
- Academic Editor
Background: Metabolic disorders, including obesity, are often
accompanied by an increased risk of cardiovascular complications. Monocytes are
the common link between obesity and cardiovascular diseases (CVDs). The bias of
innate cellular immunity towards pro-inflammatory activation stimulates the
development of diseases associated with chronic inflammation, in particular
metabolic disorders, including obesity, as well as CVDs. Disorders in the
functional state of monocytes and activation of inflammation may be associated
with mitochondrial dysfunction. Mutations accumulating in mitochondrial DNA with
age may lead to mitochondrial dysfunction and may be considered a potential
marker for developing chronic inflammatory diseases. Methods: The
present study aimed to study the relationship between mitochondrial heteroplasmy
in CD14
Cardiovascular diseases (CVDs) remain the leading cause of disability and mortality worldwide [1]. Cardiovascular events are complications in many pathological conditions, including metabolic disorders [2]. The impaired lipid and carbohydrate metabolisms in obesity lead to hyperinsulinemia, insulin resistance, increased cholesterol levels, hormonal regulation disorders, and chronic systemic inflammation, which are important pathogenetic mechanisms in developing diabetes mellitus and atherosclerosis underlying the development of CVD [3]. In the development of obesity and CVD, an important role is assigned to cells of the immune system, particularly mononuclear phagocytes, including monocytes, which are key cells in the development of inflammatory processes associated with metabolic and cardiovascular disorders [4]. Circulating blood monocytes are transformed into tissue macrophages that secrete pro-inflammatory molecules and phagocytize lipids, forming foam cells, an important stage in chronic inflammation and the formation of atherosclerotic lesions in the vascular wall [5].
Currently, mitochondrial dysfunction is considered a possible factor in the disturbance of the functional status of monocytes, contributing to the pro-inflammatory activation of monocytes and the development of metabolic disorders, particularly obesity and CVD [6]. Mitochondria are involved in reactive oxygen species (ROS) production, glucose-derived pyruvate degradation, fatty acid oxidation, signaling pathway regulation, and immune cell differentiation, thus regulating various biological processes [7, 8]. Numerous studies have demonstrated the role mitochondrial DNA (mtDNA) mutations play in the development of mitochondrial dysfunction [6]. mtDNA polymorphisms are associated with certain metabolic diseases. For example, mitochondrial variants MT-16320 are associated with blood glucose levels, MT-8706 and MT-8898 with waist–hip ratio, and MT-8414 and MT-16189 polymorphisms may increase the risk of type 2 diabetes [9]. As a result of the conducted studies on the complete mtDNA genome of patients with CVD, 11 heterogeneous “hot spots” were identified, which cover the main genes, such as nicotinamide adenine dinucleotide (NADH) dehydrogenase (ND) subunits 1, 4, 5, and 6, cytochrome C oxidase I (CoI), 16S rRNA, and the D-loop [10]. Mutations in the ND and CoI genes directly affect adenosine triphosphate (ATP) production. In addition, mtDNA mutations can affect the processes of mitochondrial autophagy, leading to unstable mitochondrial division and an increased risk of myocardial infarction [11].
Previous studies have demonstrated variants of mtDNA heteroplasmy associated
with atherosclerosis and CVD, particularly mitochondrial mutations A11467G,
576insC, and 10958ins [12, 13]. At the same time, the single-nucleotide variation
(SNV) A1811G was identified as anti-atherogenic since the A1811G heteroplasmy
level was higher in the control group than in patients with atherosclerosis [14].
This study aimed to investigate the level of heteroplasmic mtDNA mutations in
CD14
The present study included 22 patients (9 men and 13 women) aged 50 to 70 years
with a high body mass index (BMI)
| Control | Group of patients with obesity and CHD | Significance, p | |
| Age, years | 58.1 (3.5) | 61.2 (4.6) | |
| Gender, w/m | 10/12 | 9/13 | |
| BMI, kg/m |
23.3 (4.1) | 39.6 (5.4) | |
| Arterial hypertension, % | 23% | 91% | |
| Statin administration, % | 32% | 86% | |
| Glucose, mmol/L | 5.0 (0.6) | 5.1 (0.5) | |
| Total cholesterol, mmol/L | 5.2 (0.6) | 5.8 (0.8) | |
| HDL, mmol/L | 1.5 (0.8) | 1.34 (0.4) | |
| LDL, mmol/L | 2.3 (1.5) | 2.5 (1.3) | |
| Triglycerides, mmol/L | 3.2 (1.8) | 2.4 (1.0) | |
| cIMT, mm | 0.7 (0.2) | 0.8 (0.1) |
CHD, coronary heart disease; BMI, body mass index; HDL, high-density lipoprotein cholesterol; LDL, low-density lipoprotein cholesterol; cIMT, thickness of the intima–medial layer of the common carotid artery.
A total of 9 mL of whole blood was collected in a vacutainer containing
K
Total DNA was isolated according to a standard protocol using phenol–chloroform
extraction [17]. The concentration and quality of the obtained DNA from
CD14
The following mitochondrial genome mutations associated with mitochondrial dysfunction were selected for PCR analysis (Table 2). These mutations were analyzed by digital PCR. To measure the level of relative DNA expression, primers specific to the transcripts of the corresponding genes were selected using Primer-BLAST (National Center for Biotechnology Information, NIH, Bethesda, MD, USA). Primers were ordered from Evrogen (Evrogen, Moscow, Russia). Currently, the primer sequences are at the patent stage, meaning they cannot be published in Open Access. The level of mtDNA heteroplasmy for the selected mutations was determined by digital PCR (QIAcuity Eight, Qiagen, Hilden, Germany). The QuantiFast SYBR® Green RT-PCR kit (Qiagen, Hilden, Germany) was used to amplify the reaction mixture. The reaction was carried out according to the manufacturer’s protocol. The level of mtDNA heteroplasmy was calculated as a percentage of the mutant mitochondrial DNA templates relative to the total amount of mitochondrial DNA templates [18, 19].
| Mutation | Localization | Mutation characteristics | Exerted effect |
| 576insC | Region D-loops | Cytosine insertion (C) | Reading frame shift |
| A1811G | rRNA–16S | Change adenine (A) to guanine (G) | Impaired translation process |
| 10958ins | ND4—NADH dehydrogenase subunit 4 | Insertion of adenine–thymine–cytosine–adenine (ATCA) | Impaired respiratory chain complex I function |
| A11467G | ND4—NADH dehydrogenase subunit 4 | Change adenine (A) to guanine (G) | Impaired respiratory chain complex I function |
Statistical analysis was performed using the R project software (version
2023.03.1+446, R Foundation for Statistical Computing, Vienna, Austria) for
statistical computing. Cochran’s equation was used to estimate sample size. The
Shapiro–Wilk criterion was applied to check the normality of the distribution of
quantitative features. Methods of variational statistics, such as the mean (M)
and standard deviation (SD), were used for the analysis. The difference between
groups was assessed using the Mann–Whitney analysis. Differences were considered
significant at p
The level of the studied variants of mtDNA heteroplasmy in the monocytes of
study participants is presented in Table 3. The level of the SNV A11467G was
significantly higher in the control group than in the study participants with
obesity and CHD (p
| mtDNA heteroplasmy, % | Control | Group of patients with obesity and CHD | Significance, p |
| A11467G | 53 (8) | 44 (14) | |
| 576insC | 37 (6) | 78 (23) | |
| A1811G | 40 (22) | 48 (18) | |
| 10958ins | 52 (17) | 59 (25) |
*statistically significant differences compared to the control group p
The data are presented as mean and standard deviation (mean (SD)).
A statistical analysis of the relationship between the studied variants of mtDNA heteroplasmy and traditional CVD risk factors, in particular, age, BMI, blood lipid profile indicators, and cIMT, was carried out. Fig. 1 demonstrates the result of the relationship using Spearman’s rank correlation coefficient between the levels of heteroplasmic mtDNA variants and the age, BMI, total cholesterol, HDL, LDL, and mean cIMT of the study participants.
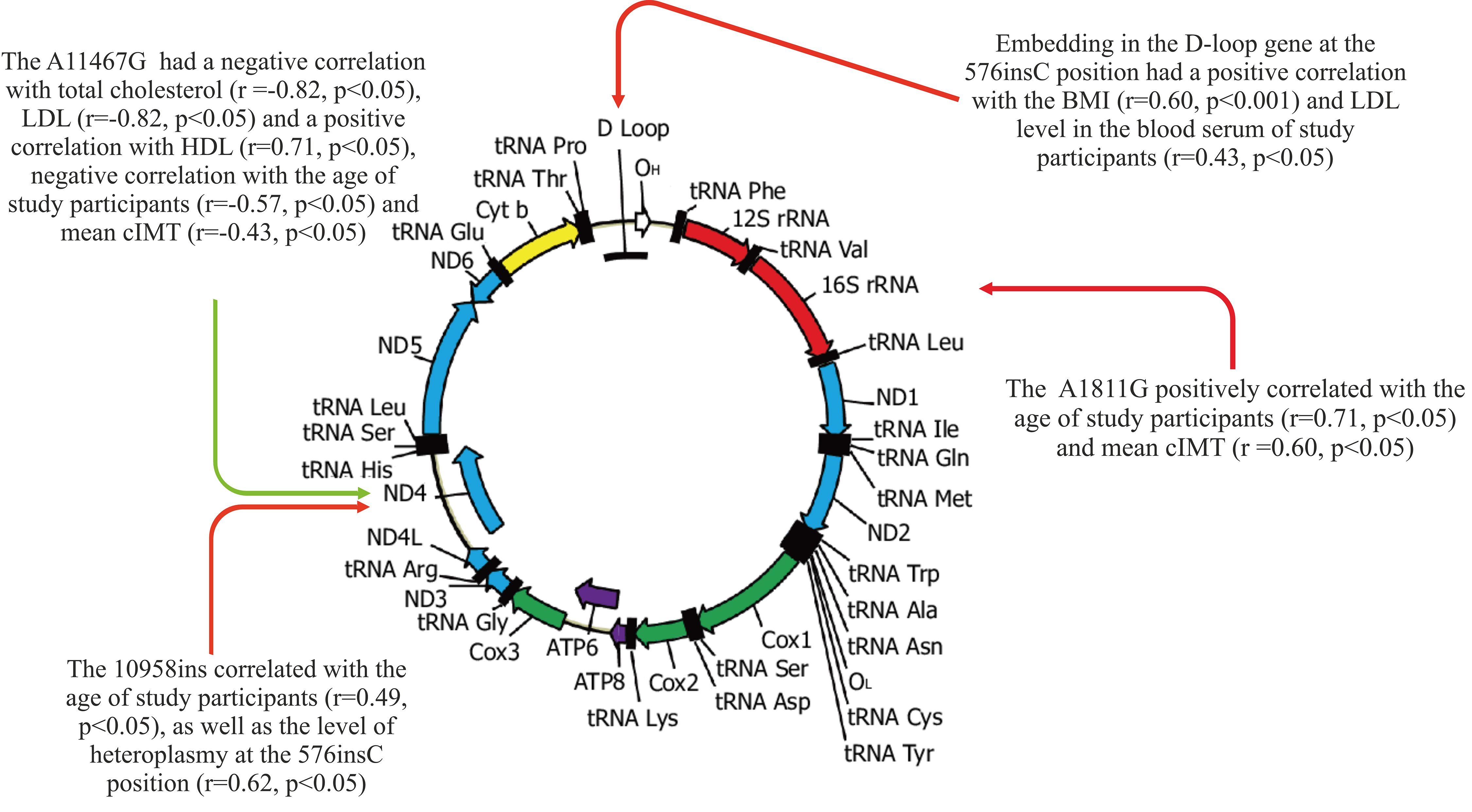 Fig. 1.
Fig. 1.The relationship between the studied mtDNA mutations and traditional CVD risk factors. CVD, cardiovascular disease.
The results of the correlation analysis showed that the A11467G mutation in the
ND4 gene had a negative correlation with total cholesterol (r = –0.82,
p
Embedding in the D-loop gene at the 576insC position had a positive correlation
with the BMI (r = 0.60, p
A significant relationship was revealed between the mitochondrial mutation
10958ins, which is located in the ND4 gene, and the age of the study
participants (r = 0.49, p
Multivariable logistic regression analysis was conducted to reveal the potential contribution of independent variables in the development of obesity and CHD. It was demonstrated that the heteroplasmy level of the mitochondrial genome variant 576insC is a significant predictor of CHD (odds ratio: 1.93; 95% confidence interval: 1.50–2.62).
In this study, mtDNA heteroplasmic variants associated with CHD and obesity were
identified for the first time in the CD14
Since mtDNA encodes several proteins involved in the functioning of the respiratory chain, mutations in mtDNA can disrupt the functioning of the respiratory chain, which is the process of energy production in mitochondria [22]. This can lead to an increase in the formation of ROS and a change in the metabolism of glucose and lipids. Changes in metabolism can contribute to the development of atherosclerosis, as elevated lipid levels and oxidative stress lead to cell damage, inflammation, and the formation of atherosclerotic plaques [23]. For example, it was previously found that mitochondrial mutations A1811G and G9477A correlate with the degree of pro-inflammatory activation of monocytes [24]. Activation of monocytes may be associated with various immunological responses and inflammatory processes. This study showed that the A1811G mutation in the ribosomal RNA gene rRNA–16S positively correlates with the age of study participants and cIMT, confirming the important role of immuno-inflammatory mechanisms in atherogenesis.
Previous studies have identified many mtDNA mutations associated with the development of CHD [25]. Some studies confirm the possible involvement of mtDNA mutations in the development of atherosclerosis [26]. These mutations can be classified into several main categories based on different mechanisms of action. The first category includes mutations in transport RNAs (tRNAs) that lead to the destabilization of base pairing and a change in the secondary structure of tRNAs, which can lead to accelerated degradation of tRNAs and a subsequent decrease in the level of mitochondrial proteins [27]. The second category includes mutations in the components of phosphorylating oxidation (OXPHOS), resulting in a reduction in ATP synthesis and an increase in the formation of ROS, which in turn contributes to the dysfunction of energy metabolism and mitochondrial damage. The third category involves mutations in the mtDNA D-loop, which can disrupt the normal replication process and decrease the number of mtDNA copies [28]. However, some studies have shown that mtDNA heteroplasmy can have a protective effect since various mutations in mtDNA can lead to intergenomic competition and intergenomic complementary interaction. Most of the detected mtDNA mutations are non-pathogenic. However, the combination of various factors, including genetic and environmental, can contribute to the development of clinical manifestations of the disease [25].
The mtDNA heteroplasmic variants identified in this study, associated with traditional CVD risk factors in CHD and obesity, can be used to develop personalized therapeutic strategies in patients with high cardiovascular risk against the background of metabolic disorders. Mitochondrial genome editing may be considered a possible therapeutic approach for atherosclerosis and CVD in the future [29]. Moreover, the purpose of such an intervention may be both the removal of genetic variants in the mitochondrial genome associated with pathology and the introduction of potentially beneficial changes in the functioning of mitochondria. Thus, the SNV A11467G in the ND4 gene can be considered a potential target for editing the mitochondrial genome in the treatment of CVD since an increase in the level of A11467G heteroplasmy is associated with positive changes in a number of traditional CVD risk factors. The study of the main molecular mechanisms in each specific mtDNA mutation will help to develop more accessible diagnostic tools for the timely detection of patients with high risk of atherosclerosis and the prevention of metabolic disorders and concomitant CVD. However, the widespread and successful use of mtDNA decoding methods in clinical practice requires a standardized approach to interpreting mtDNA variability and the level of heteroplasmy, which is the subject of further research.
Data are available from the corresponding author upon request.
AMM and VAK designed the research study. AIB, TVT performed the research. TVK and YVM analyzed the data. TVT and TVK wrote the manuscript. TVK and VAK revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This work was performed in accordance with the Declaration of Helsinki of 1975 and its revised version of 2013. The study was approved by the Local Ethics Committee of the Petrovsky National Research Centre of Surgery at December, 13, 2022 (approval number # 3).
Not applicable.
This work was supported by the Ministry of Science and Higher Education of the Russian Federation—the state task of Petrovsky National Research Centre of Surgery (Project # 123030700026-8).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.