON demonstrates mtDNA inheritance and
initially presents with vision loss in one eye, followed by the other. Later in
life, LHON is associated with an increased mortality risk due to stroke,
demyelinating disorders, epilepsy, atherosclerosis, alcohol-induced disorders,
and dementia [21]. Due to sex hormone differences, approximately 50% of males
and 10% of females demonstrate phenotypic manifestations. Other genetic
components or environmental factors (e.g., smoking, alcohol) may define
phenotypic expression. Ocular phenotypes can mimic autosomal dominant optic
atrophy due to OPA1 (nDNA) variants. OPA1 codes for a
mitochondrial fusion protein, cristae integrity, and apoptotic mechanisms. The
m.11778G
Case 4: A 4-year-old boy died from respiratory failure secondary to
hypertrophic cardiomyopathy (HCM) and lactic acidosis. He had a mtDNA mutation
(m.8993T
DPMS 4: Leigh syndrome (LS). LS mainly demonstrates autosomal recessive
inheritance, with
LS and NARP phenotypes have different clinical presentations; however, both can
be caused by the same mtDNA variant or by various mutations in one of
Case 5: A female who died in an automobile accident at 48 years of age with no phenotypic features of a mitochondrial disorder had given birth to three males (1M–3M) during her first marriage and three females (1F–3F) during her second marriage. All children, except for 1F, had learning disabilities before 20 years and seizures before 35 years. 1M, 2M, 2F, and 3F experienced recurrent headaches with vomiting, peripheral neuropathy, and mild to moderate muscle weakness accompanied by exercise intolerance that manifested periodically before 16 years old. 1M died of dementia eight years after his first seizure. 3M had left ventricular hypertrophy, mild hearing, and cortical vision loss, and he died at 35 years old from a myocardial infarction the same year his brother (2M) died.
Chart reviews indicated lactic academia in several offspring during various ED
visits. 1F is taller than and has no shared phenotypic features with any of her
five biological siblings/half-siblings. However, her daughter had a seizure at 5
years old, exhibited RRF on a muscle biopsy, and had high lactic acid levels.
Genetic testing reported a heteroplasmic mtDNA mutation in MT-TL1
(m.3243A
DPMS 5: Diagnosis: Mitochondrial encephalomyopathy, lactic acidosis,
and stroke-like episodes (MELAS). MELAS presents with a broad range of variable
expressivity, and
Case 6: A 14-year-old male, whose fraternal twin sister died at 5 weeks
old, was phenotypically normal until 11 years old when he had a systemic
infection and developed sporadic myoclonic seizures. The physical examination
revealed remarkable short stature and dysmorphic facial features (large ears),
bilateral optic nerve atrophy, and SNHL. Serum lactate was elevated. Other urine
and blood studies were normal; a brain MRI was normal. Genetic testing revealed a
pathogenic MT-TK mutation (m.8344A
DPMS 6: Diagnosis: myoclonic epilepsy associated with RRFs (MERFF).
Patients with MERFF typically develop normally before presenting with features
characterized by this case. While phenotypes vary, presentations usually include
myoclonus, ataxia, generalized epilepsy, and RRFs observed in the muscle biopsy
[24]. Many tRNA
Case 7: An 8-month-old female presented with anemia and hemoglobin (Hb) of 7.8 g/dL. She was admitted to ICU 2-months later with a 1-week history of emesis and fever. Laboratory testing revealed neutropenia, thrombocytopenia, anemia, elevated liver enzymes and lipase levels, and low pancreatic amylase. Bone marrow (BM) aspiration illustrated prominent myeloproliferative features and immature megakaryocytes. Ferritin, glucose, and ammonia levels were normal. After being discharged on day 20, she began experiencing increased intestinal motility. The patient was admitted again, aged 21 months, for vomiting and seizures. The laboratory results were similar to the first admission, with increased levels of B-type natriuretic peptide, cardiac troponin, vacuolization in erythroid and myeloid precursor vacuoles with BM ring sideroblasts, weak pupil reflexes, reduced pancreatic function, and abnormal occipital lobe abnormalities on MRI. The patient died from sepsis 1 month after hospitalization. nDNA NGS was negative for pathogenic or likely pathogenic variants; however, mtDNA analysis in the proband revealed an 83% heteroplasmic deletion of 4977 bp, which spanned NADH dehydrogenase, cytochrome oxidase, and Complex V.
DPMS 7: Diagnosis: Pearson syndrome. Pearson syndrome is a multi-system disorder presenting with anemia following episodic fever and emesis. Clinical phenotypes show significant variability among cases, and BM failure and death are usually associated with pancreatic insufficiency [26]. Early molecular testing maximizes supportive care and therapy, such as transfusions. Increased heteroplasmy corresponds to poor prognosis.
Case 8: A 35-year-old cachectic female with consanguineous parents died
from complications due to intestinal perforation and cirrhosis. She had two
older, unaffected siblings. Fourteen years earlier, she presented with an
18-month history of progressive abdominal pain, borborygmi, and recent onset of
diarrhea and weight loss. At that time, she had a small bowel diverticulosis,
denied dieting and drug use, and had an otherwise negative clinical history. In
subsequent years, she developed progressive ptosis, demyelinating peripheral
neuropathy, ophthalmoplegia, and hearing loss. She was pre-diabetic at 30 years
old and had elevated triglycerides and plasma lactate. Crohn’s and Whipple’s
diseases were ruled out. A brain MRI was positive for leukoencephalopathy. White
blood cell thymidine phosphorylase (TP) activity was 2%, and molecular testing
revealed a nonsense homozygous TYMP variant (p.858C
DPMS 8: Diagnosis: Mitochondrial neurogastrointestinal
encephalomyopathy (MNGIE). MNGIE is an autosomal recessive disorder caused by
Pathogenic variants in nDNA genes (e.g., NDUFA, NDUFB, ACAD9, FOXRED1, TMEM126B, ACAD9) and mtDNA genes (e.g., MT-ND1-6, MT-NDL4, NT-TL1) are usually fatal and associated with neonatal onset, lactic acidosis, encephalomyopathy, optic nerve atrophy, leukoencephalopathy, hypertrophic cardiomyopathy, and exercise intolerance, as well as conditions such as LS, MELAS, and LHON [27]. Pathogenic variants in four nDNA-encoded SDH subunits have been implicated in KSS and LS, the most common phenotypes being encephalomyopathy with rhabdomyolysis.
Mitochondrial MT-CYB and nDNA CYC1, UBCRB, BCS1L, and LYRM7 pathogenic variants often mimic glycogen storage diseases [27]. The affected patients present with hypoglycemia, lactic acidosis, elevated creatine kinase, and urine organic acidemias. MT-CYB variants often result in LHON, encephalopathy, cardiomyopathy, and myopathy [27]. BCS1L variants can cause SNHL, encephalopathy, liver failure, and tubulopathies [16, 28]. Mitochondrial MT-CO1–3 variants and several nDNA gene COX gene mutations have been implicated in various presentations, including failure to thrive, encephalopathy, hypotonia, LS, cardiac conduction defects, and hepatic failure. Common metabolic findings in these patients include aminoaciduria, anemia, decreased COX activity, glycosuria, hypercalcuria, and lactic acidosis [29]. NARP and LS can arise in patients with MT-ATP6 variants and nDNA mutations involving ATP5A1, SUMG5, ATPAF2, and TMEM70. For example, MT-ATP6 mutations may manifest as familial bilateral striatal necrosis, NARP, or LS, and mutations in MT-ATP6 are usually associated with cardiomyopathy and neuropathy [30]. nDNA ACO2 codes for aconitase. ACO2 mutations resulting in aconitase deficiency disrupt the mitochondrial conversion of citrate to isocitrate and are characterized by optic neuropathy with early childhood presentation [30]. Infantile cerebellar-retinal degeneration also results from ACO2 mutations and presents with hypotonia, metabolic acidosis, and hyperglycemia [30].
The aforementioned discussion in the context of OXPHOS defects was previously attributed to the redundant proton-centric ETC model, which included mitochondrial complexes to describe proton pumps working in conjunction with trans-membrane potentials to enable chemiosmotic rotary ATP synthesis. However, a more recent thermodynamically and kinetically valid model based on the Murburn concept was explained by Manoj [31]. The Murburn concept proposed a closed oxidation system composed of molecules, unbound ions, and radicals. This oxygen-centric model employs diffusible reactive oxygen species (DROS) that allow electron transfers outside active sites and operate through simple collision models. ADP/Pi is activated by DROS produced through oxidation of NADPH. In this context, the Murburn model allows direct coupling of substrate oxidation with the phosphorylation of ADP. In contrast to the ETC model, where oxygen remains bound to Complex IV to generate water, the Murburn model proposes that oxygen interacts with all mitochondrial complexes without relying on coenzyme Q (CoQ) and cytochrome c (cyt c) to serve as mobile carriers of electrons. Instead, CoQ and cyt c in the Murburn model serve as milieu-associated transient charged storage capacitors [31, 32]. This new insight allows interpretation with a novel perspective, wherein DROS are seen as essential intermediates of metabolism instead of merely agents of chaos.
At the initial presentation, variable expressivity and phenotypic heterogeneity often present diagnostic challenges. These obstacles are further confounded by overlapping biochemical, anthropomorphic, and systemic phenotypes observed in different mitochondrial disorders. Multi-pronged laboratory investigations accompanied by a detailed clinical history are optimal approaches in diagnosing mitochondrial disorders (Fig. 6). Algorithms include biochemical, histology, imaging, and genetic testing combinations. Biochemical tests on blood, urine, and CSF can narrow differential diagnoses of mitochondrial disorders, most of which are associated with increased blood levels of CK, lactate, and pyruvate [33]. Since lactate levels may be normal in some mitochondrial disorders, serum and CSF lactate levels should be used to support diagnoses. For example, lactate elevations are more likely to increase in LS and MELAS than CPEO and LHON; both are associated with glucose dysregulation. Organic acid analysis and amino acid profiles often provide additional diagnostic evidence. While blood testing can identify mutations causing many mitochondrial disorders, mtDNA mutations, such as complex rearrangements, may be detected only in skeletal muscle.
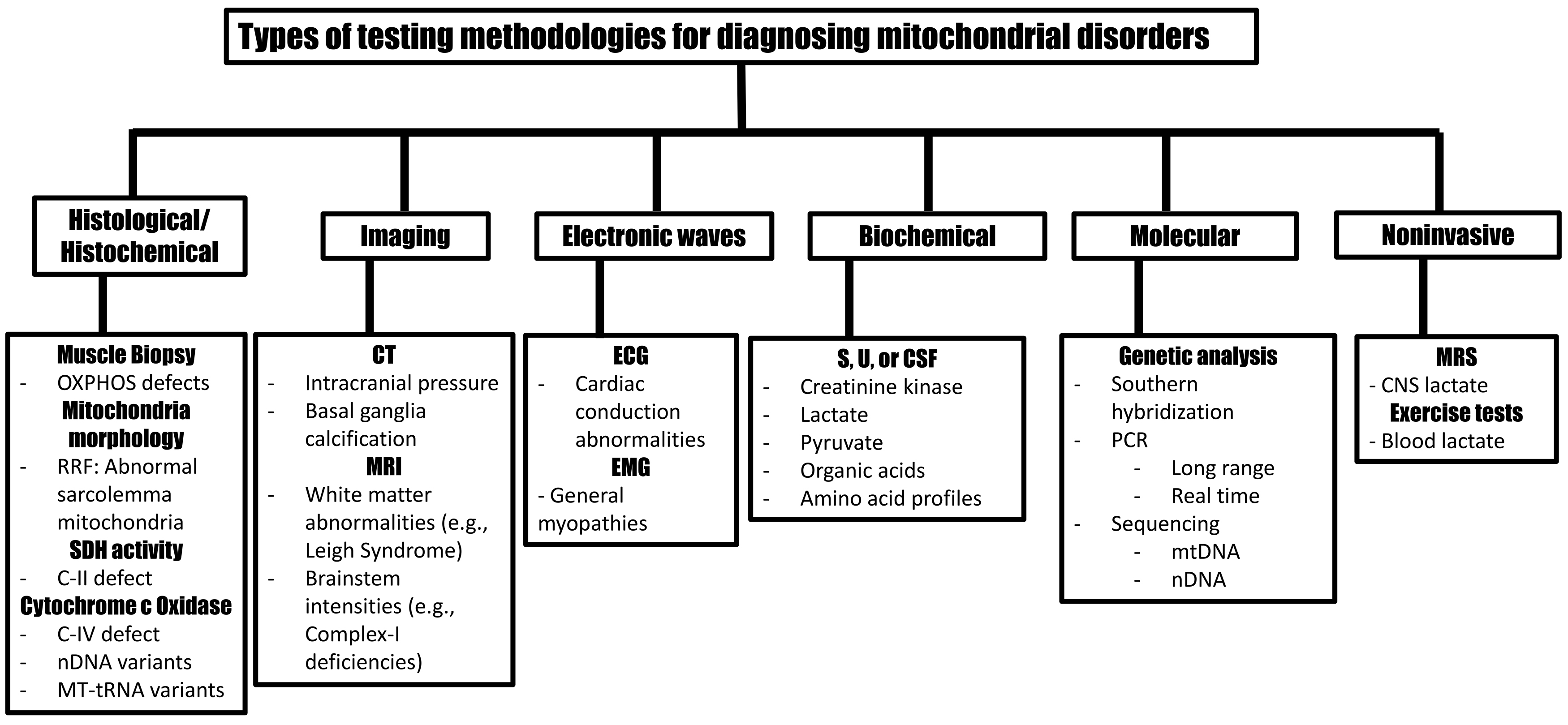 Fig. 6.
Fig. 6.Testing methodologies to support mitochondrial disorder diagnosis. OXPHOS, oxidative phosphorylation; RRF, ragged red fibers; SDH, succinate dehydrogenase; C, complex; nDNA, nuclear DNA; mtDNA, mitochondrial DNA; tRNA, transfer RNA; CT, computer tomography; MRI, magnetic resonance image; ECG, electrocardiogram; EMG, electromyography; S, serum; U, urine; CSF, cerebrospinal fluid; PCR, polymerase chain reaction; CNS, central nervous system.
Histological and histochemical features in muscle biopsies can identify OXPHOS defects and further support the diagnosis of mitochondrial disorders. For example, modified Gomorie trichome stains can reveal RRF to indicate subsarcolemmal sequestering of mitochondria [34]. SDH positivity suggests coenzyme Q oxidoreductase disorders, and COX-negative fibers, usually mosaic/heteroplasmic, imply decreased cytochrome c oxidase activity [33]. Homogenous decreases in COX activity implicate nDNA mutations, such as SURF1, while complete COX depletion suggests large deletions or mitochondrial tRNA mutations [35].
Age-related decreases in COX activity can confound diagnoses because mutated mtDNA accumulates in cells as part of cellular aging [34, 35, 36]. This clonal expansion of mtDNA is evident in tumor cells undergoing rapid proliferation and mitotic segregation. Fusion is programmed to occur between the G1 and S cell cycle phases to ensure sufficient mitochondrial replication for transmission to daughter cells [36, 37]. Normal cellular aging processes are associated with diminished mitochondrial function, and these have been especially noted in skeletal muscle and brain [34]. Accumulation of age-related mtDNA deletions, particularly cytochrome c oxidase deficiencies, is implicated in neurodegeneration and Parkinsonism [38, 39]. Further discussion on mitochondrial dysfunction related to malignancies is outside the scope of this review.
While supportive care may temporarily benefit patients with mitochondrial
disorders, treatments do not correct/cure multi-systemic disease phenotypes.
However, endurance training in these patients has been shown to improve quality
of life through increased physical abilities, and this is attributed to increased
proportions of healthy mitochondria that partially restore OXPHOS. Additionally,
ketogenic diets targeting caloric needs can improve life quality. Diets high in
fat and low in carbohydrates promote mitochondrial
Diagnostic and therapeutic interventions are in the infancy stages; however, efforts to mitigate or prevent mitochondrial disorders are promising. For example, mitochondrial genetic replacement therapy significantly decreases the transfer of mtDNA mutations to embryos. Such replacement therapy incorporates maternal nDNA and healthy donor mitochondria into the resulting embryo. Subsequently, wild-type components are introduced into the embryos, and selecting those embryos for implementation mitigates the risk of mitochondrial disorders. Preimplantation genetic testing for mtDNA disorders can identify well-characterized point mutations and is interpreted more clearly than prenatal genomic testing (PGT) [41]. PGT can be performed on chorionic villus sampling (CVS) or amniotic fluid to identify pathogenic mtDNA variants. Amniocentesis provides more homogenous samples than CVS and mitigates potential confounders related to placental mosaicism. While multiple sampling during pregnancy increases accuracy in estimating proportions of pathogenic variants, it also increases the risk of fetal loss [42]. Through further investigative studies of mtDNA disorders, treatments will continue to advance and may result in more effective management during pregnancy [16, 42].
As the accuracy of molecular testing increases with advancing technology, diagnoses and clinical management will become more precise. One problem with PCR-restriction fragment length polymorphism and Sanger sequencing is the production of false negative results due to low heteroplasmy in peripheral blood. Therefore, quantitative PCR (qPCR) is a common method for measuring mtDNA copy number. qPCR technology uses a reference gene and reports the mtDNA-to-nDNA ratio. This ratio can be biased and have poor sensitivities due to variances in PCR reactions, especially with errors compounded in target genes [43, 44, 45]. Droplet digital PCR (ddPCR) is reported to be a more sensitive and quantitative methodology that uses an oil emulsion, generating a sample made of several thousand microsphere droplets, each representing an isolated, partitioned PCR system [46]. Since thousands of individual PCR reactions reach a plateau independently, qPCR demonstrates increased efficiency because it minimizes PCR inhibitors. In this methodology, the qPCR fluoresce signal is measured using flow cytometry to count the total number of droplets and those that fluoresce. This produces a distribution ratio to quantify the mtDNA copy number [47].
While real-time PCR uses standard curves to calculate the means of multiple reactions individually, ddPCR surveys arrays of simultaneous reactions to generate more accurate quantification of nucleic acid targets. With advancing technology, ddPCR may become an economical method to measure heteroplasmy at very low analytical sensitivities accurately; however, clinical sensitivity will continue to be interpreted in the context of presenting phenotypes, disease thresholds, and actionable results.
Broad genetic heterogeneity and variable phenotypic presentations of mitochondrial disorders often present diagnostic challenges. Practices of initially performing testing on muscle biopsies have shifted to quicker NGS diagnostics using blood. NGS approaches have expedited testing for nDNA analyses and increased heteroplasmy detection in mtDNA. However, due to increased sensitivities, interpretation may become more complicated as additional VUS results are introduced in the molecular pathology report. Other considerations include pathogenic variants in mtDNA that vary in size and can be identified as single nucleotide variants, indels, or large deletions.
NGS often incorporates multi-gene panels with gene variants categorized by various clinical phenotypes. Therefore, nuclear gene panels interrogating Mendelian mitochondrial diseases have high pre-test probabilities in cases with presentations that suggest a mitochondrial disorder caused by a nuclear defect. Suspected enzyme deficiencies or mtDNA maintenance defects usually warrant targeted nuclear gene panel testing [48]. Examples include defects in OXPHOS, infants with acute liver failure, and cofactor biosynthesis defects, such as pyruvate dehydrogenase deficiency. Additional familial testing should be considered in cases associated with nDNA variants that lead to mitochondrial disease.
Maternal relatives may benefit from testing with genetic counseling and risk assessment when mtDNA variants are implicated. Various methodologies, such as real-time PCR, NGS, capillary electrophoresis, and Sanger sequencing, can test for mtDNA single-nucleotide variants. Indeed, variants detected by one of these methods should be confirmed by an alternate method to identify false positives, which some polymorphisms may cause. In addition to real-time PCR and NGS, rearrangements can be identified using long-range PCR and Southern blotting. The whole mitochondrial genome may be interrogated using whole genome sequencing (WGS), while the diagnostic quality depends on the assay’s ability to maintain adequate read depth throughout the mitochondrial genome. Thus, since nuclear-encoded mitochondrial DNA sequences can lead to false positive results, an alternate method should be used for confirmation [49].
The American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) provide updated, peer-reviewed guidelines for nDNA variant calling. ACMG and AMP also guide mtDNA variant interpretation [50]. MITOMAP is a mtDNA comprehensive tool that is routinely updated with information on loci, phenotypes, GenBank frequencies, and publication links [51]. With continued advancements in testing methodologies and confirmation of variant calls, a more robust and accurate interpretation process may allow the reclassification of previous results from ‘VUS’ to ‘likely pathogenic’ or ‘pathogenic’. This is especially evident when used with clinical presentation, family history, and clinical practice guidelines. When evaluated in the context of phased testing, molecular diagnostic results can strengthen the evidence for pathogenicity, especially in cases with high pre-test probabilities [36].
Phased testing may include initial genetic testing followed by genome sequencing
that targets well-known variants in nDNA and mtDNA. If NGS is inconclusive,
biopsies from post-mitotic tissue, such as muscle, can be employed for
biochemical, histochemical, and/or further genetic testing that could identify
deletions and rearrangements. This approach is useful in detecting mtDNA copy
number in mtDNA depletion syndromes when suspected nDNA mutations are not
identified. Interpretation can be confounded if age-matched normal control
samples are not used for reference range comparisons [52]. Additional
considerations could include parent–child trios for cases with high pre-test
probabilities that yielded previous negative test results. Genomic testing can
generate false negative results when molecular lesions include del/dups that are
Clinical applications in diagnosing mitochondrial disorders continue to advance with advances in genomic and transcriptomics. Whole-genome sequencing currently provides analysis of both nDNA and mtDNA in blood. Long-read technology advances are promising for identifying point mutations and rearrangements in mtDNA, identifying phasing and new variant types in nDNA, and identifying epigenetic variations [11].
In addition to databases that support diagnostics of mitochondrial disorders, machine learning is emerging as a tool to predict pathology associated with mitochondrial dysfunction. While machine learning and artificial intelligence (AI) are in the infancy stages for diagnosing mitochondrial disorders, advances have been made in identifying systemic mitochondrial dysfunction in predicting the development of Alzheimer’s disease (AD). Proteome analysis of the AD brain demonstrates dysregulated mitochondrial Complex V [54]. Specifically, Finney et al. [55] demonstrated that AI can identify dysregulation in genes responsible for OXPHOS and ATP synthesis associated with dysfunctional mitochondrial ribosomes in the frontal cortex and cerebellum in patients before the onset of AD. Through machine learning investigations, two Complex V genes (ATP5L and ATP5H) have been implicated in a common pathway that codes for Complex V components [55, 56]. These AI studies are beginning to demonstrate that altered gene pathways can lead to targeted mitochondrial dysfunction that can be implicated in disease processes.
Mitochondria homeostasis is essential for cellular metabolism, and dysregulation of homeostasis through disruption of mtDNA or nDNA genes encoding for mitochondrial function can be associated with cancer [57]. Emerging studies continue to illustrate how mtDNA mutations can serve therapeutic and prognostic roles in managing some cancers [58, 59]. These studies propose that mtDNA and nDNA genes may communicate with each other to maintain cellular homeostasis. Networks modulating mitochondrial functions in cancers are evident at the DNA (mtDNA SNPs and mutations), RNA, and protein levels (mtDNA and nDNA encoded genes). Advancing molecular technologies continues to elucidate functional roles for target genes. Outcomes from this technology are being translated into novel findings in areas of omics (transcriptomics, proteomics, and metabolomics). Hopefully, technical and clinical integration in these areas will further reveal how knowledge of molecular pathways can be leveraged into novel diagnostics and treatments.
The combination of salient features for the two-genome (mtDNA and nDNA) portrayal and maternal genomic landscape lends to the unique specificity and complexities of mitochondrial disorders in the context of mutations that disrupt mitochondrial function and ATP production. As illustrated by the eight case presentations and in contrast to most non-mitochondrial disorders, mutations associated with mitochondrial alterations often lead to various phenotypic and systemic manifestations. Defects in mitochondrial energy production can be measured in biochemical deviations, such as lactate/pyruvate ratios, ketone body production, altered amino acid status, discordant TCA cycle intermediates, and biochemical examination of tissue enzymes. Because DROS should no longer be interpreted as only pathological manifestations (per Murburn insights), the correlation between mutations in mitochondrial proteins and milieu DROS dynamics is important to understand the normal and pathological functions better. While more accurate biochemical diagnoses may direct better therapy, newly identified variants should be meticulously curated for safe clinical patient management.
While mitochondrial diseases remain incurable, evolving robust visual analyses of the mitochondrial genome, various phenotypes continue to elucidate more granular disease classifications. Clinical sequelae from mitochondrial disorders debilitate patients and devastate families. Therefore, novel, multi-pronged investigational approaches, diagnostic advancements, and robust management of patients with mitochondrial disorders are essential to improve clinical outcomes of comprehensive genomic testing and non-genetic assessments using biochemical analyses, histochemical stains, and imaging studies. In the context of the cases presented in this review, advances in RNA sequencing will continue to improve functional molecular diagnostics in the most challenging cases. Furthermore, such cases could also benefit from confirmatory methods that may employ NGS, tRNA methodology, and machine learning through advances in AI technology.
These advancements, in conjunction with a deeper molecular understanding of the two-genomic landscape, will improve the management of mitochondrial disorders by providing accurate and timely diagnoses to reduce disease burden and improve the lives of patients and their families.
JA contributed to the concept, design, acquisition, analysis, interpretation, drafting of the manuscript, ethical considerations, and final approval of the submitted document, and was accountable for the accuracy of this investigation. JA contributed to editorial changes in the manuscript.
Not applicable.
I am grateful to Dr. Frank Wians for his support in preparation of Fig. 6.
This research received no external funding.
The author declares no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.